Glycogen Synthase Kinase 3β in Cancer Biology and Treatment


Abstract
1. GSK3β Biology in Normal Cells and Disease
2. Overview of GSK3β Biology in Cancer
3. Tumor-Promoting Roles of GSK3β in Various Cancer Types
4. Aberrant GSK3β and the Hallmark Properties of Cancer
4.1. GSK3β and Tumor Cell Survival, Evasion of Apoptosis and Proliferation
4.2. GSK3β and Tumor Invasion
4.3. GSK3β and Therapy Resistance
4.4. GSK3β, Cancer Stem Cells and the “Stemness” Phenotype
5. Protection of Normal Cells during Cancer Therapy by Targeting GSK3β
5.1. GSK3β and Cancer Immunotherapy
5.2. GSK3β and Cancer Therapy-Induced Hematotoxicity
5.3. GSK3β and Therapy-Induced Central and Peripheral Neuropathy
5.4. GSK3β and Opioid-Induced Analgesic Tolerance and Withdrawal Syndrome
5.5. GSK3β and Normal Tissue Damage Associated with Surgery for Cancer
6. Future Perspectives on GSK3β in Cancer Treatment
6.1. GSK3β and the Regulation of Immune Checkpoints in Cancer
6.2. GSK3β and the Regulation of IL-17/Th17 Immunity
6.3. GSK3β and the Therapeutic Targeting of K-Ras Mutant Tumors
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 4EBP1 | eukaryotic translation initiation factor 4E-binding protein 1 |
| 5-FU | 5-fluorouracil |
| ACNU | nimustine hydrochloride |
| AMP | adenosine monophosphate |
| AMPK | AMP-activated protein kinase |
| Ara-C | cytosine arabinoside |
| ARHGAP | Rho GTPase-activating protein |
| ATRA | all-trans retinoic acid |
| Bax | Bcl-2-assosicated X protein |
| Bcl-2 | B-cell lymphoma 2 |
| Bmi1 | B cell-specific Moloney murine leukemia virus integration site 1 |
| CDK | cyclin-dependent kinase |
| C/EBPα | CCAAT/enhancer binding protein α |
| CLOVA | combined cimetidine, lithium chloride, olanzapine and valproate regimen |
| c-Myb | avian myeloblastosis virus oncogene cellular homolog |
| c-Myc | cellular myelocytomatosis |
| CXCR4 | C-X-C chemokine receptor type 4 |
| eIF4E | eukaryotic initiation factor-4E |
| ESCC | esophageal SCC |
| FAK | focal adhesion kinase |
| FasL | Fas ligand |
| FKHR | Forkhead in rhabdomyosarcoma |
| FOLFOX | combined folate, 5-fluorouracil and oxaliplatin regimen |
| FOXO | Forkhead box O |
| Gli | glioma-associated oncogene homologue |
| GSK3β | glycogen synthase kinase 3β |
| GTP | guanine triphosphate |
| HCC | hepatocellular carcinoma |
| hnRNPA1 | heterogeneous nuclear ribonucleoprotein A1 |
| HNSCC | head and neck SCC |
| HOX | homeobox |
| HPV | human papillomavirus |
| HSP | heat shock protein |
| hTERT | human telomerase reverse transcriptase |
| IRS-1 | insulin receptor substrate 1 |
| JNK | c-jun N-terminal kinase |
| KDM1A | lysine-specific demethylase 1A |
| LKB1 | liver kinase B1 |
| MAPK | mitogen-activated protein kinase |
| Mdm | mouse double minute |
| MMP-2 | matrix metalloproteinase-2 |
| mTOR | mammalian target of rapamycin |
| NFAT | nuclear factor of activated T cells |
| NF-κB | nuclear factor kappa B |
| NSCLC | non-small-cell lung cancer |
| PAX3 | paired box 3 |
| PI3K | phosphoinositide 3-kinase |
| PKC | protein kinase C |
| PTEN | phosphatase and tensin homolog deleted from chromosome 10 |
| Pyk2 | proline-rich tyrosine kinase 2 |
| Rac1 | RAS-related C3 botulinus toxin substrate 1 |
| RAR | retinoic acid receptor |
| Rb | retinoblastoma |
| Rho | Ras homologous |
| SCC | squamous cell carcinoma |
| Src | Rous sarcoma oncogene cellular homolog |
| STAT3 | signal transducer and activator of transcription 3 |
| T-BET | T-box protein expressed in T cells |
| TFEB | transcription factor EB |
| TLR | Toll-like receptor |
| TNF | tumor necrosis factor |
| TP53INP1 | tumor protein p53-inducible nuclear protein 1 |
| TRAIL | TNF-related apoptosis-inducing ligand |
| VDR | vitamin D receptor |
| WASP | Wiskott–Aldrich syndrome protein |
| WAVE2 | WASP-family verprolin homologous protein 2 |
| XIAP | X-linked inhibitor of apoptosis protein |
References
- Xu, C.; Kim, N.G.; Gumbiner, B.M. Regulation of protein stability by GSK3 mediated phosphorylation. Cell Cycle 2009, 8, 4032–4039. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Wang, J.Y.; Gao, L.M.; Yin, X.F.; Liu, L. Identification and analysis of glycogen synthase kinase 3 β1 interactome. Cell Biol. Int. 2013, 37, 768–779. [Google Scholar] [CrossRef]
- Cormier, K.W.; Woodgett, J.R. Recent advances in understanding the cellular roles of GSK-3. F1000Res 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.; Woodgett, J.R. Glycogen synthase kinase 3: A kinase for all pathways? Curr. Top. Dev. Biol. 2017, 123, 277–302. [Google Scholar] [CrossRef] [PubMed]
- Amar, S.; Belmaker, R.H.; Agam, G. The possible involvement of glycogen synthase kinase-3 (GSK-3) in diabetes, cancer and central nervous system diseases. Curr. Pharm. Des. 2011, 17, 2264–2277. [Google Scholar] [CrossRef] [PubMed]
- Takahashi-Yanaga, F. Activator or inhibitor? GSK-3 as a new drug target. Biochem. Pharmacol. 2013, 86, 191–199. [Google Scholar] [CrossRef]
- Beurel, E.; Grieco, S.F.; Jope, R.S. Glycogen synthase kinase-3 (GSK3): Regulation, actions, and diseases. Pharmacol. Ther. 2015, 148, 114–131. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Cocco, L. GSK-3 signaling in health. Adv. Biol. Regul. 2017, 65, 1–4. [Google Scholar] [CrossRef]
- Khan, I.; Tantray, M.A.; Alam, M.S.; Hamid, H. Natural and synthetic bioactive inhibitors of glycogen synthase kinase. Eur. J. Med. Chem. 2017, 125, 464–477. [Google Scholar] [CrossRef]
- Palomo, V.; Martinez, A. Glycogen synthase kinase 3 (GSK-3) inhibitors: A patent update (2014–2015). Expert Opin. Ther. Pat. 2017, 27, 657–666. [Google Scholar] [CrossRef]
- Saraswati, A.P.; Ali Hussaini, S.M.; Krishna, N.H.; Babu, B.N.; Kamal, A. Glycogen synthase kinase-3 and its inhibitors: Potential target for various therapeutic conditions. Eur. J. Med. Chem. 2018, 144, 843–858. [Google Scholar] [CrossRef] [PubMed]
- Nagini, S.; Sophia, J.; Mishra, R. Glycogen synthase kinases: Moonlighting proteins with theranostic potential in cancer. Semin. Cancer Biol. 2019, 56, 25–36. [Google Scholar] [CrossRef]
- Luo, J. Glycogen synthase kinase 3β (GSK3β) in tumorigenesis and cancer chemotherapy. Cancer Lett. 2009, 273, 194–200. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Davis, N.M.; Abrams, S.L.; Montalto, G.; Cervello, M.; Basecke, J.; Libra, M.; Nicoletti, F.; Cocco, L.; Martelli, A.M.; et al. Diverse roles of GSK-3: Tumor promoter-tumor suppressor, target in cancer therapy. Adv. Biol. Regul. 2014, 54, 176–196. [Google Scholar] [CrossRef] [PubMed]
- Domoto, T.; Pyko, I.V.; Furuta, T.; Miyashita, K.; Uehara, M.; Shimasaki, T.; Nakada, M.; Minamoto, T. Glycogen synthase kinase-3β is a pivotal mediator of cancer invasion and resistance to therapy. Cancer Sci. 2016, 107, 1363–1372. [Google Scholar] [CrossRef]
- Malhi, G.S. Lithium therapy in bipolar disorder: A balancing act? The Lancet 2015, 386, 415–416. [Google Scholar] [CrossRef]
- Shine, B.; McKnight, R.F.; Leaver, L.; Geddes, J.R. Long-term effects of lithium on renal, thyroid, and parathyroid function: A retrospective analysis of laboratory data. The Lancet 2015, 386, 461–468. [Google Scholar] [CrossRef]
- Martinsson, L.; Westman, J.; Hällgren, J.; Ösby, U.; Backlund, L. Lithium treatment and cancer incidence in bipolar disorder. Bipolar Disord. 2016, 18, 33–40. [Google Scholar] [CrossRef]
- Huang, R.Y.; Hsieh, K.P.; Huang, W.W.; Yang, Y.H. Use of lithium and cancer risk in patients with bipolar disorder: Population-based cohort study. Br. J. Psychiatry 2016, 209, 393–399. [Google Scholar] [CrossRef]
- Ge, W.; Jakobsson, E. Systems biology understanding of the effects of lithium on cancer. Front. Oncol. 2019, 9, 296. [Google Scholar] [CrossRef]
- Miyashita, K.; Nakada, M.; Shakoori, A.; Ishigaki, Y.; Shimasaki, T.; Motoo, Y.; Kawakami, K.; Minamoto, T. An emerging strategy for cancer treatment targeting aberrant glycogen synthase kinase 3β. Anticancer Agents Med. Chem. 2009, 9, 1114–1122. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Steelman, L.S.; Bertrand, F.E.; Davis, N.M.; Sokolosky, M.; Abrams, S.L.; Montalto, G.; D’Assoro, A.B.; Libra, M.; Nicoletti, F.; et al. GSK-3 as potential target for therapeutic intervention in cancer. Oncotarget 2014, 5, 2881–2911. [Google Scholar] [CrossRef] [PubMed]
- Walz, A.; Ugolkov, A.; Chandra, S.; Kozikowski, A.; Carneiro, B.A.; O’Halloran, T.V.; Giles, F.J.; Billadeau, D.D.; Mazar, A.P. Molecular pathways: Revisiting glycogen synthase kinase-3β as a target for the treatment of cancer. Clin. Cancer Res. 2017, 23, 1891–1897. [Google Scholar] [CrossRef] [PubMed]
- Osolodkin, D.I.; Palyulin, V.A.; Zefirov, N.S. Glycogen synthase kinase 3 as an anticancer drug target: Novel experimental findings and trends in the design of inhibitors. Curr. Pharm. Des. 2013, 19, 665–679. [Google Scholar] [CrossRef] [PubMed]
- Sahin, I.; Eturi, A.; De Souza, A.; Pamarthy, S.; Tavora, F.; Giles, F.J.; Carneiro, B.A. Glycogen synthase kinase-3β inhibitors as novel cancer treatments and modulators of antitumor immune responses. Cancer Biol. Ther. 2019, 20, 1047–1056. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.S.; Wang, C.L.; Wen, J.F.; Wang, Y.J.; Hu, Y.B.; Ren, H.Z. Lithium inhibits proliferation of human esophageal cancer cell line Eca-109 by inducing a G2/M cell cycle arrest. World J. Gastroenterol. 2008, 14, 3982–3989. [Google Scholar] [CrossRef]
- Gao, S.; Li, S.; Duan, X.; Gu, Z.; Ma, Z.; Yuan, X.; Feng, X.; Wang, H. Inhibition of glycogen synthase kinase 3β (GSK3β) suppresses the progression of esophageal squamous cell carcinoma by modifying STAT3 activity. Mol. Carcinog. 2017, 56, 2301–2316. [Google Scholar] [CrossRef]
- Mai, W.; Miyashita, K.; Shakoori, A.; Zhang, B.; Yu, Z.W.; Takahashi, Y.; Motoo, Y.; Kawakami, K.; Minamoto, T. Detection of active fraction of glycogen synthase kinase 3β in cancer cells by nonradioisotopic in vitro kinase assay. Oncology 2006, 71, 297–305. [Google Scholar] [CrossRef]
- Mai, W.; Kawakami, K.; Shakoori, A.; Kyo, S.; Miyashita, K.; Yokoi, K.; Jin, M.; Shimasaki, T.; Motoo, Y.; Minamoto, T. Deregulated GSK3β sustains gastrointestinal cancer cells survival by modulating human telomerase reverse transcriptase and telomerase. Clin. Cancer Res. 2009, 15, 6810–6819. [Google Scholar] [CrossRef]
- Yoon, J.; Ko, Y.S.; Cho, S.J.; Park, J.; Choi, Y.S.; Choi, Y.; Pyo, J.S.; Ye, S.K.; Youn, H.D.; Lee, J.S.; et al. Signal transducers and activators of transcription 3-induced metastatic potential in gastric cancer cells is enhanced by glycogen synthase kinase-3β. APMIS 2015, 123, 373–382. [Google Scholar] [CrossRef]
- Zhang, J.Y.; Zhao, Y.L.; Lv, Y.P.; Cheng, P.; Chen, W.; Duan, M.; Teng, Y.S.; Wang, T.T.; Peng, L.S.; Mao, F.Y.; et al. Modulation of CD8(+) memory stem T cell activity and glycogen synthase kinase 3β inhibition enhances anti-tumoral immunity in gastric cancer. Oncoimmunology 2018, 7, e1412900. [Google Scholar] [CrossRef]
- Gould, T.D.; Gray, N.A.; Manji, H.K. Effects of a glycogen synthase kinase-3 inhibitor, lithium, in adenomatous polyposis coli mutant mice. Pharmacol. Res. 2003, 48, 49–53. [Google Scholar] [CrossRef]
- Shakoori, A.; Ougolkov, A.; Yu, Z.W.; Zhang, B.; Modarressi, M.H.; Billadeau, D.D.; Mai, M.; Takahashi, Y.; Minamoto, T. Deregulated GSK3β activity in colorectal cancer: Its association with tumor cell survival and proliferation. Biochem. Biophys. Res. Commun. 2005, 334, 1365–1373. [Google Scholar] [CrossRef]
- Ghosh, J.C.; Altieri, D.C. Activation of p53-dependent apoptosis by acute ablation of glycogen synthase kinase-3β in colorectal cancer cells. Clin. Cancer Res. 2005, 11, 4580–4588. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tan, J.; Zhuang, L.; Leong, H.S.; Iyer, N.G.; Liu, E.T.; Yu, Q. Pharmacologic modulation of glycogen synthase kinase-3β promotes p53-dependent apoptosis through a direct Bax-mediated mitochondrial pathway in colorectal cancer cells. Cancer Res. 2005, 65, 9012–9020. [Google Scholar] [CrossRef] [PubMed]
- Rottmann, S.; Wang, Y.; Nasoff, M.; Deveraux, Q.L.; Quon, K.C. A TRAIL receptor-dependent synthetic lethal relationship between MYC activation and GSK3β/FBW7 loss of function. Proc. Natl. Acad. Sci. USA 2005, 102, 15195–15200. [Google Scholar] [CrossRef] [PubMed]
- Shakoori, A.; Mai, W.; Miyashita, K.; Yasumoto, K.; Takahashi, Y.; Ooi, A.; Kawakami, K.; Minamoto, T. Inhibition of GSK-3β activity attenuates proliferation of human colon cancer cells in rodents. Cancer Sci. 2007, 98, 1388–1393. [Google Scholar] [CrossRef]
- Bilsland, A.E.; Hoare, S.; Stevenson, K.; Plumb, J.; Gomez-Roman, N.; Cairney, C.; Burns, S.; Lafferty-Whyte, K.; Roffey, J.; Hammonds, T.; et al. Dynamic telomerase gene suppression via network effects of GSK3 inhibition. PLoS ONE 2009, 4, e6459. [Google Scholar] [CrossRef]
- Mayes, P.A.; Dolloff, N.G.; Daniel, C.J.; Liu, J.J.; Hart, L.S.; Kuribayashi, K.; Allen, J.E.; Jee, D.I.; Dorsey, J.F.; Liu, Y.Y.; et al. Overcoming hypoxia-induced apoptotic resistance through combinatorial inhibition of GSK-3β and CDK1. Cancer Res. 2011, 71, 5265–5275. [Google Scholar] [CrossRef]
- Deevi, R.; Fatehullah, A.; Jagan, I.; Nagaraju, M.; Bingham, V.; Campbell, F.C. PTEN regulates colorectal epithelial apoptosis through Cdc42 signalling. Br. J. Cancer 2011, 105, 1313–1321. [Google Scholar] [CrossRef]
- Grassilli, E.; Narloch, R.; Federzoni, E.; Ianzano, L.; Pisano, F.; Giovannoni, R.; Romano, G.; Masiero, L.; Leone, B.E.; Bonin, S.; et al. Inhibition of GSK3B bypass drug resistance of p53-null colon carcinomas by enabling necroptosis in response to chemotherapy. Clin. Cancer Res. 2013, 19, 3820–3831. [Google Scholar] [CrossRef] [PubMed]
- Turano, M.; Costabile, V.; Cerasuolo, A.; Duraturo, F.; Liccardo, R.; Delrio, P.; Pace, U.; Rega, D.; Dodaro, C.A.; Milone, M.; et al. Characterisation of mesenchymal colon tumour-derived cells in tumourspheres as a model for colorectal cancer progression. Int. J. Oncol. 2018, 53, 2379–2396. [Google Scholar] [CrossRef] [PubMed]
- Trnski, D.; Sabol, M.; Gojević, A.; Martinić, M.; Ozretić, P.; Musani, V.; Ramić, S.; Levanat, S. GSK3β and Gli3 play a role in activation of Hedgehog-Gli pathway in human colon cancer—Targeting GSK3β downregulates the signaling pathway and reduces cell proliferation. Biochim. Biophys. Acta 2015, 1852, 2574–2584. [Google Scholar] [CrossRef]
- Yoshino, Y.; Suzuki, M.; Takahashi, H.; Ishioka, C. Inhibition of invasion by glycogen synthase kinase-3β inhibitors through dysregulation of actin re-organisation via down-regulation of WAVE2. Biochem. Biophys. Res. Commun. 2015, 464, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Costabile, V.; Duraturo, F.; Delrio, P.; Rega, D.; Pace, U.; Liccardo, R.; Rossi, G.B.; Genesio, R.; Nitsch, L.; Izzo, P.; et al. Lithium chloride induces mesenchymal-to-epithelial reverting transition in primary colon cancer cell cultures. Int. J. Oncol. 2015, 46, 1913–1923. [Google Scholar] [CrossRef]
- Gao, C.; Chen, G.; Kuan, S.F.; Zhang, D.H.; Schlaepfer, D.D.; Hu, J. FAK/PYK2 promotes the Wnt/β-catenin pathway and intestinal tumorigenesis by phosphorylating GSK3β. Elife 2015, 4. [Google Scholar] [CrossRef]
- Yoshino, Y.; Ishioka, C. Inhibition of glycogen synthase kinase-3β induces apoptosis and mitotic catastrophe by disrupting centrosome regulation in cancer cells. Sci. Rep. 2015, 5, 13249. [Google Scholar] [CrossRef]
- Saud, S.M.; Li, W.; Gray, Z.; Matter, M.S.; Colburn, N.H.; Young, M.R.; Kim, Y.S. Diallyl disulfide (DADS), a constituent of garlic, inactivates NF-κB and prevents colitis-induced colorectal cancer by inhibiting GSK-3β. Cancer Prev. Res. 2016, 9, 607–615. [Google Scholar] [CrossRef]
- Ishida, R.; Koyanagi-Aoi, M.; Oshima, N.; Kakeji, Y.; Aoi, T. The tissue-reconstructing ability of colon CSCs is enhanced by FK506 and suppressed by GSK3 inhibition. Mol. Cancer Res. 2017, 15, 1455–1466. [Google Scholar] [CrossRef]
- Dewi, F.R.P.; Domoto, T.; Hazawa, M.; Kobayashi, A.; Douwaki, T.; Minamoto, T.; Wong, R.W. Colorectal cancer cells require glycogen synthase kinase-3β for sustaining mitosis via translocated promoter region (TPR)-dynein interaction. Oncotarget 2018, 9, 13337–13352. [Google Scholar] [CrossRef]
- Kazi, A.; Xiang, S.; Yang, H.; Delitto, D.; Trevino, J.; Jiang, R.H.Y.; Ayaz, M.; Lawrence, H.R.; Kennedy, P.; Sebti, S.M. GSK3 suppression upregulates β-catenin and c-Myc to abrogate KRas-dependent tumors. Nat. Commun. 2018, 9, 5154. [Google Scholar] [CrossRef] [PubMed]
- Ougolkov, A.V.; Fernandez-Zapico, M.E.; Savoy, D.N.; Urrutia, R.A.; Billadeau, D.D. Glycogen synthase kinase-3β participates in nuclear factor κB-mediated gene transcription and cell survival in pancreatic cancer cells. Cancer Res. 2005, 65, 2076–2081. [Google Scholar] [CrossRef] [PubMed]
- Ougolkov, A.V.; Fernandez-Zapico, M.E.; Bilim, V.N.; Smyrk, T.C.; Chari, S.T.; Billadeau, D.D. Aberrant nuclear accumulation of glycogen synthase kinase-3β in human pancreatic cancer: Association with kinase activity and tumor dedifferentiation. Clin. Cancer Res. 2006, 12, 5074–5081. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.; Baldwin, A.S. Maintenance of constitutive IκB kinase activity by glycogen synthase kinase-3α/β in pancreatic cancer. Cancer Res. 2008, 68, 8156–8163. [Google Scholar] [CrossRef] [PubMed]
- Mamaghani, S.; Patel, S.; Hedley, D.W. Glycogen synthase kinase-3 inhibition disrupts nuclear factor-κB activity in pancreatic cancer, but fails to sensitize to gemcitabine chemotherapy. BMC Cancer 2009, 9, 132. [Google Scholar] [CrossRef] [PubMed]
- Gaisina, I.N.; Gallier, F.; Ougolkov, A.V.; Kim, K.H.; Kurome, T.; Guo, S.; Holzle, D.; Luchini, D.N.; Blond, S.Y.; Billadeau, D.D.; et al. From a natural product lead to the identification of potent and selective benzofuran-3-yl-(indol-3-yl)maleimides as glycogen synthase kinase 3β inhibitors that suppress proliferation and survival of pancreatic cancer cells. J. Med. Chem. 2009, 52, 1853–1863. [Google Scholar] [CrossRef]
- Guzmán, E.A.; Johnson, J.D.; Linley, P.A.; Gunasekera, S.E.; Wright, A.E. A novel activity from an old compound: Manzamine A reduces the metastatic potential of AsPC-1 pancreatic cancer cells and sensitizes them to TRAIL-induced apoptosis. Investig. New Drugs 2011, 29, 777–785. [Google Scholar] [CrossRef]
- Zhang, J.S.; Koenig, A.; Harrison, A.; Ugolkov, A.V.; Fernandez-Zapico, M.E.; Couch, F.J.; Billadeau, D.D. Mutant K-Ras increases GSK-3β gene expression via an ETS-p300 transcriptional complex in pancreatic cancer. Oncogene 2011, 30, 3705–3715. [Google Scholar] [CrossRef]
- Shimasaki, T.; Ishigaki, Y.; Nakamura, Y.; Takata, T.; Nakaya, N.; Nakajima, H.; Sato, I.; Zhao, X.; Kitano, A.; Kawakami, K.; et al. Glycogen synthase kinase 3β inhibition sensitizes pancreatic cancer cells to gemcitabine. J. Gastroenterol. 2012, 47, 321–333. [Google Scholar] [CrossRef]
- Marchand, B.; Tremblay, I.; Cagnol, S.; Boucher, M.J. Inhibition of glycogen synthase kinase-3 activity triggers an apoptotic response in pancreatic cancer cells through JNK-dependent mechanisms. Carcinogenesis 2012, 33, 529–537. [Google Scholar] [CrossRef]
- Mamaghani, S.; Simpson, C.D.; Cao, P.M.; Cheung, M.; Chow, S.; Bandarchi, B.; Schimmer, A.D.; Hedley, D.W. Glycogen synthase kinase-3 inhibition sensitizes pancreatic cancer cells to TRAIL-induced apoptosis. PLoS ONE 2012, 7, e41102. [Google Scholar] [CrossRef] [PubMed]
- Kitano, A.; Shimasaki, T.; Chikano, Y.; Nakada, M.; Hirose, M.; Higashi, T.; Ishigaki, Y.; Endo, Y.; Takino, T.; Sato, H.; et al. Aberrant glycogen synthase kinase 3β is involved in pancreatic cancer cell invasion and resistance to therapy. PLoS ONE 2013, 8, e55289. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.S.; Herreros-Villanueva, M.; Koenig, A.; Deng, Z.; de Narvajas, A.A.; Gomez, T.S.; Meng, X.; Bujanda, L.; Ellenrieder, V.; Li, X.K.; et al. Differential activity of GSK-3 isoforms regulates NF-κB and TRAIL- or TNFα induced apoptosis in pancreatic cancer cells. Cell Death Dis. 2014, 5, e1142. [Google Scholar] [CrossRef] [PubMed]
- Ying, X.; Jing, L.; Ma, S.; Li, Q.; Luo, X.; Pan, Z.; Feng, Y.; Feng, P. GSK3β mediates pancreatic cancer cell invasion in vitro via the CXCR4/MMP-2 Pathway. Cancer Cell Int. 2015, 15, 70. [Google Scholar] [CrossRef]
- Kunnimalaiyaan, S.; Gamblin, T.C.; Kunnimalaiyaan, M. Glycogen synthase kinase-3 inhibitor AR-A014418 suppresses pancreatic cancer cell growth via inhibition of GSK-3-mediated Notch1 expression. HPB 2015, 17, 770–776. [Google Scholar] [CrossRef]
- Ma, S.; Li, Q.; Pan, F. CXCR4 promotes GSK3β expression in pancreatic cancer cells via the Akt pathway. Int. J. Clin. Oncol. 2015, 20, 525–530. [Google Scholar] [CrossRef]
- Marchand, B.; Arsenault, D.; Raymond-Fleury, A.; Boisvert, F.M.; Boucher, M.J. Glycogen synthase kinase-3 (GSK3) inhibition induces prosurvival autophagic signals in human pancreatic cancer cells. J. Biol. Chem. 2015, 290, 5592–5605. [Google Scholar] [CrossRef]
- Baumgart, S.; Chen, N.M.; Zhang, J.S.; Billadeau, D.D.; Gaisina, I.N.; Kozikowski, A.P.; Singh, S.K.; Fink, D.; Ströbel, P.; Klindt, C.; et al. GSK-3β governs inflammation-induced NFATc2 signaling hubs to promote pancreatic cancer progression. Mol. Cancer Ther. 2016, 15, 491–502. [Google Scholar] [CrossRef]
- Edderkaoui, M.; Chheda, C.; Soufi, B.; Zayou, F.; Hu, R.W.; Ramanujan, V.K.; Pan, X.; Boros, L.G.; Tajbakhsh, J.; Madhav, A.; et al. An inhibitor of GSK3B and HDACs kills pancreatic cancer cells and slows pancreatic tumor growth and metastasis in mice. Gastroenterology 2018, 155, 1985–1998. [Google Scholar] [CrossRef]
- Nesteruk, K.; Smits, R.; Bruno, M.; Peppelenbosch, M.P.; Fuhler, G.M. Upregulated β-catenin signaling does not affect survival of pancreatic cancer cells during dual inhibition of GSK3B and HDAC. Pancreatology 2020, 20, 558–561. [Google Scholar] [CrossRef]
- Beurel, E.; Blivet-Van Eggelpoël, M.J.; Kornprobst, M.; Moritz, S.; Delelo, R.; Paye, F.; Housset, C.; Desbois-Mouthon, C. Glycogen synthase kinase-3 inhibitors augment TRAIL-induced apoptotic death in human hepatoma cells. Biochem. Pharmacol. 2009, 77, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, M.; Sakai, H.; Shirakami, Y.; Yasuda, Y.; Kubota, M.; Terakura, D.; Baba, A.; Ohno, T.; Hara, Y.; Tanaka, T.; et al. Preventive effects of (-)-epigallocatechin gallate on diethylnitrosamine-induced liver tumorigenesis in obese and diabetic C57BL/KsJ-db/db mice. Cancer Prev. Res 2011, 4, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Ninomiya, S.; Shimizu, M.; Imai, K.; Takai, K.; Shiraki, M.; Hara, T.; Tsurumi, H.; Ishizaki, S.; Moriwaki, H. Possible role of visfatin in hepatoma progression and the effects of branched-chain amino acids on visfatin-induced proliferation in human hepatoma cells. Cancer Prev. Res. 2011, 4, 2092–2100. [Google Scholar] [CrossRef] [PubMed]
- Fu, K.; Pan, H.; Liu, S.; Lv, J.; Wan, Z.; Li, J.; Sun, Q.; Liang, J. Glycogen synthase kinase-3β regulates tumor necrosis factor-related apoptosis inducing ligand (TRAIL)-induced apoptosis via the NF-κB pathway in hepatocellular carcinoma. Oncol. Lett. 2015, 10, 3557–3564. [Google Scholar] [CrossRef]
- Shigeishi, H.; Biddle, A.; Gammon, L.; Emich, H.; Rodini, C.O.; Gemenetzidis, E.; Fazil, B.; Sugiyama, M.; Kamata, N.; Mackenzie, I.C. Maintenance of stem cell self-renewal in head and neck cancers requires actions of GSK3β influenced by CD44 and RHAMM. Stem Cells 2013, 31, 2073–2083. [Google Scholar] [CrossRef]
- Schulz, L.; Pries, R.; Lanka, A.S.; Drenckhan, M.; Rades, D.; Wollenberg, B. Inhibition of GSK3α/β impairs the progression of HNSCC. Oncotarget 2018, 9, 27630–27644. [Google Scholar] [CrossRef]
- Zeng, J.; Liu, D.; Qiu, Z.; Huang, Y.; Chen, B.; Wang, L.; Xu, H.; Huang, N.; Liu, L.; Li, W. GSK3β overexpression indicates poor prognosis and its inhibition reduces cell proliferation and survival of non-small cell lung cancer cells. PLoS ONE 2014, 9, e91231. [Google Scholar] [CrossRef]
- Vincent, E.E.; Elder, D.J.; O’Flaherty, L.; Pardo, O.E.; Dzien, P.; Phillips, L.; Morgan, C.; Pawade, J.; May, M.T.; Sohail, M.; et al. Glycogen synthase kinase 3 protein kinase activity is frequently elevated in human non-small cell lung carcinoma and supports tumour cell proliferation. PLoS ONE 2014, 9, e114725. [Google Scholar] [CrossRef]
- O’Flaherty, L.; Shnyder, S.D.; Cooper, P.A.; Cross, S.J.; Wakefield, J.G.; Pardo, O.E.; Seckl, M.J.; Tavaré, J.M. Tumor growth suppression using a combination of taxol-based therapy and GSK3 inhibition in non-small cell lung cancer. PLoS ONE 2019, 14, e0214610. [Google Scholar] [CrossRef]
- Shin, S.; Wolgamott, L.; Tcherkezian, J.; Vallabhapurapu, S.; Yu, Y.; Roux, P.P.; Yoon, S.O. Glycogen synthase kinase-3β positively regulates protein synthesis and cell proliferation through the regulation of translation initiation factor 4E-binding protein 1. Oncogene 2014, 33, 1690–1699. [Google Scholar] [CrossRef]
- Gupta, C.; Kaur, J.; Tikoo, K. Regulation of MDA-MB-231 cell proliferation by GSK-3β involves epigenetic modifications under high glucose conditions. Exp. Cell Res. 2014, 324, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Yuan, X.; Jiang, Z.; Gan, D.; Ding, L.; Sun, Y.; Zhou, J.; Xu, L.; Liu, Y.; Wang, G. Regulation of AKT phosphorylation by GSK3β and PTEN to control chemoresistance in breast cancer. Breast Cancer Res. Treat. 2019, 176, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.; Zhang, L.; Thrasher, J.B.; Du, J.; Li, B. Glycogen synthase kinase-3β suppression eliminates tumor necrosis factor-related apoptosis-inducing ligand resistance in prostate cancer. Mol. Cancer Ther. 2003, 2, 1215–1222. [Google Scholar] [PubMed]
- Mazor, M.; Kawano, Y.; Zhu, H.; Waxman, J.; Kypta, R.M. Inhibition of glycogen synthase kinase-3 represses androgen receptor activity and prostate cancer cell growth. Oncogene 2004, 23, 7882–7892. [Google Scholar] [CrossRef] [PubMed]
- Sun, A.; Shanmugam, I.; Song, J.; Terranova, P.F.; Thrasher, J.B.; Li, B. Lithium suppresses cell proliferation by interrupting E2F-DNA interaction and subsequently reducing S-phase gene expression in prostate cancer. Prostate 2007, 67, 976–988. [Google Scholar] [CrossRef] [PubMed]
- Rinnab, L.; Schütz, S.V.; Diesch, J.; Schmid, E.; Küfer, R.; Hautmann, R.E.; Spindler, K.D.; Cronauer, M.V. Inhibition of glycogen synthase kinase-3 in androgen-responsive prostate cancer cell lines: Are GSK inhibitors therapeutically useful? Neoplasia 2008, 10, 624–634. [Google Scholar] [CrossRef]
- Schütz, S.V.; Cronauer, M.V.; Rinnab, L. Inhibition of glycogen synthase kinase-3β promotes nuclear export of the androgen receptor through a CRM1-dependent mechanism in prostate cancer cell lines. J. Cell Biochem. 2010, 109, 1192–1200. [Google Scholar] [CrossRef]
- Schütz, S.V.; Schrader, A.J.; Zengerling, F.; Genze, F.; Cronauer, M.V.; Schrader, M. Inhibition of glycogen synthase kinase-3β counteracts ligand-independent activity of the androgen receptor in castration resistant prostate cancer. PLoS ONE 2011, 6, e25341. [Google Scholar] [CrossRef]
- Zhu, Q.; Yang, J.; Han, S.; Liu, J.; Holzbeierlein, J.; Thrasher, J.B.; Li, B. Suppression of glycogen synthase kinase 3 activity reduces tumor growth of prostate cancer in vivo. Prostate 2011, 71, 835–845. [Google Scholar] [CrossRef]
- Darrington, R.S.; Campa, V.M.; Walker, M.M.; Bengoa-Vergniory, N.; Gorrono-Etxebarria, I.; Uysal-Onganer, P.; Kawano, Y.; Waxman, J.; Kypta, R.M. Distinct expression and activity of GSK-3α and GSK-3β in prostate cancer. Int. J. Cancer 2012, 131, E872–E883. [Google Scholar] [CrossRef]
- Goc, A.; Al-Husein, B.; Katsanevas, K.; Steinbach, A.; Lou, U.; Sabbineni, H.; DeRemer, D.L.; Somanath, P.R. Targeting Src-mediated Tyr216 phosphorylation and activation of GSK-3 in prostate cancer cells inhibit prostate cancer progression in vitro and in vivo. Oncotarget 2014, 5, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Kroon, J.; in ′t Veld, L.S.; Buijs, J.T.; Cheung, H.; van der Horst, G.; van der Pluijm, G. Glycogen synthase kinase-3β inhibition depletes the population of prostate cancer stem/progenitor-like cells and attenuates metastatic growth. Oncotarget 2014, 5, 8986–8994. [Google Scholar] [CrossRef] [PubMed]
- Sun, A.; Li, C.; Chen, R.; Huang, Y.; Chen, Q.; Cui, X.; Liu, H.; Thrasher, J.B.; Li, B. GSK-3β controls autophagy by modulating LKB1-AMPK pathway in prostate cancer cells. Prostate 2016, 76, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Bilim, V.; Ougolkov, A.; Yuuki, K.; Naito, S.; Kawazoe, H.; Muto, A.; Oya, M.; Billadeau, D.; Motoyama, T.; Tomita, Y. Glycogen synthase kinase-3: A new therapeutic target in renal cell carcinoma. Br. J. Cancer 2009, 101, 2005–2014. [Google Scholar] [CrossRef]
- Tsukigi, M.; Bilim, V.; Yuuki, K.; Ugolkov, A.; Naito, S.; Nagaoka, A.; Kato, T.; Motoyama, T.; Tomita, Y. Re-expression of miR-199a suppresses renal cancer cell proliferation and survival by targeting GSK-3β. Cancer Lett. 2012, 315, 189–197. [Google Scholar] [CrossRef]
- Kawazoe, H.; Bilim, V.N.; Ugolkov, A.V.; Yuuki, K.; Naito, S.; Nagaoka, A.; Kato, T.; Tomita, Y. GSK-3 inhibition in vitro and in vivo enhances antitumor effect of sorafenib in renal cell carcinoma (RCC). Biochem. Biophys. Res. Commun. 2012, 423, 490–495. [Google Scholar] [CrossRef]
- Pal, K.; Cao, Y.; Gaisina, I.N.; Bhattacharya, S.; Dutta, S.K.; Wang, E.; Gunosewoyo, H.; Kozikowski, A.P.; Billadeau, D.D.; Mukhopadhyay, D. Inhibition of GSK-3 induces differentiation and impaired glucose metabolism in renal cancer. Mol. Cancer Ther. 2014, 13, 285–296. [Google Scholar] [CrossRef]
- Naito, S.; Bilim, V.; Yuuki, K.; Ugolkov, A.; Motoyama, T.; Nagaoka, A.; Kato, T.; Tomita, Y. Glycogen synthase kinase-3β: A prognostic marker and a potential therapeutic target in human bladder cancer. Clin. Cancer Res. 2010, 16, 5124–5132. [Google Scholar] [CrossRef]
- Yohn, N.L.; Bingaman, C.N.; DuMont, A.L.; Yoo, L.I. Phosphatidylinositol 3′-kinase, mTOR, and glycogen synthase kinase-3β mediated regulation of p21 in human urothelial carcinoma cells. BMC Urol. 2011, 11, 19. [Google Scholar] [CrossRef]
- Guo, X.; Huang, H.; Jin, H.; Xu, J.; Risal, S.; Li, J.; Li, X.; Yan, H.; Zeng, X.; Xue, L.; et al. ISO, via upregulating miR-137 transcription, inhibits GSK3β-HSP70-MMP-2 axis, resulting in attenuating urothelial cancer invasion. Mol. Ther. Nucleic Acids 2018, 12, 337–349. [Google Scholar] [CrossRef]
- Cao, Q.; Lu, X.; Feng, Y.J. Glycogen synthase kinase-3β positively regulates the proliferation of human ovarian cancer cells. Cell Res. 2006, 16, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Hilliard, T.S.; Gaisina, I.N.; Muehlbauer, A.G.; Gaisin, A.M.; Gallier, F.; Burdette, J.E. Glycogen synthase kinase 3β inhibitors induce apoptosis in ovarian cancer cells and inhibit in-vivo tumor growth. Anticancer Drugs 2011, 22, 978–985. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Abdelmohsen, K.; Morin, P.J.; Gorospe, M. Novel microRNA reporter uncovers repression of let-7 by GSK-3β. PLoS ONE 2013, 8, e66330. [Google Scholar] [CrossRef]
- Mathuram, T.L.; Ravikumar, V.; Reece, L.M.; Sasikumar, C.S.; Cherian, K.M. Correlative studies unravelling the possible mechanism of cell death in tideglusib-treated human ovarian teratocarcinoma-derived PA-1 cells. J. Environ. Pathol. Toxicol. Oncol. 2017, 36, 321–344. [Google Scholar] [CrossRef]
- Yin, Y.; Kizer, N.T.; Thaker, P.H.; Chiappinelli, K.B.; Trinkaus, K.M.; Goodfellow, P.J.; Ma, L. Glycogen synthase kinase 3β inhibition as a therapeutic approach in the treatment of endometrial cancer. Int. J. Mol. Sci. 2013, 14, 16617–16637. [Google Scholar] [CrossRef]
- Ma, C.; Zeng, C.; Jin, L.; Yang, Y.; Li, P.; Chen, L.; Wang, J. GSK3β mediates the carcinogenic effect of HPV16 in cervical cancer. Sci. Rep. 2015, 5, 16555. [Google Scholar] [CrossRef] [PubMed]
- Kotliarova, S.; Pastorino, S.; Kovell, L.C.; Kotliarov, Y.; Song, H.; Zhang, W.; Bailey, R.; Maric, D.; Zenklusen, J.C.; Lee, J.; et al. Glycogen synthase kinase-3 inhibition induces glioma cell death through c-MYC, nuclear factor-κB, and glucose regulation. Cancer Res. 2008, 68, 6643–6651. [Google Scholar] [CrossRef] [PubMed]
- Nowicki, M.O.; Dmitrieva, N.; Stein, A.M.; Cutter, J.L.; Godlewski, J.; Saeki, Y.; Nita, M.; Berens, M.E.; Sander, L.M.; Newton, H.B.; et al. Lithium inhibits invasion of glioma cells; possible involvement of glycogen synthase kinase-3. Neuro Oncol. 2008, 10, 690–699. [Google Scholar] [CrossRef] [PubMed]
- Miyashita, K.; Kawakami, K.; Nakada, M.; Mai, W.; Shakoori, A.; Fujisawa, H.; Hayashi, Y.; Hamada, J.; Minamoto, T. Potential therapeutic effect of glycogen synthase kinase 3β inhibition against human glioblastoma. Clin. Cancer Res. 2009, 15, 887–897. [Google Scholar] [CrossRef]
- Korur, S.; Huber, R.M.; Sivasankaran, B.; Petrich, M.; Morin, P., Jr.; Hemmings, B.A.; Merlo, A.; Lino, M.M. GSK3β regulates differentiation and growth arrest in glioblastoma. PLoS ONE 2009, 4, e7443. [Google Scholar] [CrossRef]
- Li, Y.; Lu, H.; Huang, Y.; Xiao, R.; Cai, X.; He, S.; Yan, G. Glycogen synthase kinases-3β controls differentiation of malignant glioma cells. Int. J. Cancer 2010, 127, 1271–1282. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.P.; Nowicki, M.O.; Liu, F.; Press, R.; Godlewski, J.; Abdel-Rasoul, M.; Kaur, B.; Fernandez, S.A.; Chiocca, E.A.; Lawler, S.E. Indirubins decrease glioma invasion by blocking migratory phenotypes in both the tumor and stromal endothelial cell compartments. Cancer Res. 2011, 71, 5374–5380. [Google Scholar] [CrossRef] [PubMed]
- Pyko, I.V.; Nakada, M.; Sabit, H.; Teng, L.; Furuyama, N.; Hayashi, Y.; Kawakami, K.; Minamoto, T.; Fedulau, A.S.; Hamada, J. Glycogen synthase kinase 3β inhibition sensitizes human glioblastoma cells to temozolomide by affecting O6-methylguanine DNA methyltransferase promoter methylation via c-Myc signaling. Carcinogenesis 2013, 34, 2206–2217. [Google Scholar] [CrossRef] [PubMed]
- Zou, Q.; Hou, Y.; Shen, F.; Wang, Y. Polarized regulation of glycogen synthase kinase-3β is important for glioma cell invasion. PLoS ONE 2013, 8, e81814. [Google Scholar] [CrossRef]
- Yadav, A.K.; Vashishta, V.; Joshi, N.; Taneja, P. AR-A014418 used against GSK3β downregulates expression of hnRNPA1 and SF2/ASF splicing factors. J. Oncol. 2014, 2014, 695325. [Google Scholar] [CrossRef]
- Chikano, Y.; Domoto, T.; Furuta, T.; Sabit, H.; Kitano-Tamura, A.; Pyko, I.V.; Takino, T.; Sai, Y.; Hayashi, Y.; Sato, H.; et al. Glycogen synthase kinase 3β sustains invasion of glioblastoma via the focal adhesion kinase, Rac1, and c-Jun N-terminal kinase-mediated pathway. Mol. Cancer Ther. 2015, 14, 564–574. [Google Scholar] [CrossRef]
- Zhou, A.; Lin, K.; Zhang, S.; Chen, Y.; Zhang, N.; Xue, J.; Wang, Z.; Aldape, K.D.; Xie, K.; Woodgett, J.R.; et al. Nuclear GSK3β promotes tumorigenesis by phosphorylating KDM1A and inducing its deubiquitylation by USP22. Nat. Cell Biol. 2016, 18, 954–966. [Google Scholar] [CrossRef]
- Han, S.; Meng, L.; Jiang, Y.; Cheng, W.; Tie, X.; Xia, J.; Wu, A. Lithium enhances the antitumour effect of temozolomide against TP53 wild-type glioblastoma cells via NFAT1/FasL signalling. Br. J. Cancer 2017, 116, 1302–1311. [Google Scholar] [CrossRef]
- Furuta, T.; Sabit, H.; Dong, Y.; Miyashita, K.; Kinoshita, M.; Uchiyama, N.; Hayashi, Y.; Hayashi, Y.; Minamoto, T.; Nakada, M. Biological basis and clinical study of glycogen synthase kinase-3β-targeted therapy by drug repositioning for glioblastoma. Oncotarget 2017, 8, 22811–22824. [Google Scholar] [CrossRef]
- Bruning-Richardson, A.; Droop, A.; Tams, D.; Boissinot, M.; Hayes, J.; Cheng, V.; Cockle, J.; Ismail, A.; Morton, R.; Esteves, F.; et al. Identification of transcriptional targets of GSK3 involved in glioblastoma invasion. Neuro Oncol. 2018, 20, i26. [Google Scholar] [CrossRef]
- Sengupta, S.; Katz, S.C.; Sengupta, S.; Sampath, P. Glycogen synthase kinase 3 inhibition lowers PD-1 expression, promotes long-term survival and memory generation in antigen-specific CAR-T cells. Cancer Lett. 2018, 433, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Kitabayashi, T.; Dong, Y.; Furuta, T.; Sabit, H.; Jiapaer, S.; Zhang, J.; Zhang, G.; Hayashi, Y.; Kobayashi, M.; Domoto, T.; et al. Identification of GSK3β inhibitor kenpaullone as a temozolomide enhancer against glioblastoma. Sci. Rep. 2019, 9, 10049. [Google Scholar] [CrossRef]
- Ito, H.; Watari, K.; Shibata, T.; Miyamoto, T.; Murakami, Y.; Nakahara, Y.; Izumi, H.; Wakimoto, H.; Kuwano, M.; Abe, T.; et al. Bidirectional regulation between NDRG1 and GSK3β controls tumor growth and is targeted by differentiation inducing factor-1 in glioblastoma. Cancer Res. 2020, 80, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Kappes, A.; Vaccaro, A.; Kunnimalaiyaan, M.; Chen, H. Lithium ions: A novel treatment for pheochromocytomas and paragangliomas. Surgery 2007, 141, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Pizarro, J.G.; Folch, J.; Esparza, J.L.; Jordan, J.; Pallàs, M.; Camins, A. A molecular study of pathways involved in the inhibition of cell proliferation in neuroblastoma B65 cells by the GSK-3 inhibitors lithium and SB-415286. J. Cell Mol. Med. 2009, 13, 3906–3917. [Google Scholar] [CrossRef]
- Duffy, D.J.; Krstic, A.; Schwarzl, T.; Higgins, D.G.; Kolch, W. GSK3 inhibitors regulate MYCN mRNA levels and reduce neuroblastoma cell viability through multiple mechanisms, including p53 and Wnt signaling. Mol. Cancer Ther. 2014, 13, 454–467. [Google Scholar] [CrossRef]
- Mathuram, T.L.; Ravikumar, V.; Reece, L.M.; Karthik, S.; Sasikumar, C.S.; Cherian, K.M. Tideglusib induces apoptosis in human neuroblastoma IMR32 cells, provoking sub-G0/G1 accumulation and ROS generation. Environ. Toxicol. Pharmacol. 2016, 46, 194–205. [Google Scholar] [CrossRef]
- De Toni, F.; Racaud-Sultan, C.; Chicanne, G.; Mas, V.M.; Cariven, C.; Mesange, F.; Salles, J.P.; Demur, C.; Allouche, M.; Payrastre, B.; et al. A crosstalk between the Wnt and the adhesion-dependent signaling pathways governs the chemosensitivity of acute myeloid leukemia. Oncogene 2006, 25, 3113–3122. [Google Scholar] [CrossRef]
- Ougolkov, A.V.; Bone, N.D.; Fernandez-Zapico, M.E.; Kay, N.E.; Billadeau, D.D. Inhibition of glycogen synthase kinase-3 activity leads to epigenetic silencing of nuclear factor κB target genes and induction of apoptosis in chronic lymphocytic leukemia B cells. Blood 2007, 110, 735–742. [Google Scholar] [CrossRef]
- Holmes, T.; O’Brien, T.A.; Knight, R.; Lindeman, R.; Shen, S.; Song, E.; Symonds, G.; Dolnikov, A. Glycogen synthase kinase-3β inhibition preserves hematopoietic stem cell activity and inhibits leukemic cell growth. Stem Cells 2008, 26, 1288–1297. [Google Scholar] [CrossRef]
- Wang, Z.; Smith, K.S.; Murphy, M.; Piloto, O.; Somervaille, T.C.; Cleary, M.L. Glycogen synthase kinase 3 in MLL leukaemia maintenance and targeted therapy. Nature 2008, 455, 1205–1209. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Iwasaki, M.; Ficara, F.; Lin, C.; Matheny, C.; Wong, S.H.; Smith, K.S.; Cleary, M.L. GSK-3 promotes conditional association of CREB and its coactivators with MEIS1 to facilitate HOX-mediated transcription and oncogenesis. Cancer Cell 2010, 17, 597–608. [Google Scholar] [CrossRef]
- De Toni-Costes, F.; Despeaux, M.; Bertrand, J.; Bourogaa, E.; Ysebaert, L.; Payrastre, B.; Racaud-Sultan, C. A New α5β1 integrin-dependent survival pathway through GSK3β activation in leukemic cells. PLoS ONE 2010, 5, e9807. [Google Scholar] [CrossRef] [PubMed]
- Song, E.Y.; Palladinetti, P.; Klamer, G.; Ko, K.H.; Lindeman, R.; O’Brien, T.A.; Dolnikov, A. Glycogen synthase kinase-3β inhibitors suppress leukemia cell growth. Exp. Hematol. 2010, 38, 908–921. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Zhang, L.; van Laar, T.; van Dam, H.; Ten Dijke, P. GSK3β inactivation induces apoptosis of leukemia cells by repressing the function of c-Myb. Mol. Biol. Cell 2011, 22, 3533–3540. [Google Scholar] [CrossRef] [PubMed]
- Bourogaa, E.; Bertrand, J.; Despeaux, M.; Jarraya, R.; Fabre, N.; Payrastre, L.; Demur, C.; Fournié, J.J.; Damak, M.; Feki, A.E.; et al. Hammada scoparia flavonoids and rutin kill adherent and chemoresistant leukemic cells. Leuk. Res. 2011, 35, 1093–1101. [Google Scholar] [CrossRef]
- Bertrand, J.; Despeaux, M.; Joly, S.; Bourogaa, E.; Gallay, N.; Demur, C.; Bonnevialle, P.; Louache, F.; Maguer-Satta, V.; Vergnolle, N.; et al. Sex differences in the GSK3β-mediated survival of adherent leukemic progenitors. Oncogene 2012, 31, 694–705. [Google Scholar] [CrossRef]
- Mirlashari, M.R.; Randen, I.; Kjeldsen-Kragh, J. Glycogen synthase kinase-3 (GSK-3) inhibition induces apoptosis in leukemic cells through mitochondria-dependent pathway. Leuk. Res. 2012, 36, 499–508. [Google Scholar] [CrossRef]
- Gupta, K.; Gulen, F.; Sun, L.; Aguilera, R.; Chakrabarti, A.; Kiselar, J.; Agarwal, M.K.; Wald, D.N. GSK3 is a regulator of RAR-mediated differentiation. Leukemia 2012, 26, 1277–1285. [Google Scholar] [CrossRef]
- Kretzschmar, C.; Roolf, C.; Langhammer, T.S.; Sekora, A.; Pews-Davtyan, A.; Beller, M.; Frech, M.J.; Eisenlöffel, C.; Rolfs, A.; Junghanss, C. The novel arylindolylmaleimide PDA-66 displays pronounced antiproliferative effects in acute lymphoblastic leukemia cells. BMC Cancer 2014, 14, 71. [Google Scholar] [CrossRef]
- Gupta, K.; Stefan, T.; Ignatz-Hoover, J.; Moreton, S.; Parizher, G.; Saunthararajah, Y.; Wald, D.N. GSK-3 inhibition sensitizes acute myeloid leukemia cells to 1,25D-mediated differentiation. Cancer Res. 2016, 76, 2743–2753. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Ueda, M.; Stetson, L.; Ignatz-Hoover, J.; Moreton, S.; Chakrabarti, A.; Xia, Z.; Karan, G.; de Lima, M.; Agrawal, M.K.; et al. A novel glycogen synthase kinase-3 inhibitor optimized for acute myeloid leukemia differentiation activity. Mol. Cancer Ther. 2016, 15, 1485–1494. [Google Scholar] [CrossRef] [PubMed]
- Parameswaran, R.; Ramakrishnan, P.; Moreton, S.A.; Xia, Z.; Hou, Y.; Lee, D.A.; Gupta, K.; de Lima, M.; Beck, R.C.; Wald, D.N. Repression of GSK3 restores NK cell cytotoxicity in AML patients. Nat. Commun. 2016, 7, 11154. [Google Scholar] [CrossRef] [PubMed]
- Cichocki, F.; Valamehr, B.; Bjordahl, R.; Zhang, B.; Rezner, B.; Rogers, P.; Gaidarova, S.; Moreno, S.; Tuininga, K.; Dougherty, P.; et al. GSK3 inhibition drives maturation of NK cells and enhances their antitumor activity. Cancer Res. 2017, 77, 5664–5675. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Uddin, S.; Zimmerman, T.; Kang, J.A.; Ulaszek, J.; Wickrema, A. Growth control of multiple myeloma cells through inhibition of glycogen synthase kinase-3. Leuk. Lymphoma 2008, 49, 1945–1953. [Google Scholar] [CrossRef] [PubMed]
- Kunnimalaiyaan, M.; Vaccaro, A.M.; Ndiaye, M.A.; Chen, H. Inactivation of glycogen synthase kinase-3β, a downstream target of the raf-1 pathway, is associated with growth suppression in medullary thyroid cancer cells. Mol. Cancer Ther. 2007, 6, 1151–1158. [Google Scholar] [CrossRef]
- Adler, J.T.; Cook, M.; Luo, Y.; Pitt, S.C.; Ju, J.; Li, W.; Shen, B.; Kunnimalaiyaan, M.; Chen, H. Tautomycetin and tautomycin suppress the growth of medullary thyroid cancer cells via inhibition of glycogen synthase kinase-3β. Mol. Cancer Ther. 2009, 8, 914–920. [Google Scholar] [CrossRef]
- Chen, J.Y.; Cook, M.R.; Pinchot, S.N.; Kunnimalaiyaan, M.; Chen, H. MG-132 inhibits carcinoid growth and alters the neuroendocrine phenotype. J. Surg. Res. 2010, 158, 15–19. [Google Scholar] [CrossRef][Green Version]
- Aristizabal Prada, E.T.; Weis, C.; Orth, M.; Lauseker, M.; Spöttl, G.; Maurer, J.; Grabowski, P.; Grossman, A.; Auernhammer, C.J.; Nölting, S. GSK3α/β: A novel therapeutic target for neuroendocrine tumors. Neuroendocrinology 2018, 106, 335–351. [Google Scholar] [CrossRef]
- Aristizabal Prada, E.T.; Spöttl, G.; Maurer, J.; Lauseker, M.; Koziolek, E.J.; Schrader, J.; Grossman, A.; Pacak, K.; Beuschlein, F.; Auernhammer, C.J.; et al. The role of GSK3 and its reversal with GSK3 antagonism in everolimus resistance. Endocr. Relat. Cancer 2018, 25, 893–908. [Google Scholar] [CrossRef]
- Tang, Q.L.; Xie, X.B.; Wang, J.; Chen, Q.; Han, A.J.; Zou, C.Y.; Yin, J.Q.; Liu, D.W.; Liang, Y.; Zhao, Z.Q.; et al. Glycogen synthase kinase-3β, NF-κB signaling, and tumorigenesis of human osteosarcoma. J. Natl. Cancer Inst. 2012, 104, 749–763. [Google Scholar] [CrossRef] [PubMed]
- Shimozaki, S.; Yamamoto, N.; Domoto, T.; Nishida, H.; Hayashi, K.; Kimura, H.; Takeuchi, A.; Miwa, S.; Igarashi, K.; Kato, T.; et al. Efficacy of glycogen synthase kinase-3β targeting against osteosarcoma via activation of β-catenin. Oncotarget 2016, 7, 77038–77051. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nishimura, H.; Nakamura, O.; Yamagami, Y.; Mori, M.; Horie, R.; Fukuoka, N.; Yamamoto, T. GSK-3 inhibitor inhibits cell proliferation and induces apoptosis in human osteosarcoma cells. Oncol. Rep. 2016, 35, 2348–2354. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.; Wang, X.; Chen, Y.; Liang, D.; Luo, H.; Long, L.; Hu, Z.; Bao, J. Identification of two potential glycogen synthase kinase 3β inhibitors for the treatment of osteosarcoma. Acta Biochim. Biophys. Sin. 2018, 50, 456–464. [Google Scholar] [CrossRef] [PubMed]
- Zeng, F.Y.; Dong, H.; Cui, J.; Liu, L.; Chen, T. Glycogen synthase kinase 3 regulates PAX3-FKHR-mediated cell proliferation in human alveolar rhabdomyosarcoma cells. Biochem. Biophys. Res. Commun. 2010, 391, 1049–1055. [Google Scholar] [CrossRef]
- Chen, E.Y.; DeRan, M.T.; Ignatius, M.S.; Grandinetti, K.B.; Clagg, R.; McCarthy, K.M.; Lobbardi, R.M.; Brockmann, J.; Keller, C.; Wu, X.; et al. Glycogen synthase kinase 3 inhibitors induce the canonical WNT/β-catenin pathway to suppress growth and self-renewal in embryonal rhabdomyosarcoma. Proc. Natl. Acad. Sci. USA 2014, 111, 5349–5354. [Google Scholar] [CrossRef]
- Abe, K.; Yamamoto, N.; Domoto, T.; Bolidong, D.; Hayashi, K.; Takeuchi, A.; Miwa, S.; Igarashi, K.; Inatani, H.; Aoki, Y.; et al. Glycogen synthase kinase 3β as a potential therapeutic target in synovial sarcoma and fibrosarcoma. Cancer Sci. 2020, 111, 429–440. [Google Scholar] [CrossRef]
- Smalley, K.S.; Contractor, R.; Haass, N.K.; Kulp, A.N.; Atilla-Gokcumen, G.E.; Williams, D.S.; Bregman, H.; Flaherty, K.T.; Soengas, M.S.; Meggers, E.; et al. An organometallic protein kinase inhibitor pharmacologically activates p53 and induces apoptosis in human melanoma cells. Cancer Res. 2007, 67, 209–217. [Google Scholar] [CrossRef]
- Kubic, J.D.; Mascarenhas, J.B.; Iizuka, T.; Wolfgeher, D.; Lang, D. GSK-3 promotes cell survival, growth, and PAX3 levels in human melanoma cells. Mol. Cancer Res. 2012, 10, 1065–1076. [Google Scholar] [CrossRef]
- Atkinson, J.M.; Rank, K.B.; Zeng, Y.; Capen, A.; Yadav, V.; Manro, J.R.; Engler, T.A.; Chedid, M. Activating the Wnt/β-catenin pathway for the treatment of melanoma-application of LY2090314, a novel selective inhibitor of glycogen synthase kinase-3. PLoS ONE 2015, 10, e0125028. [Google Scholar] [CrossRef]
- Minamoto, T.; Kotake, M.; Nakada, M.; Shimasaki, T.; Motoo, Y.; Kawakami, K. Distinct pathologic role for glycogen synthase kinase 3β in colorectal cancer progression. In Colorectal Cancer Biology—From Genes to Tumor; IntechOpen: London, UK, 2012. [Google Scholar] [CrossRef]
- Garcea, G.; Manson, M.M.; Neal, C.P.; Pattenden, C.J.; Sutton, C.D.; Dennison, A.R.; Berry, D.P. Glycogen synthase kinase-3β; a new target in pancreatic cancer? Curr. Cancer Drug Targets 2007, 7, 209–215. [Google Scholar] [CrossRef]
- Motoo, Y.; Shimasaki, T.; Ishigaki, Y.; Nakajima, H.; Kawakami, K.; Minamoto, T. Metabolic disorder, inflammation, and deregulated molecular pathways converging in pancreatic cancer development: Implications for new therapeutic strategies. Cancers 2011, 3, 446–460. [Google Scholar] [CrossRef]
- Shimasaki, T.; Kitano, A.; Motoo, Y.; Minamoto, T. Aberrant glycogen synthase kinase 3β in the development of pancreatic cancer. J. Carcinog. 2012, 11, 15. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Bhojani, M.S.; Ben-Josef, E.; Spalding, A.C.; Kuick, R.; Sun, Y.; Morgan, M.A. Glycogen synthase kinase 3β in pancreatic cancer and its implications in chemotherapy and radiation therapy. J. Carcinog. Mutagenes. 2013, 4, 147. [Google Scholar] [CrossRef]
- Li, B.; Thrasher, J.B.; Terranova, P. Glycogen synthase kinase-3: A potential preventive target for prostate cancer management. Urol. Oncol. 2015, 33, 456–463. [Google Scholar] [CrossRef] [PubMed]
- Nakada, M.; Minamoto, T.; Pyko, I.; Hayashi, Y.; Hamada, J. The pivotal role of GSK3β in glioma biology. In Molecular Targets of CNS Tumors; IntechOpen: London, UK, 2011. [Google Scholar] [CrossRef]
- Nakada, M.; Furuta, T.; Hayashi, Y.; Minamoto, T.; Hamada, J. The strategy for enhancing temozolomide against malignant glioma. Front. Oncol. 2012, 2, 98. [Google Scholar] [CrossRef] [PubMed]
- Atkins, R.J.; Stylli, S.S.; Luwor, R.B.; Kaye, A.H.; Hovens, C.M. Glycogen synthase kinase-3β (GSK-3β) and its dysregulation in glioblastoma multiforme. J. Clin. Neurosci. 2013, 20, 1185–1192. [Google Scholar] [CrossRef] [PubMed]
- Holmes, T.; O′Brien, T.A.; Knight, R.; Lindeman, R.; Symonds, G.; Dolnikov, A. The role of glycogen synthase kinase-3β in normal haematopoiesis, angiogenesis and leukaemia. Curr. Med. Chem. 2008, 15, 1493–1499. [Google Scholar] [CrossRef]
- Birch, N.W.; Zeleznik-Le, N.J. Glycogen synthase kinase-3 and leukemia: Restoring the balance. Cancer Cell 2010, 17, 529–531. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Steelman, L.S.; Bertrand, F.E.; Davis, N.M.; Abrams, S.L.; Montalto, G.; D′Assoro, A.B.; Libra, M.; Nicoletti, F.; Maestro, R.; et al. Multifaceted roles of GSK-3 and Wnt/β-catenin in hematopoiesis and leukemogenesis: Opportunities for therapeutic intervention. Leukemia 2014, 28, 15–33. [Google Scholar] [CrossRef]
- Bang, D.; Wilson, W.; Ryan, M.; Yeh, J.J.; Baldwin, A.S. GSK-3α promotes oncogenic KRAS function in pancreatic cancer via TAK1-TAB stabilization and regulation of noncanonical NF-κB. Cancer Discov. 2013, 3, 690–703. [Google Scholar] [CrossRef] [PubMed]
- Banerji, V.; Frumm, S.M.; Ross, K.N.; Li, L.S.; Schinzel, A.C.; Hahn, C.K.; Kakoza, R.M.; Chow, K.T.; Ross, L.; Alexe, G.; et al. The intersection of genetic and chemical genomic screens identifies GSK-3α as a target in human acute myeloid leukemia. J. Clin. Investig. 2012, 122, 935–947. [Google Scholar] [CrossRef] [PubMed]
- Wagner, F.F.; Benajiba, L.; Campbell, A.J.; Weïwer, M.; Sacher, J.R.; Gale, J.P.; Ross, L.; Puissant, A.; Alexe, G.; Conway, A.; et al. Exploiting an Asp-Glu “switch” in glycogen synthase kinase 3 to design paralog-selective inhibitors for use in acute myeloid leukemia. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Duda, P.; Akula, S.M.; Abrams, S.L.; Steelman, L.S.; Martelli, A.M.; Cocco, L.; Ratti, S.; Candido, S.; Libra, M.; Montalto, G.; et al. Targeting GSK3 and associated signaling pathways involved in cancer. Cells 2020, 9, 1110. [Google Scholar] [CrossRef]
- Rizzieri, D.A.; Cooley, S.; Odenike, O.; Moonan, L.; Chow, K.H.; Jackson, K.; Wang, X.; Brail, L.; Borthakur, G. An open-label phase 2 study of glycogen synthase kinase-3 inhibitor LY2090314 in patients with acute leukemia. Leuk. Lymphoma 2016, 57, 1800–1806. [Google Scholar] [CrossRef]
- Gray, J.E.; Infante, J.R.; Brail, L.H.; Simon, G.R.; Cooksey, J.F.; Jones, S.F.; Farrington, D.L.; Yeo, A.; Jackson, K.A.; Chow, K.H.; et al. A first-in-human phase I dose-escalation, pharmacokinetic, and pharmacodynamic evaluation of intravenous LY2090314, a glycogen synthase kinase 3 inhibitor, administered in combination with pemetrexed and carboplatin. Investig. New Drugs 2015, 33, 1187–1196. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Fuchs, S.Y.; Ougolkov, A.V.; Spiegelman, V.S.; Minamoto, T. Oncogenic β-catenin signaling networks in colorectal cancer. Cell Cycle 2005, 4, 1522–1539. [Google Scholar] [CrossRef]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef]
- Albuquerque, C.; Breukel, C.; van der Luijt, R.; Fidalgo, P.; Lage, P.; Slors, F.J.; Leitão, C.N.; Fodde, R.; Smits, R. The ‘just-right’ signaling model: APC somatic mutations are selected based on a specific level of activation of the β-catenin signaling cascade. Hum. Mol. Genet. 2002, 11, 1549–1560. [Google Scholar] [CrossRef]
- Kim, K.; Pang, K.M.; Evans, M.; Hay, E.D. Overexpression of β-catenin induces apoptosis independent of its transactivation function with LEF-1 or the involvement of major G1 cell cycle regulators. Mol. Biol. Cell 2000, 11, 3509–3523. [Google Scholar] [CrossRef]
- Cho, Y.J.; Yoon, J.; Ko, Y.S.; Kim, S.Y.; Cho, S.J.; Kim, W.H.; Park, J.W.; Youn, H.D.; Kim, J.H.; Lee, B.L. Glycogen synthase kinase-3β does not correlate with the expression and activity of β-catenin in gastric cancer. APMIS 2010, 118, 782–790. [Google Scholar] [CrossRef] [PubMed]
- Al-Aynati, M.M.; Radulovich, N.; Riddell, R.H.; Tsao, M.S. Epithelial-cadherin and β-catenin expression changes in pancreatic intraepithelial neoplasia. Clin. Cancer Res. 2004, 10, 1235–1240. [Google Scholar] [CrossRef] [PubMed]
- Erdal, E.; Ozturk, N.; Cagatay, T.; Eksioglu-Demiralp, E.; Ozturk, M. Lithium-mediated downregulation of PKB/Akt and cyclin E with growth inhibition in hepatocellular carcinoma cells. Int. J. Cancer 2005, 115, 903–910. [Google Scholar] [CrossRef] [PubMed]
- Brack, A.S.; Conboy, I.M.; Conboy, M.J.; Shen, J.; Rando, T.A. A temporal switch from notch to Wnt signaling in muscle stem cells is necessary for normal adult myogenesis. Cell Stem Cell 2008, 2, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Brauer, C.; Thu, K.L.; Mason, J.M.; Blaser, H.; Bray, M.R.; Mak, T.W. Targeting mitosis in cancer: Emerging strategies. Mol. Cell 2015, 60, 524–536. [Google Scholar] [CrossRef]
- Gönczy, P. Centrosomes and cancer: Revisiting a long-standing relationship. Nat. Rev. Cancer 2015, 15, 639–652. [Google Scholar] [CrossRef]
- Mc Gee, M.M. Targeting the mitotic catastrophe signaling pathway in cancer. Mediat. Inflamm. 2015, 2015, 146282. [Google Scholar] [CrossRef]
- Ferreira, L.M.; Hebrant, A.; Dumont, J.E. Metabolic reprogramming of the tumor. Oncogene 2012, 31, 3999–4011. [Google Scholar] [CrossRef]
- Cantor, J.R.; Sabatini, D.M. Cancer cell metabolism: One hallmark, many faces. Cancer Discov. 2012, 2, 881–898. [Google Scholar] [CrossRef]
- Lu, J. The Warburg metabolism fuels tumor metastasis. Cancer Metastasis Rev. 2019, 38, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, S.; Miyamoto, S.; Kikuchi, O.; Goto, T.; Amanuma, Y.; Muto, M. Recent advances from basic and clinical studies of esophageal squamous cell carcinoma. Gastroenterology 2015, 149, 1700–1715. [Google Scholar] [CrossRef] [PubMed]
- Stuelten, C.H.; Parent, C.A.; Montell, D.J. Cell motility in cancer invasion and metastasis: Insights from simple model organisms. Nat. Rev. Cancer 2018, 18, 296–312. [Google Scholar] [CrossRef]
- Welch, D.R.; Hurst, D.R. Defining the hallmarks of metastasis. Cancer Res. 2019, 79, 3011–3027. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef]
- Brabletz, T.; Kalluri, R.; Nieto, M.A.; Weinberg, R.A. EMT in cancer. Nat. Rev. Cancer 2018, 18, 128–134. [Google Scholar] [CrossRef]
- Bunz, F. EMT and back again: Visualizing the dynamic phenotypes of metastasis. Cancer Res. 2020, 80, 153–155. [Google Scholar] [CrossRef]
- Williams, E.D.; Gao, D.; Redfern, A.; Thompson, E.W. Controversies around epithelial-mesenchymal plasticity in cancer metastasis. Nat. Rev. Cancer 2019, 19, 716–732. [Google Scholar] [CrossRef]
- Bachelder, R.E.; Yoon, S.O.; Franci, C.; de Herreros, A.G.; Mercurio, A.M. Glycogen synthase kinase-3 is an endogenous inhibitor of Snail transcription: Implications for the epithelial-mesenchymal transition. J. Cell Biol. 2005, 168, 29–33. [Google Scholar] [CrossRef]
- Sun, T.; Rodriguez, M.; Kim, L. Glycogen synthase kinase 3 in the world of cell migration. Dev. Growth Differ. 2009, 51, 735–742. [Google Scholar] [CrossRef]
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Primers 2016, 2, 16022. [Google Scholar] [CrossRef] [PubMed]
- Lapointe, S.; Perry, A.; Butowski, N.A. Primary brain tumours in adults. The Lancet 2018, 392, 432–446. [Google Scholar] [CrossRef]
- Machesky, L.M. Lamellipodia and filopodia in metastasis and invasion. FEBS Lett. 2008, 582, 2102–2111. [Google Scholar] [CrossRef]
- Paz, H.; Pathak, N.; Yang, J. Invading one step at a time: The role of invadopodia in tumor metastasis. Oncogene 2014, 33, 4193–4202. [Google Scholar] [CrossRef]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Vasan, N.; Baselga, J.; Hyman, D.M. A view on drug resistance in cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef]
- Alexander, S.; Friedl, P. Cancer invasion and resistance: Interconnected processes of disease progression and therapy failure. Trends Mol. Med. 2012, 18, 13–26. [Google Scholar] [CrossRef]
- Al-Lazikani, B.; Banerji, U.; Workman, P. Combinatorial drug therapy for cancer in the post-genomic era. Nat. Biotechnol. 2012, 30, 679–692. [Google Scholar] [CrossRef]
- Papadatos-Pastos, D.; De Miguel Luken, M.J.; Yap, T.A. Combining targeted therapeutics in the era of precision medicine. Br. J. Cancer 2015, 112, 1–3. [Google Scholar] [CrossRef][Green Version]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef]
- Soteriou, D.; Fuchs, Y. A matter of life and death: Stem cell survival in tissue regeneration and tumour formation. Nat. Rev. Cancer 2018, 18, 187–201. [Google Scholar] [CrossRef] [PubMed]
- Lytle, N.K.; Barber, A.G.; Reya, T. Stem cell fate in cancer growth, progression and therapy resistance. Nat. Rev. Cancer 2018, 18, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Clarke, M.F. Clinical and therapeutic implications of cancer stem cells. N. Engl. J. Med. 2019, 380, 2237–2245. [Google Scholar] [CrossRef]
- Marcucci, F.; Rumio, C.; Lefoulon, F. Anti-cancer stem-like cell compounds in clinical development—An overview and critical appraisal. Front. Oncol. 2016, 6, 115. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, J. The cancer stem cell gamble. Science 2015, 347, 226–229. [Google Scholar] [CrossRef] [PubMed]
- Prestegarden, L.; Enger, P. Cancer stem cells in the central nervous system—A critical review. Cancer Res. 2010, 70, 8255–8258. [Google Scholar] [CrossRef]
- Bahr, C.; Correia, N.C.; Trumpp, A. Stem cells make leukemia grow again. EMBO J. 2017, 36, 2667–2669. [Google Scholar] [CrossRef]
- Sato, N.; Meijer, L.; Skaltsounis, L.; Greengard, P.; Brivanlou, A.H. Maintenance of pluripotency in human and mouse embryonic stem cells through activation of Wnt signaling by a pharmacological GSK-3-specific inhibitor. Nat. Med. 2004, 10, 55–63. [Google Scholar] [CrossRef]
- Trowbridge, J.J.; Xenocostas, A.; Moon, R.T.; Bhatia, M. Glycogen synthase kinase-3 is an in vivo regulator of hematopoietic stem cell repopulation. Nat. Med. 2006, 12, 89–98. [Google Scholar] [CrossRef]
- Ko, K.H.; Holmes, T.; Palladinetti, P.; Song, E.; Nordon, R.; O’Brien, T.A.; Dolnikov, A. GSK-3β inhibition promotes engraftment of ex vivo-expanded hematopoietic stem cells and modulates gene expression. Stem Cells 2011, 29, 108–118. [Google Scholar] [CrossRef]
- Lapid, K.; Itkin, T.; D′Uva, G.; Ovadya, Y.; Ludin, A.; Caglio, G.; Kalinkovich, A.; Golan, K.; Porat, Z.; Zollo, M.; et al. GSK3β regulates physiological migration of stem/progenitor cells via cytoskeletal rearrangement. J. Clin. Investig. 2013, 123, 1705–1717. [Google Scholar] [CrossRef] [PubMed]
- Farkona, S.; Diamandis, E.P.; Blasutig, I.M. Cancer immunotherapy: The beginning of the end of cancer? BMC Med. 2016, 14, 73. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Wang, G.; Huang, D.; Sui, M.; Xu, Y. Cancer immunotherapy based on natural killer cells: Current progress and new opportunities. Front. Immunol. 2019, 10, 1205. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Correa, B.; Lopez-Sejas, N.; Duran, E.; Labella, F.; Alonso, C.; Solana, R.; Tarazona, R. Modulation of NK cells with checkpoint inhibitors in the context of cancer immunotherapy. Cancer Immunol. Immunother. 2019, 68, 861–870. [Google Scholar] [CrossRef]
- Suen, W.C.; Lee, W.Y.; Leung, K.T.; Pan, X.H.; Li, G. Natural killer cell-based cancer immunotherapy: A review on 10 years completed clinical trials. Cancer Investig. 2018, 36, 431–457. [Google Scholar] [CrossRef]
- Ben-Shmuel, A.; Biber, G.; Barda-Saad, M. Unleashing natural killer cells in the tumor microenvironment-The next generation of immunotherapy? Front. Immunol. 2020, 11, 275. [Google Scholar] [CrossRef]
- Gattinoni, L.; Zhong, X.S.; Palmer, D.C.; Ji, Y.; Hinrichs, C.S.; Yu, Z.; Wrzesinski, C.; Boni, A.; Cassard, L.; Garvin, L.M.; et al. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat. Med. 2009, 15, 808–813. [Google Scholar] [CrossRef]
- June, C.H.; Sadelain, M. Chimeric antigen receptor therapy. N. Engl. J. Med. 2018, 379, 64–73. [Google Scholar] [CrossRef]
- Ghimire, S.; Weber, D.; Mavin, E.; Wang, X.N.; Dickinson, A.M.; Holler, E. Pathophysiology of GvHD and other HSCT-related major complications. Front. Immunol. 2017, 8, 79. [Google Scholar] [CrossRef]
- Kanfar, S.; Al-Anazi, K. Autologous graft versus host disease: An updated review. Ann. Stem Cells Regen. Med. 2018, 1, 1002. [Google Scholar]
- Klamer, G.; Shen, S.; Song, E.; Rice, A.M.; Knight, R.; Lindeman, R.; O’Brien, T.A.; Dolnikov, A. GSK3 inhibition prevents lethal GVHD in mice. Exp. Hematol. 2013, 41, 39–55.e10. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Klamer, G.; Xu, N.; O′Brien, T.A.; Dolnikov, A. GSK-3β inhibition preserves naive T cell phenotype in bone marrow reconstituted mice. Exp. Hematol. 2013, 41, 1016–1027.e1011. [Google Scholar] [CrossRef] [PubMed]
- Dolnikov, A.; Xu, N.; Shen, S.; Song, E.; Holmes, T.; Klamer, G.; O’Brien, T.A. GSK-3β inhibition promotes early engraftment of ex vivo-expanded haematopoietic stem cells. Cell Prolif. 2014, 47, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Bloom, J.; Brandt, J. Chapter 11: Toxic responses of the blood. In Casarett’s & Doull’s Toxicology: The Basic Science of Poisons; The McGraw-Hill Companies: New York, NY, USA, 2008; pp. 455–484. [Google Scholar]
- Ziepert, M.; Schmits, R.; Trümper, L.; Pfreundschuh, M.; Loeffler, M. Prognostic factors for hematotoxicity of chemotherapy in aggressive non-Hodgkin’s lymphoma. Ann. Oncol. 2008, 19, 752–762. [Google Scholar] [CrossRef] [PubMed]
- Barcellos-Hoff, M.H.; Park, C.; Wright, E.G. Radiation and the microenvironment—Tumorigenesis and therapy. Nat. Rev. Cancer 2005, 5, 867–875. [Google Scholar] [CrossRef]
- Shukla, P.; Singh, R. Potential pharmacological interventions against hematotoxicity: An overview. Expert Rev. Hematol. 2015, 8, 505–514. [Google Scholar] [CrossRef]
- Barry, F.A.; Graham, G.J.; Fry, M.J.; Gibbins, J.M. Regulation of glycogen synthase kinase 3 in human platelets: A possible role in platelet function? FEBS Lett. 2003, 553, 173–178. [Google Scholar] [CrossRef]
- Ewertz, M.; Qvortrup, C.; Eckhoff, L. Chemotherapy-induced peripheral neuropathy in patients treated with taxanes and platinum derivatives. Acta Oncol. 2015, 54, 587–591. [Google Scholar] [CrossRef] [PubMed]
- Carozzi, V.A.; Canta, A.; Chiorazzi, A. Chemotherapy-induced peripheral neuropathy: What do we know about mechanisms? Neurosci. Lett. 2015, 596, 90–107. [Google Scholar] [CrossRef] [PubMed]
- Hou, S.; Huh, B.; Kim, H.K.; Kim, K.H.; Abdi, S. Treatment of chemotherapy-induced peripheral neuropathy: Systematic review and recommendations. Pain Phys. 2018, 21, 571–592. [Google Scholar]
- Hu, S.; Huang, K.M.; Adams, E.J.; Loprinzi, C.L.; Lustberg, M.B. Recent developments of novel pharmacologic therapeutics for prevention of chemotherapy-induced peripheral neuropathy. Clin. Cancer Res. 2019, 25, 6295–6301. [Google Scholar] [CrossRef] [PubMed]
- Cross, D.A.; Culbert, A.A.; Chalmers, K.A.; Facci, L.; Skaper, S.D.; Reith, A.D. Selective small-molecule inhibitors of glycogen synthase kinase-3 activity protect primary neurones from death. J. Neurochem. 2001, 77, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, L.; Kordes, S.; Reinhardt, P.; Glatza, M.; Baumann, M.; Drexler, H.C.A.; Menninger, S.; Zischinsky, G.; Eickhoff, J.; Fröb, C.; et al. Dual inhibition of GSK3β and CDK5 protects the cytoskeleton of neurons from neuroinflammatory-mediated degeneration in vitro and in vivo. Stem Cell Rep. 2019, 12, 502–517. [Google Scholar] [CrossRef] [PubMed]
- Eom, T.Y.; Roth, K.A.; Jope, R.S. Neural precursor cells are protected from apoptosis induced by trophic factor withdrawal or genotoxic stress by inhibitors of glycogen synthase kinase 3. J. Biol. Chem. 2007, 282, 22856–22864. [Google Scholar] [CrossRef] [PubMed]
- Mo, M.; Erdelyi, I.; Szigeti-Buck, K.; Benbow, J.H.; Ehrlich, B.E. Prevention of paclitaxel-induced peripheral neuropathy by lithium pretreatment. FASEB J. 2012, 26, 4696–4709. [Google Scholar] [CrossRef] [PubMed]
- Anderson, V.A.; Godber, T.; Smibert, E.; Weiskop, S.; Ekert, H. Cognitive and academic outcome following cranial irradiation and chemotherapy in children: A longitudinal study. Br. J. Cancer 2000, 82, 255–262. [Google Scholar] [CrossRef]
- Surma-aho, O.; Niemelä, M.; Vilkki, J.; Kouri, M.; Brander, A.; Salonen, O.; Paetau, A.; Kallio, M.; Pyykkönen, J.; Jääskeläinen, J. Adverse long-term effects of brain radiotherapy in adult low-grade glioma patients. Neurology 2001, 56, 1285–1290. [Google Scholar] [CrossRef]
- Thotala, D.K.; Hallahan, D.E.; Yazlovitskaya, E.M. Inhibition of glycogen synthase kinase 3β attenuates neurocognitive dysfunction resulting from cranial irradiation. Cancer Res. 2008, 68, 5859–5868. [Google Scholar] [CrossRef]
- Yang, E.S.; Nowsheen, S.; Wang, T.; Thotala, D.K.; Xia, F. Glycogen synthase kinase 3β inhibition enhances repair of DNA double-strand breaks in irradiated hippocampal neurons. Neuro Oncol. 2011, 13, 459–470. [Google Scholar] [CrossRef]
- Waber, D.P. Central nervous system late effects: A new frontier? Pediatric Blood Cancer 2011, 57, 355–356. [Google Scholar] [CrossRef]
- Elens, I.; Deprez, S.; Danckaerts, M.; Bijttebier, P.; Labarque, V.; Uyttebroeck, A.; Van Gool, S.; D′Hooge, R.; Lemiere, J. Neurocognitive sequelae in adult childhood leukemia survivors related to levels of phosphorylated tau. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef] [PubMed]
- Henson, L.A.; Maddocks, M.; Evans, C.; Davidson, M.; Hicks, S.; Higginson, I.J. Palliative care and the management of common distressing symptoms in advanced cancer: Pain, breathlessness, nausea and vomiting, and fatigue. J. Clin. Oncol. 2020, 38, 905–914. [Google Scholar] [CrossRef] [PubMed]
- Degenhardt, L.; Grebely, J.; Stone, J.; Hickman, M.; Vickerman, P.; Marshall, B.D.L.; Bruneau, J.; Altice, F.L.; Henderson, G.; Rahimi-Movaghar, A.; et al. Global patterns of opioid use and dependence: Harms to populations, interventions, and future action. The Lancet 2019, 394, 1560–1579. [Google Scholar] [CrossRef]
- Li, Y.; Eitan, S.; Wu, J.; Evans, C.J.; Kieffer, B.; Sun, X.; Polakiewicz, R.D. Morphine induces desensitization of insulin receptor signaling. Mol. Cell Biol. 2003, 23, 6255–6266. [Google Scholar] [CrossRef][Green Version]
- Muller, D.L.; Unterwald, E.M. In vivo regulation of extracellular signal-regulated protein kinase (ERK) and protein kinase B (Akt) phosphorylation by acute and chronic morphine. J. Pharmacol. Exp. Ther. 2004, 310, 774–782. [Google Scholar] [CrossRef]
- Johnston, I.N.; Westbrook, R.F. Inhibition of morphine analgesia by lithium: Role of peripheral and central opioid receptors. Behav. Brain Res. 2004, 151, 151–158. [Google Scholar] [CrossRef]
- Parkitna, J.R.; Obara, I.; Wawrzczak-Bargiela, A.; Makuch, W.; Przewlocka, B.; Przewlocki, R. Effects of glycogen synthase kinase 3β and cyclin-dependent kinase 5 inhibitors on morphine-induced analgesia and tolerance in rats. J. Pharmacol. Exp. Ther. 2006, 319, 832–839. [Google Scholar] [CrossRef]
- Liao, W.W.; Tsai, S.Y.; Liao, C.C.; Chen, K.B.; Yeh, G.C.; Chen, J.Y.; Wen, Y.R. Coadministration of glycogen-synthase kinase 3 inhibitor with morphine attenuates chronic morphine-induced analgesic tolerance and withdrawal syndrome. J. Chin. Med. Assoc. 2014, 77, 31–37. [Google Scholar] [CrossRef]
- The ESMO/European Sarcoma Network Working Group. Bone sarcomas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2014, 25 (Suppl. 3), iii113–iii123. [Google Scholar] [CrossRef]
- Gilbert, N.F.; Cannon, C.P.; Lin, P.P.; Lewis, V.O. Soft-tissue sarcoma. J. Am. Acad. Orthopeadic Surg. 2009, 17, 40–47. [Google Scholar] [CrossRef]
- Botter, S.M.; Neri, D.; Fuchs, B. Recent advances in osteosarcoma. Curr. Opin. Pharmacol. 2014, 16, 15–23. [Google Scholar] [CrossRef]
- Kawai, A.; Yonemori, K.; Takahashi, S.; Araki, N.; Ueda, T. Systemic therapy for soft tissue sarcoma: Proposals for the optimal use of pazopanib, trabectedin, and eribulin. Adv. Ther. 2017, 34, 1556–1571. [Google Scholar] [CrossRef] [PubMed]
- Kansara, M.; Teng, M.W.; Smyth, M.J.; Thomas, D.M. Translational biology of osteosarcoma. Nat. Rev. Cancer 2014, 14, 722–735. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Poulin, N.M.; Nielsen, T.O. New strategies in sarcoma: Linking genomic and immunotherapy approaches to molecular subtype. Clin. Cancer Res. 2015, 21, 4753–4759. [Google Scholar] [CrossRef] [PubMed]
- Dufresne, A.; Brahmi, M.; Karanian, M.; Blay, J.Y. Using biology to guide the treatment of sarcomas and aggressive connective-tissue tumours. Nat. Rev. Clin. Oncol. 2018, 15, 443–458. [Google Scholar] [CrossRef]
- Cai, Y.; Mohseny, A.B.; Karperien, M.; Hogendoorn, P.C.; Zhou, G.; Cleton-Jansen, A.M. Inactive Wnt/β-catenin pathway in conventional high-grade osteosarcoma. J. Pathol. 2010, 220, 24–33. [Google Scholar] [CrossRef]
- Thomas, D.M. Wnts, bone and cancer. J. Pathol. 2010, 220, 1–4. [Google Scholar] [CrossRef]
- Matushansky, I.; Hernando, E.; Socci, N.D.; Mills, J.E.; Matos, T.A.; Edgar, M.A.; Singer, S.; Maki, R.G.; Cordon-Cardo, C. Derivation of sarcomas from mesenchymal stem cells via inactivation of the Wnt pathway. J. Clin. Investig. 2007, 117, 3248–3257. [Google Scholar] [CrossRef]
- Hartmann, C. A Wnt canon orchestrating osteoblastogenesis. Trends Cell Biol. 2006, 16, 151–158. [Google Scholar] [CrossRef]
- Krishnan, V.; Bryant, H.U.; Macdougald, O.A. Regulation of bone mass by Wnt signaling. J. Clin. Investig. 2006, 116, 1202–1209. [Google Scholar] [CrossRef]
- Bodine, P.V.; Komm, B.S. Wnt signaling and osteoblastogenesis. Rev. Endocr. Metab. Disord. 2006, 7, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Ralston, S.H.; de Crombrugghe, B. Genetic regulation of bone mass and susceptibility to osteoporosis. Genes Dev. 2006, 20, 2492–2506. [Google Scholar] [CrossRef] [PubMed]
- Baron, R.; Kneissel, M. WNT signaling in bone homeostasis and disease: From human mutations to treatments. Nat. Med. 2013, 19, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Doble, B.W.; MacAulay, K.; Sinclair, E.M.; Drucker, D.J.; Woodgett, J.R. Tissue-specific role of glycogen synthase kinase 3β in glucose homeostasis and insulin action. Mol. Cell Biol. 2008, 28, 6314–6328. [Google Scholar] [CrossRef] [PubMed]
- Ragozzino, E.; Brancaccio, M.; Di Costanzo, A.; Scalabrì, F.; Andolfi, G.; Wanderlingh, L.G.; Patriarca, E.J.; Minchiotti, G.; Altamura, S.; Varrone, F.; et al. 6-Bromoindirubin-3′-oxime intercepts GSK3 signaling to promote and enhance skeletal muscle differentiation affecting miR-206 expression in mice. Sci. Rep. 2019, 9, 18091. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Chu, Y.; Lv, X.; Qiu, P.; Liu, C.; Zhang, H.; Li, D.; Peng, S.; Dou, Z.; Hua, J. GSK3 inhibitor-BIO regulates proliferation of immortalized pancreatic mesenchymal stem cells (iPMSCs). PLoS ONE 2012, 7, e31502. [Google Scholar] [CrossRef]
- Tatullo, M.; Makeeva, I.; Rengo, S.; Rengo, C.; Spagnuolo, G.; Codispoti, B. Small molecule GSK-3 antagonists play a pivotal role in reducing the local inflammatory response, in promoting resident stem cell activation and in improving tissue repairing in regenerative dentistry. Histol. Histopathol. 2019, 34, 1195–1203. [Google Scholar] [CrossRef]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef]
- Atkins, M.B.; Larkin, J. Immunotherapy combined or sequenced with targeted therapy in the treatment of solid tumors: Current perspectives. J. Natl. Cancer Inst. 2016, 108, djv414. [Google Scholar] [CrossRef]
- Cogdill, A.P.; Andrews, M.C.; Wargo, J.A. Hallmarks of response to immune checkpoint blockade. Br. J. Cancer 2017, 117, 1–7. [Google Scholar] [CrossRef]
- Kim, J.M.; Chen, D.S. Immune escape to PD-L1/PD-1 blockade: Seven steps to success (or failure). Ann. Oncol. 2016, 27, 1492–1504. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Postow, M.A.; Sidlow, R.; Hellmann, M.D. Immune-related adverse events associated with immune checkpoint blockade. N. Engl. J. Med. 2018, 378, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Park, R.; Lopes, L.; Cristancho, C.R.; Riano, I.M.; Saeed, A. Treatment-related adverse events of combination immune checkpoint inhibitors: Systematic review and meta-analysis. Front. Oncol. 2020, 10, 258. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.; Harker, J.A.; Chanthong, K.; Stevenson, P.G.; Zuniga, E.I.; Rudd, C.E. Glycogen synthase kinase 3 inactivation drives T-bet-mediated downregulation of co-receptor PD-1 to enhance CD8+ cytolytic T cell responses. Immunity 2016, 44, 274–286. [Google Scholar] [CrossRef]
- Taylor, A.; Rothstein, D.; Rudd, C.E. Small-molecule inhibition of PD-1 transcription is an effective alternative to antibody blockade in cancer therapy. Cancer Res. 2018, 78, 706–717. [Google Scholar] [CrossRef]
- Jiao, S.; Xia, W.; Yamaguchi, H.; Wei, Y.; Chen, M.K.; Hsu, J.M.; Hsu, J.L.; Yu, W.H.; Du, Y.; Lee, H.H.; et al. PARP inhibitor upregulates PD-L1 expression and enhances cancer-associated immunosuppression. Clin. Cancer Res. 2017, 23, 3711–3720. [Google Scholar] [CrossRef]
- Taylor, A.; Rudd, C.E. Glycogen synthase kinase 3 inactivation compensates for the lack of CD28 in the priming of CD8+ cytotoxic T-cells: Implications for anti-PD-1 immunotherapy. Front. Immunol. 2017, 8, 1653. [Google Scholar] [CrossRef]
- Kamphorst, A.O.; Wieland, A.; Nasti, T.; Yang, S.; Zhang, R.; Barber, D.L.; Konieczny, B.T.; Daugherty, C.Z.; Koenig, L.; Yu, K.; et al. Rescue of exhausted CD8 T cells by PD-1-targeted therapies is CD28-dependent. Science 2017, 355, 1423–1427. [Google Scholar] [CrossRef]
- Taylor, A.; Rudd, C.E. Small molecule inhibition of glycogen synthase kinase-3 in cancer immunotherapy. Adv. Exp. Med. Biol. 2019, 1164, 225–233. [Google Scholar] [CrossRef]
- De Simone, V.; Pallone, F.; Monteleone, G.; Stolfi, C. Role of TH17 cytokines in the control of colorectal cancer. Oncoimmunology 2013, 2, e26617. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Wu, P.; Huang, Q.; Liu, Y.; Ye, J.; Huang, J. Interleukin-17: A promoter in colorectal cancer progression. Clin. Dev. Immunol. 2013, 2013, 436307. [Google Scholar] [CrossRef]
- Housseau, F.; Wu, S.; Wick, E.C.; Fan, H.; Wu, X.; Llosa, N.J.; Smith, K.N.; Tam, A.; Ganguly, S.; Wanyiri, J.W.; et al. Redundant innate and adaptive sources of IL17 production drive colon tumorigenesis. Cancer Res. 2016, 76, 2115–2124. [Google Scholar] [CrossRef] [PubMed]
- Amicarella, F.; Muraro, M.G.; Hirt, C.; Cremonesi, E.; Padovan, E.; Mele, V.; Governa, V.; Han, J.; Huber, X.; Droeser, R.A.; et al. Dual role of tumour-infiltrating T helper 17 cells in human colorectal cancer. Gut 2017, 66, 692–704. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Fei, M.; Wu, Y.; Zheng, D.; Wan, D.; Wang, L.; Li, D. Distribution and clinical significance of Th17 cells in the tumor microenvironment and peripheral blood of pancreatic cancer patients. Int. J. Mol. Sci. 2011, 12, 7424–7437. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yi, T.; Kortylewski, M.; Pardoll, D.M.; Zeng, D.; Yu, H. IL-17 can promote tumor growth through an IL-6-Stat3 signaling pathway. J. Exp. Med. 2009, 206, 1457–1464. [Google Scholar] [CrossRef] [PubMed]
- De Simone, V.; Franzè, E.; Ronchetti, G.; Colantoni, A.; Fantini, M.C.; Di Fusco, D.; Sica, G.S.; Sileri, P.; MacDonald, T.T.; Pallone, F.; et al. Th17-type cytokines, IL-6 and TNF-α synergistically activate STAT3 and NF-kB to promote colorectal cancer cell growth. Oncogene 2015, 34, 3493–3503. [Google Scholar] [CrossRef]
- Loncle, C.; Bonjoch, L.; Folch-Puy, E.; Lopez-Millan, M.B.; Lac, S.; Molejon, M.I.; Chuluyan, E.; Cordelier, P.; Dubus, P.; Lomberk, G.; et al. IL17 functions through the novel REG3β-JAK2-STAT3 inflammatory pathway to promote the transition from chronic pancreatitis to pancreatic cancer. Cancer Res. 2015, 75, 4852–4862. [Google Scholar] [CrossRef]
- Beurel, E.; Jope, R.S. Differential regulation of STAT family members by glycogen synthase kinase-3. J. Biol. Chem. 2008, 283, 21934–21944. [Google Scholar] [CrossRef]
- Beurel, E.; Yeh, W.I.; Michalek, S.M.; Harrington, L.E.; Jope, R.S. Glycogen synthase kinase-3 is an early determinant in the differentiation of pathogenic Th17 cells. J. Immunol. 2011, 186, 1391–1398. [Google Scholar] [CrossRef]
- Prior, I.A.; Lewis, P.D.; Mattos, C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012, 72, 2457–2467. [Google Scholar] [CrossRef] [PubMed]
- Jinesh, G.G.; Sambandam, V.; Vijayaraghavan, S.; Balaji, K.; Mukherjee, S. Molecular genetics and cellular events of K-Ras-driven tumorigenesis. Oncogene 2018, 37, 839–846. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.D.; Fesik, S.W.; Kimmelman, A.C.; Luo, J.; Der, C.J. Drugging the undruggable RAS: Mission possible? Nat. Rev. Drug Discov. 2014, 13, 828–851. [Google Scholar] [CrossRef] [PubMed]
- Ostrem, J.M.; Shokat, K.M. Direct small-molecule inhibitors of KRAS: From structural insights to mechanism-based design. Nat. Rev. Drug Discov. 2016, 15, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Patricelli, M.P.; Janes, M.R.; Li, L.S.; Hansen, R.; Peters, U.; Kessler, L.V.; Chen, Y.; Kucharski, J.M.; Feng, J.; Ely, T.; et al. Selective inhibition of oncogenic KRAS output with small molecules targeting the inactive state. Cancer Discov. 2016, 6, 316–329. [Google Scholar] [CrossRef] [PubMed]
- Welsch, M.E.; Kaplan, A.; Chambers, J.M.; Stokes, M.E.; Bos, P.H.; Zask, A.; Zhang, Y.; Sanchez-Martin, M.; Badgley, M.A.; Huang, C.S.; et al. Multivalent small-molecule pan-RAS inhibitors. Cell 2017, 168, 878–889. [Google Scholar] [CrossRef] [PubMed]
- Keeton, A.B.; Salter, E.A.; Piazza, G.A. The RAS-effector interaction as a drug target. Cancer Res. 2017, 77, 221–226. [Google Scholar] [CrossRef]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef]
- Hallin, J.; Engstrom, L.D.; Hargis, L.; Calinisan, A.; Aranda, R.; Briere, D.M.; Sudhakar, N.; Bowcut, V.; Baer, B.R.; Ballard, J.A.; et al. The KRASG12C inhibitor MRTX849 provides insight toward therapeutic susceptibility of KRAS-mutant cancers in mouse models and patients. Cancer Discov. 2020, 10, 54–71. [Google Scholar] [CrossRef]
- Babina, I.S.; Turner, N.C. Advances and challenges in targeting FGFR signalling in cancer. Nat. Rev. Cancer 2017, 17, 318–332. [Google Scholar] [CrossRef]
- Harrison, P.T.; Huang, P.H. Exploiting vulnerabilities in cancer signalling networks to combat targeted therapy resistance. Essays Biochem. 2018, 62, 583–593. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.Y.; Zhao, Y.; Aronowitz, J.; Mai, T.T.; Vides, A.; Qeriqi, B.; Kim, D.; Li, C.; de Stanchina, E.; Mazutis, L.; et al. Rapid non-uniform adaptation to conformation-specific KRAS(G12C) inhibition. Nature 2020, 577, 421–425. [Google Scholar] [CrossRef] [PubMed]
- Hata, A.N.; Shaw, A.T. Resistance looms for KRASG12C inhibitors. Nat. Med. 2020, 26, 169–170. [Google Scholar] [CrossRef] [PubMed]
- Ryan, M.B.; Fece de la Cruz, F.; Phat, S.; Myers, D.T.; Wong, E.; Shahzade, H.A.; Hong, C.B.; Corcoran, R.B. Vertical pathway inhibition overcomes adaptive feedback resistance to KRASG12C inhibition. Clin. Cancer Res. 2020, 26, 1633–1643. [Google Scholar] [CrossRef] [PubMed]
- Yaeger, R.; Solit, D.B. Overcoming adaptive resistance to KRAS inhibitors through vertical pathway targeting. Clin. Cancer Res. 2020, 26, 1538–1540. [Google Scholar] [CrossRef] [PubMed]
- June, C.H. Drugging the undruggable ras—immunotherapy to the rescue? N. Engl. J. Med. 2016, 375, 2286–2289. [Google Scholar] [CrossRef]
- Chatani, P.D.; Yang, J.C. Mutated RAS: Targeting the “untargetable” with T cells. Clin. Cancer Res. 2020, 26, 537–544. [Google Scholar] [CrossRef]



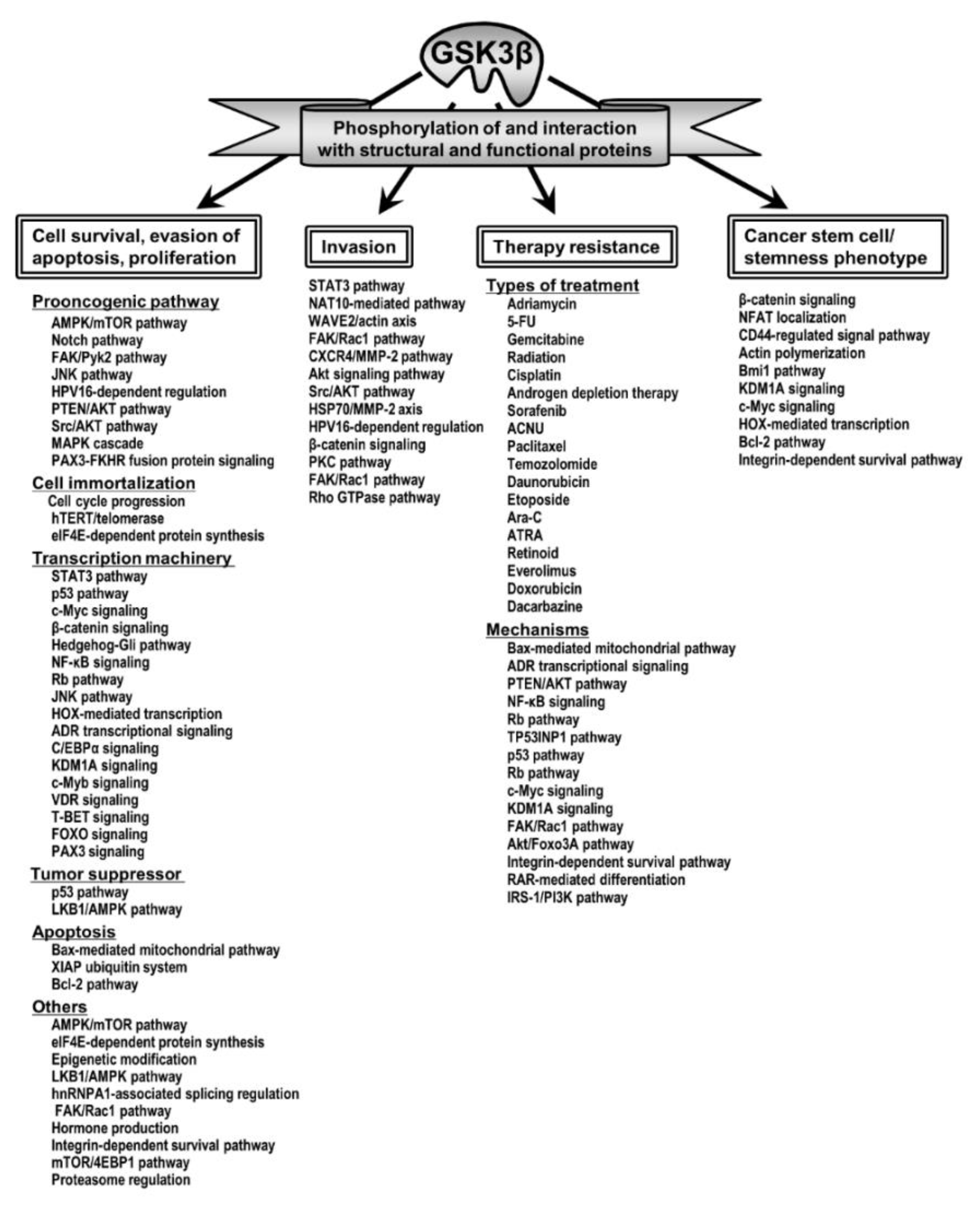{kind=link}
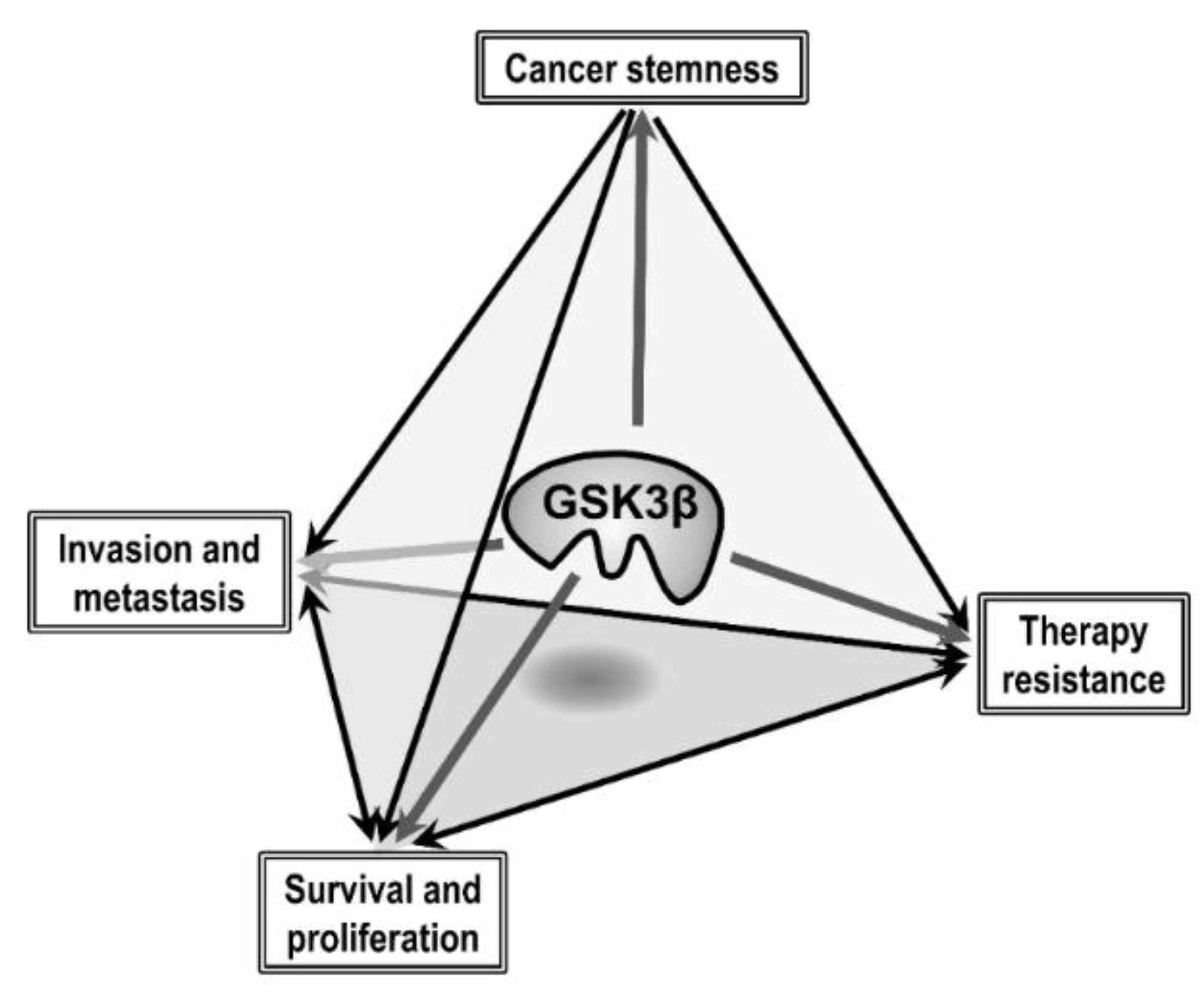{kind=link}
| Role of GSK3β in Biological Characteristics of Cancer | Reference | ||||
|---|---|---|---|---|---|
| Cell survival, evasion of apoptosis, and proliferation | Invasion | Therapy resistance *1 | Cancer stem cell/stemness phenotype | ||
| Digestive system | |||||
| Esophageal cancer(ESCC) | cell cycle progression; STAT3 pathway | [26,27] | |||
| Stomach cancer | hTERT/telomerase | STAT3 pathway | [28,29,30,31] | ||
| Colorectal cancer | p53 pathway; Bax-mediated mitochondrial pathway; TRAIL receptor-dependent synthetic lethal system; c-Myc signaling; β-catenin signaling; hTERT/telomerase; Hedgehog-Gli pathway; FAK/Pyk2 pathway; cell cycle progression; NF-κB signaling | N-acetyltransferase 10-mediated pathway; WAVE2/actin axis | adriamycin; 5-FU; Bax-mediated mitochondrial pathway; p53 pathway | β-catenin signaling; NFAT localization | [28,29,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51] |
| Pancreatic cancer | NF-κB signaling; hTERT/telomerase; XIAP ubiquitin system; TRAIL receptor-dependent synthetic lethal system; JNK pathway; Rb pathway; Notch pathway; TFEB signaling; STAT3 pathway; c-Myc signaling; β-catenin signaling; cell cycle progression | FAK/Rac1 pathway; CXCR4/MMP-2 axis; Akt signaling pathway | TP53INP1 pathway; Rb pathway | [28,29,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70] | |
| Liver cancer (HCC) | Rb pathway; hTERT/telomerase; TRAIL receptor-dependent synthetic lethal system; cell cycle progression; NF-κB signaling | [28,29,71,72,73,74] | |||
| Head and neck cancer | |||||
| HNSCC | TLR-induced cytokine signaling | CD44-regulated signaling pathway | [75,76] | ||
| Lung cancer | |||||
| NSCLC | hTERT/telomerase; β-catenin signaling; NF-κB signaling | [38,77,78,79] | |||
| Breast cancer | eIF4E-dependent protein synthesis; epigenetic modification; cell cycle progression; PTEN/AKT pathway | PTEN/AKT pathway | [47,80,81,82] | ||
| Prostate cancer | TRAIL receptor-dependent synthetic lethal system; androgen receptor transcriptional signaling; cell cycle progression; C/EBPα signaling; Src/AKT pathway; LKB1/AMPK pathway | Src/AKT pathway | androgen receptor transcriptional signaling | actin polymerization | [83,84,85,86,87,88,89,90,91,92,93] |
| Urinary system | |||||
| Renal cell carcinoma | NF-κB signaling; AMPK/mTOR pathway, cell cycle progression | NF-κB signaling | [94,95,96,97] | ||
| Bladder cancer | hTERT/telomerase; NF-κB signaling; cell cycle progression | HSP70/MMP-2 axis | [38,98,99,100] | ||
| Female genital system | |||||
| Ovarian cancer | cell cycle progression; hTERT/telomerase; p53 pathway | [38,101,102,103,104] | |||
| Endometrial cancer | cell cycle progression | p53 pathway | [105] | ||
| Cervical cancer | hTERT/telomerase; HPV16-dependent regulation | HPV16-dependent regulation | [38,106] | ||
| Central and peripheral nervous system | |||||
| Glioblastoma | TRAIL receptor-dependent synthetic lethal system; c-Myc signaling; NF-κB signaling; Bax-mediated mitochondrial pathway; cell cycle progression; hnRNPA1-associated splicing regulation; KDM1A signaling; FAK/Rac1 pathway | β-catenin signaling; PKC pathway; FAK/Rac1 pathway; Rho GTPase pathway | p53 pathway; Rb pathway; c-Myc signaling; KDM1A signaling; FAK/Rac1 pathway; NFAT/FasL signaling | Bmi1 pathway; KDM1A signaling; c-Myc signaling | [107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123] |
| Neuronal tumors | hormone production; cell cycle progression; Myc signaling; p53 pathway | [124,125,126,127] | |||
| Hematopoietic system | |||||
| Leukemia | NF-κB signaling; β-catenin signaling; cell cycle progression; HOX-mediated transcription; integrin-dependent survival pathway; Bcl-2 pathway; c-Myb signaling; cell cycle progression; mTOR/4EBP1 pathway; MAPK cascade; VDR signaling; T-BET signaling | NF-κB signaling; Akt/Foxo3A pathway; integrin-dependent survival pathway; RAR-mediated differentiation | HOX-mediated transcription; Bcl-2 pathway; integrin-dependent survival pathway | [128,129,130,131,132,133,134,135,136,137,138,139,140,141,142,143,144] | |
| Myeloma | FOXO signaling | [145] | |||
| Endocrine and neuroendocrine system | |||||
| Thyroid cancer | cell cycle progression; hormone production | [146,147] | |||
| Neuroendocrine tumors | proteasome regulation; cell cycle progression | cell cycle progression; IRS-1/PI3K pathway | [148,149,150] | ||
| Bone and soft tissue | |||||
| Osteosarcoma | NF-κB signaling; β-catenin signaling | NF-κB signaling | [151,152,153,154] | ||
| Soft tissue sarcomas | PAX3-FKHR fusion protein signaling; β-catenin signaling; cell cycle progression | [155,156,157] | |||
| Melanoma | p53 pathway; PAX3 signaling; β-catenin signaling | β-catenin signaling | [158,159,160] | ||
| GSK3β Inhibitor (Company) | Cancer Type | Trial ID and Phase | Combined Regimen | URL (Access Date: 15 May 2020) | Reference |
|---|---|---|---|---|---|
| LY2090314 (Eli Lilly) | Acute leukemia | NCT01214603 Phase II | none | https://clinicaltrials.gov/ct2/show/NCT01214603 | [177] |
| Metastatic pancreatic cancer | NCT01632306 Phase I/II | Gemcitabine, FOLFOX, or Gemcitabine + nab-paclitaxel | https://clinicaltrials.gov/ct2/show/NCT01632306 | ||
| Advanced or metastatic solid cancer | NCT01287520 Phase I | Pemetrexed + carboplatin | https://clinicaltrials.gov/show/NCT01287520 | [178] | |
| Lithium carbonate | Localized prostate cancer | NCT02198859 Phase I | none | https://clinicaltrials.gov/ct2/show/record/NCT02198859 | |
| 9-ING-41 | 29 advanced cancer types | NCT03678883 | chemotherapeutics | https://clinicaltrials.gov/ct2/show/NCT03678883 | |
| CLOVA cocktail | Advanced pancreatic cancer | UMIN000005095 Phase I/II | Gemcitabine | https://upload.umin.ac.jp/cgi-open-bin/ctr/ctr.cgi?function=brows&action=brows&type=summary&recptno=R000006032&language=E | |
| Recurrent glioblastoma | UMIN000005111 Phase I/II | Temozolomide | https://upload.umin.ac.jp/cgi-open-bin/ctr/ctr.cgi?function=brows&action=brows&type=summary&recptno=R000002506&language=E | [119] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Domoto, T.; Uehara, M.; Bolidong, D.; Minamoto, T. Glycogen Synthase Kinase 3β in Cancer Biology and Treatment. Cells 2020, 9, 1388. https://doi.org/10.3390/cells9061388
Domoto T, Uehara M, Bolidong D, Minamoto T. Glycogen Synthase Kinase 3β in Cancer Biology and Treatment. Cells. 2020; 9(6):1388. https://doi.org/10.3390/cells9061388
Chicago/Turabian StyleDomoto, Takahiro, Masahiro Uehara, Dilireba Bolidong, and Toshinari Minamoto. 2020. "Glycogen Synthase Kinase 3β in Cancer Biology and Treatment" Cells 9, no. 6: 1388. https://doi.org/10.3390/cells9061388
APA StyleDomoto, T., Uehara, M., Bolidong, D., & Minamoto, T. (2020). Glycogen Synthase Kinase 3β in Cancer Biology and Treatment. Cells, 9(6), 1388. https://doi.org/10.3390/cells9061388