Epigenetic Analysis of Circulating Tumor DNA in Localized and Metastatic Prostate Cancer: Evaluation of Clinical Biomarker Potential

 , , ,
, , , 
Abstract
1. Introduction
2. Materials and Methods
2.1. External 450K Methylation Data (Marmal-Aid)
2.2. In-House Patient Samples
2.3. DNA Isolation from Tissue and Blood
2.4. Bisulfite Conversion
2.5. Quantitative Methylation-Specific PCR (qMSP)
2.6. Methylation-Specific Droplet Digital PCR (MS-ddPCR)
2.7. cfDNA Quantification before and after Bisulfite Conversion
2.8. Statistical Analysis
2.9. Ethics Approval and Consent to Participate
3. Results
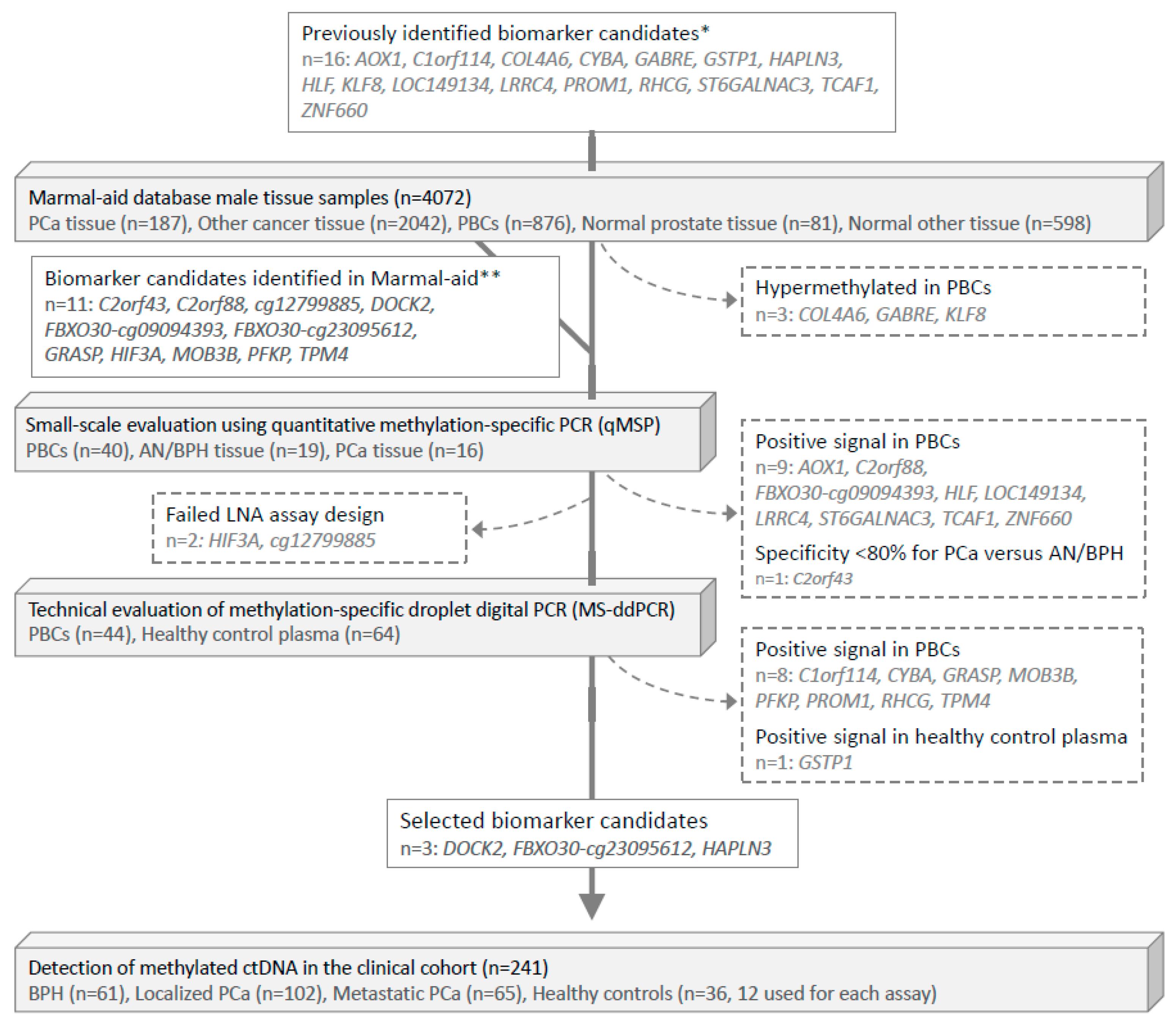
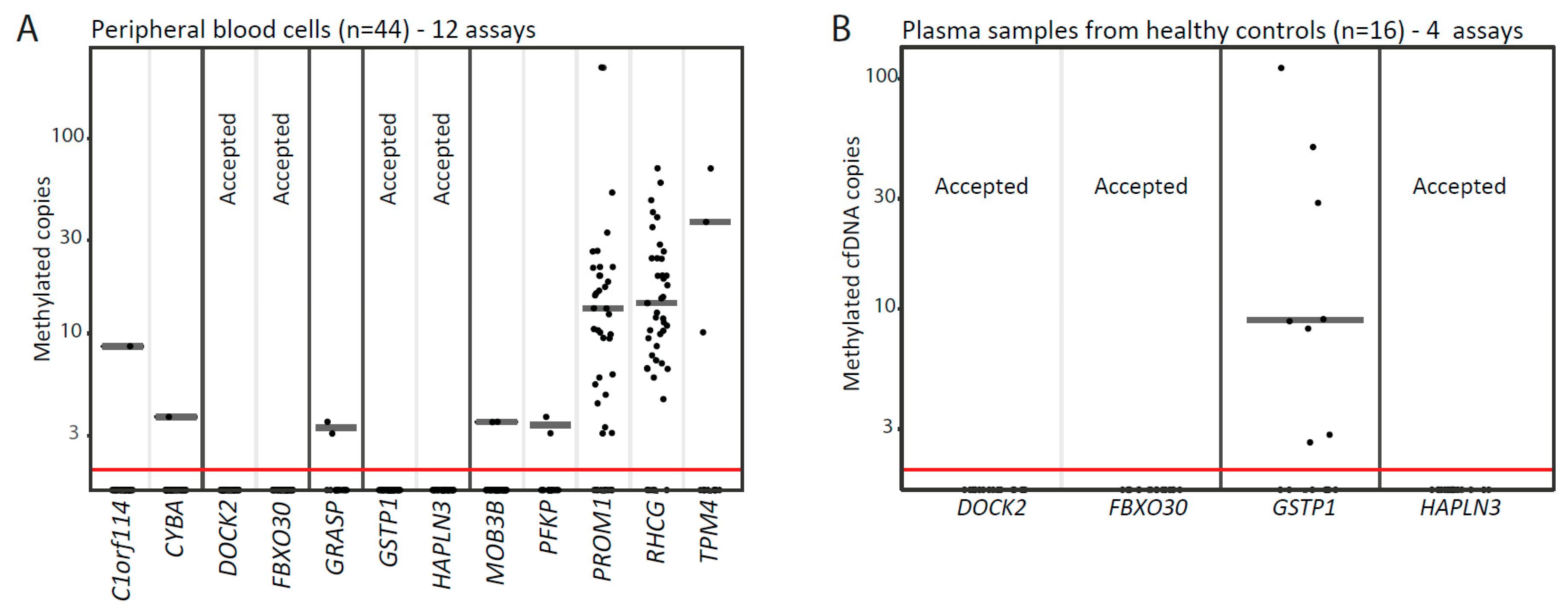
3.1. Identification of PCa-Specific DNA Methylation Biomarker Candidates Suitable for Blood-Based Analyses
3.2. CfDNA Abundance in the Clinical Cohort
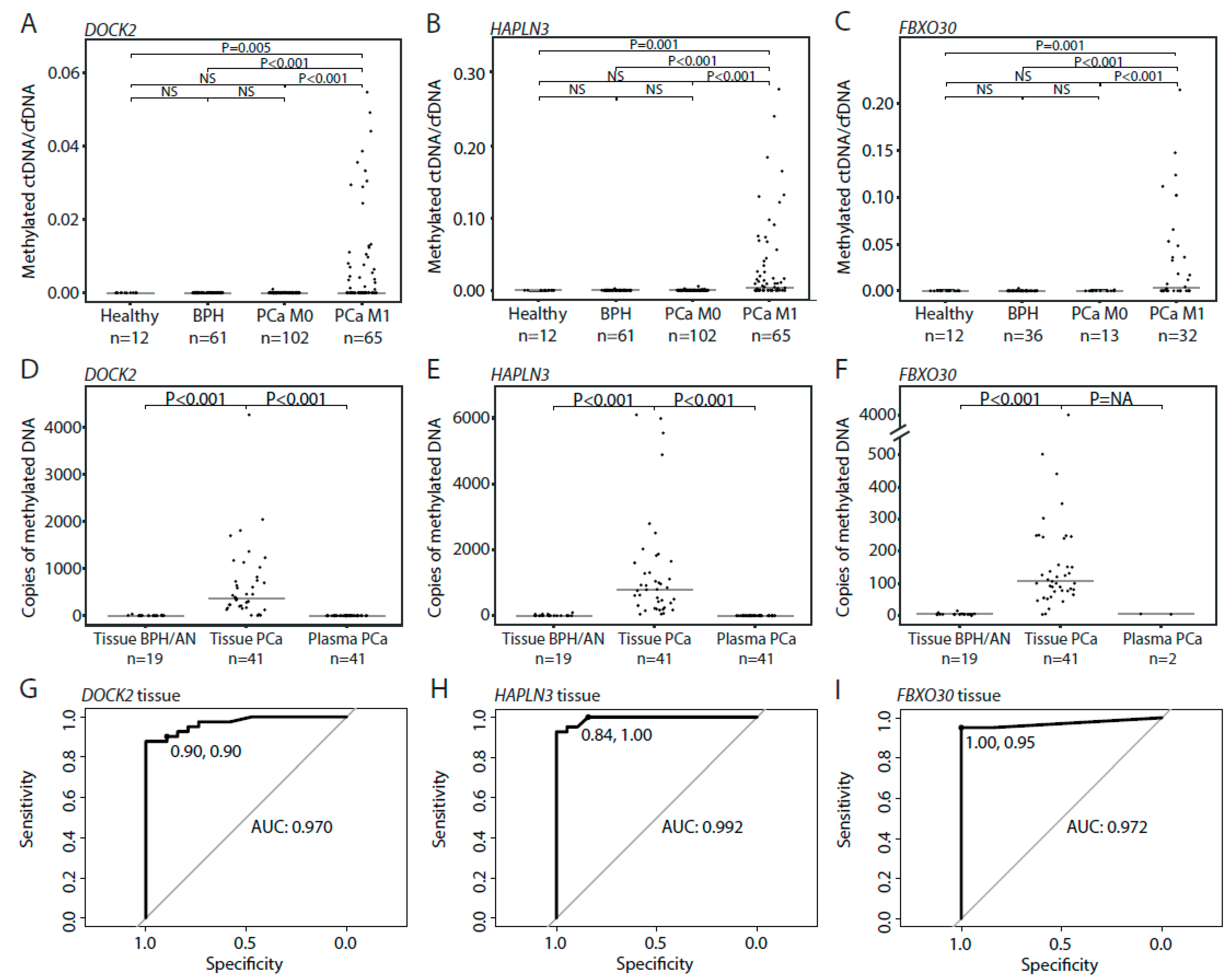
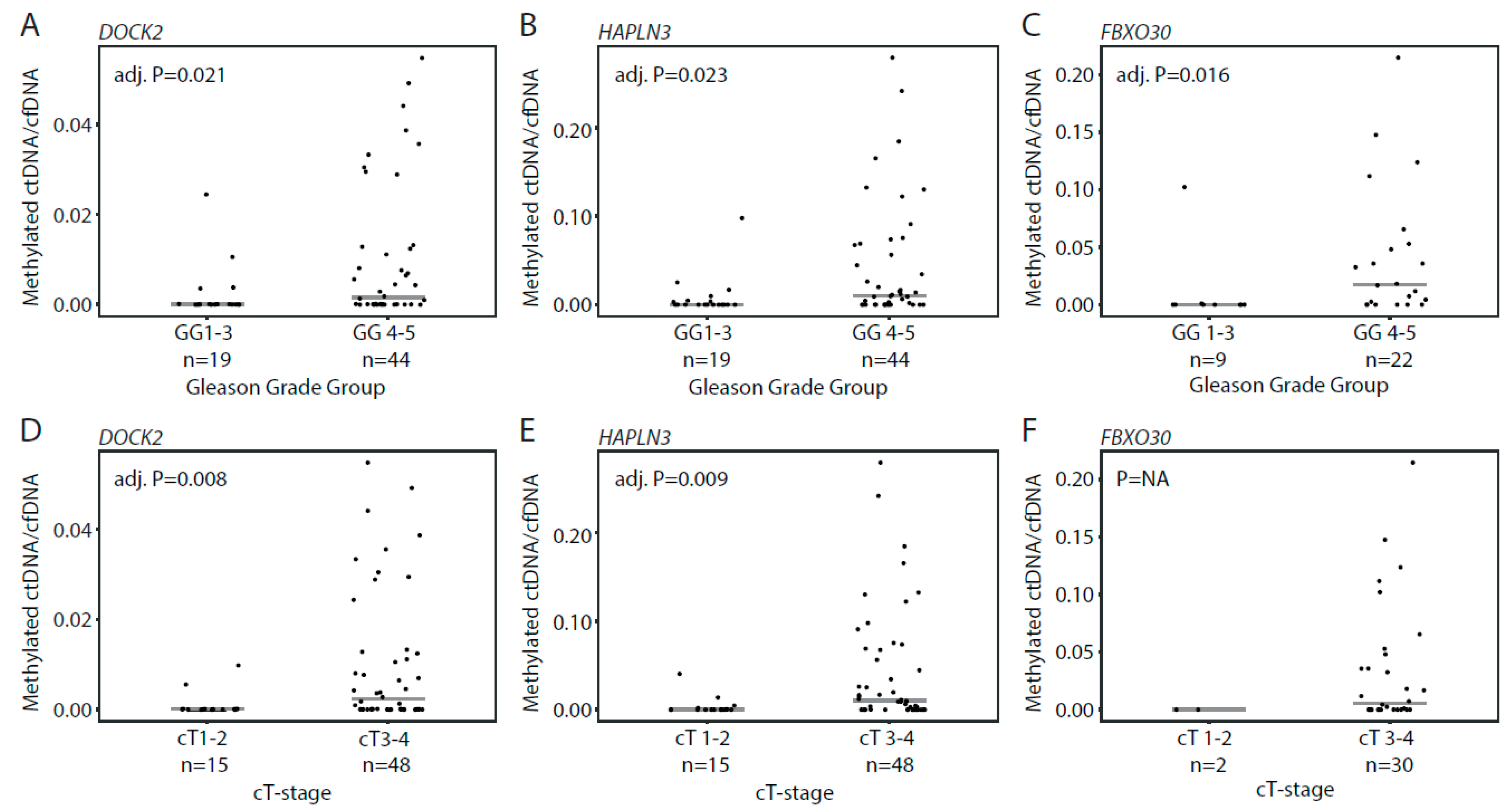
3.3. Detection of Methylated ctDNA in Plasma Samples from Patients with Localized PCa
3.4. Detection of Methylated ctDNA in Plasma Samples from Patients with De Novo mPCa
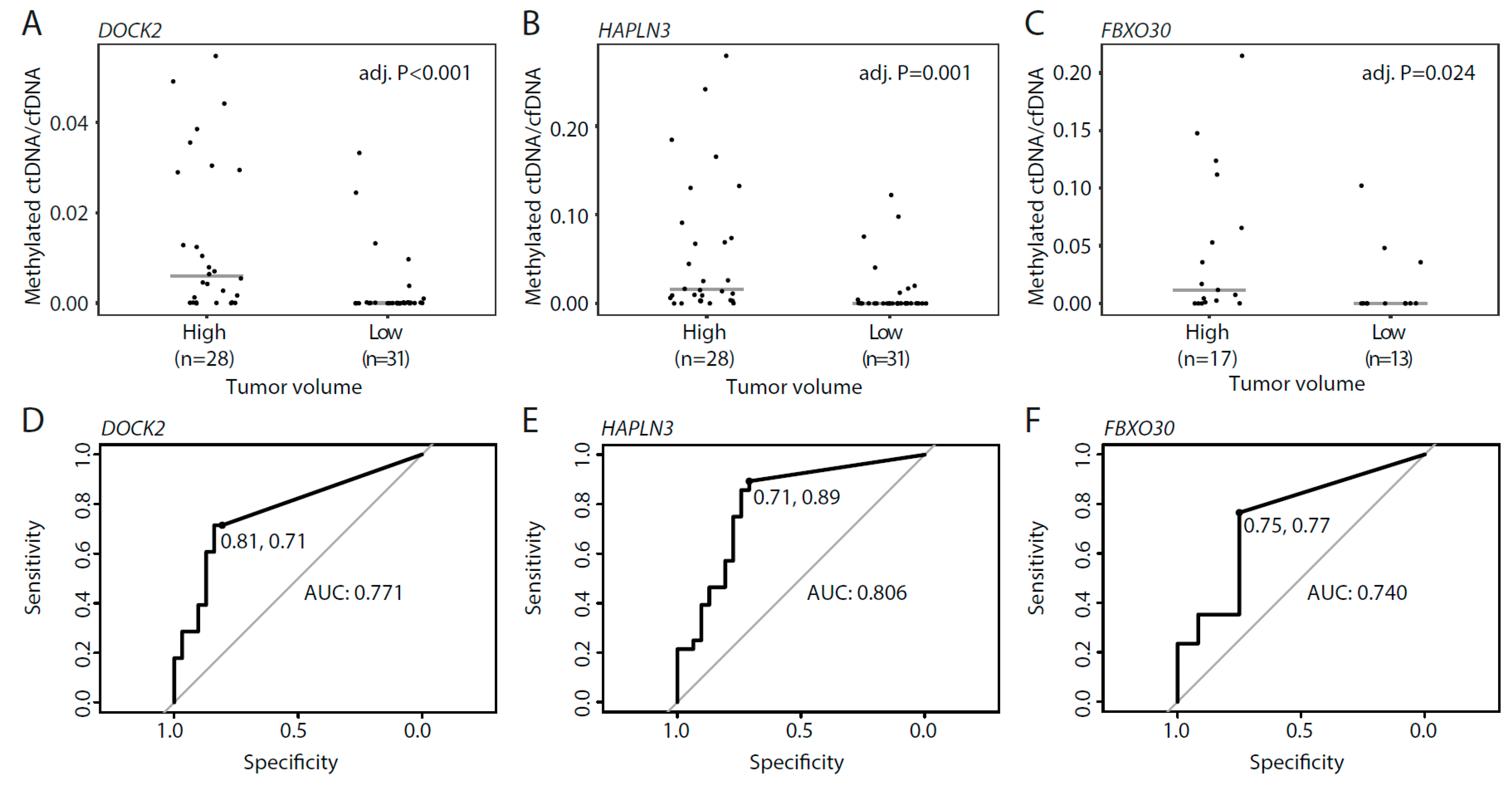
3.5. Methylated ctDNA in De Novo mPCa Patients with Low- and High Tumor-Volume
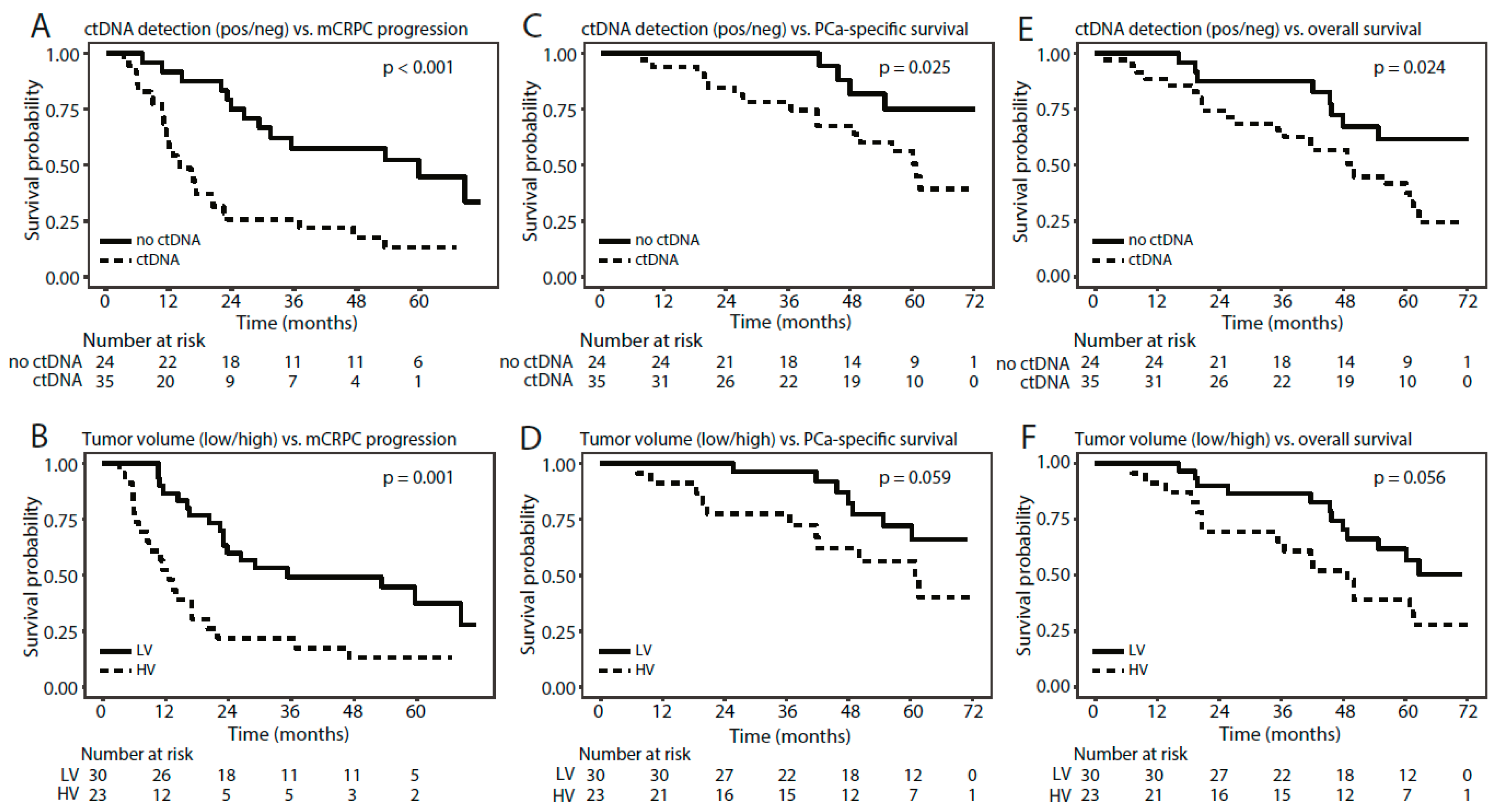
3.6. Methylated ctDNA as a Predictor of Time to mCRPC Progression, PCa-Specific Survival, and Overall Survival
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 450K | Infinium Human Methylation array 450 |
| AN | Adjacent normal |
| AUC | Area under the curve |
| BCR | Biochemical recurrence |
| BPH | Benign prostatic hyperplasia |
| cfDNA | Circulating cell-free DNA |
| ddPCR | Droplet digital polymerase chain reaction |
| FFPE | Formalin-fixed paraffin-embedded |
| LNA | Locked nucleic acids |
| mCRPC | Metastatic castration-resistant prostate cancer |
| mPCa | Metastatic prostate cancer |
| MS-ddPCR | Methylation-specific droplet digital polymerase chain reaction |
| NA | Not available |
| NS | Not significant |
| OS | Overall survival |
| PBC | Peripheral blood cells |
| PBMC | Peripheral blood mononuclear cells |
| PCa | Prostate cancer |
| qMSP | Quantitative methylation-specific polymerase chain reaction |
| ROC | Receiver operating characteristic |
| RP | Radical prostatectomy |
| TUR-P | Transurethral resection of the prostate |
References
- James, N.D.; De Bono, J.S. Abiraterone for Prostate Cancer Not Previously Treated with Hormone Therapy. N. Engl. J. Med. 2017, 377, 338–351. [Google Scholar] [CrossRef]
- James, N.D.; Sydes, M.R. Addition of docetaxel, zoledronic acid, or both to first-line long-term hormone therapy in prostate cancer (STAMPEDE): Survival results from an adaptive, multiarm, multistage, platform randomised controlled trial. Lancet 2016, 387, 1163–1177. [Google Scholar] [CrossRef]
- Kyriakopoulos, C.E.; Chen, Y.H. Chemohormonal Therapy in Metastatic Hormone-Sensitive Prostate Cancer: Long-Term Survival Analysis of the Randomized Phase III E3805 CHAARTED Trial. J. Clin. Oncol. 2018, 36, 1080–1087. [Google Scholar] [CrossRef] [PubMed]
- Fizazi, K.; Tran, N. Abiraterone plus Prednisone in Metastatic, Castration-Sensitive Prostate Cancer. N. Engl. J. Med. 2017, 377, 352–360. [Google Scholar] [CrossRef] [PubMed]
- Fizazi, K.; Tran, N. Abiraterone acetate plus prednisone in patients with newly diagnosed high-risk metastatic castration-sensitive prostate cancer (LATITUDE): Final overall survival analysis of a randomised, double-blind, phase 3 trial. Lancet Oncol. 2019, 20, 686–700. [Google Scholar] [CrossRef]
- Chi, K.N.; Agarwal, N. Apalutamide for Metastatic, Castration-Sensitive Prostate Cancer. N. Engl. J. Med. 2019, 381, 13–24. [Google Scholar] [CrossRef]
- Kristensen, H.; Haldrup, C. Hypermethylation of the GABRE~miR-452~miR-224 promoter in prostate cancer predicts biochemical recurrence after radical prostatectomy. Clin. Cancer Res. 2014, 20, 2169–2181. [Google Scholar] [CrossRef]
- Strand, S.H.; Orntoft, T.F. Prognostic DNA methylation markers for prostate cancer. Int. J. Mol. Sci. 2014, 15, 16544–16576. [Google Scholar] [CrossRef]
- Haldrup, C.; Lynnerup, A.S. Large-scale evaluation of SLC18A2 in prostate cancer reveals diagnostic and prognostic biomarker potential at three molecular levels. Mol. Oncol. 2016, 10, 825–837. [Google Scholar] [CrossRef]
- Haldrup, C.; Mundbjerg, K. DNA methylation signatures for prediction of biochemical recurrence after radical prostatectomy of clinically localized prostate cancer. J. Clin. Oncol. 2013, 31, 3250–3258. [Google Scholar] [CrossRef]
- Haldrup, C.; Pedersen, A.L. Biomarker potential of ST6GALNAC3 and ZNF660 promoter hypermethylation in prostate cancer tissue and liquid biopsies. Mol. Oncol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Strand, S.H.; Switnicki, M. RHCG and TCAF1 promoter hypermethylation predicts biochemical recurrence in prostate cancer patients treated by radical prostatectomy. Oncotarget 2017, 8, 5774–5788. [Google Scholar] [CrossRef] [PubMed]
- Norgaard, M.; Haldrup, C. Comprehensive Evaluation of TFF3 Promoter Hypomethylation and Molecular Biomarker Potential for Prostate Cancer Diagnosis and Prognosis. Int. J. Mol. Sci. 2017, 18, 2017. [Google Scholar] [CrossRef] [PubMed]
- Bjerre, M.T.; Strand, S.H. Aberrant DOCK2, GRASP, HIF3A and PKFP Hypermethylation has Potential as a Prognostic Biomarker for Prostate Cancer. Int. J. Mol. Sci. 2019, 20, 1173. [Google Scholar] [CrossRef]
- Maldonado, L.; Brait, M. GSTP1 promoter methylation is associated with recurrence in early stage prostate cancer. J. Urol. 2014, 192, 1542–1548. [Google Scholar] [CrossRef]
- Andreoiu, M.; Cheng, L. Multifocal prostate cancer: Biologic, prognostic, and therapeutic implications. Hum. Pathol. 2010, 41, 781–793. [Google Scholar] [CrossRef]
- Yadav, S.S.; Stockert, J.A. Intratumor heterogeneity in prostate cancer. Urol. Oncol. 2018, 36, 349–360. [Google Scholar] [CrossRef]
- Wyatt, A.W.; Annala, M. Concordance of Circulating Tumor DNA and Matched Metastatic Tissue Biopsy in Prostate Cancer. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef]
- Vandekerkhove, G.; Struss, W.J. Circulating Tumor DNA Abundance and Potential Utility in De novo Metastatic Prostate Cancer. Eur. Urol. 2019, 75, 667–675. [Google Scholar] [CrossRef]
- Annala, M.; Vandekerkhove, G. Circulating Tumor DNA Genomics Correlate with Resistance to Abiraterone and Enzalutamide in Prostate Cancer. Cancer Discov. 2018, 8, 444–457. [Google Scholar] [CrossRef]
- Wyatt, A.W.; Azad, A.A. Genomic Alterations in Cell-Free DNA and Enzalutamide Resistance in Castration-Resistant Prostate Cancer. JAMA Oncol. 2016, 2, 1598–1606. [Google Scholar] [CrossRef] [PubMed]
- De Laere, B.; Oeyen, S. TP53 Outperforms Other Androgen Receptor Biomarkers to Predict Abiraterone or Enzalutamide Outcome in Metastatic Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2019, 25, 1766–1773. [Google Scholar] [CrossRef] [PubMed]
- Mayrhofer, M.; De Laere, B. Cell-free DNA profiling of metastatic prostate cancer reveals microsatellite instability, structural rearrangements and clonal hematopoiesis. Genome Med. 2018, 10, 85. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Cruzata, L.; Hruby, G.W. DNA methylation changes correlate with Gleason score and tumor stage in prostate cancer. DNA Cell Biol. 2012, 31, 187–192. [Google Scholar] [CrossRef]
- Bryzgunova, O.E.; Morozkin, E.S. Methylation-specific sequencing of GSTP1 gene promoter in circulating/extracellular DNA from blood and urine of healthy donors and prostate cancer patients. Ann. N. Y. Acad. Sci. 2008, 1137, 222–225. [Google Scholar] [CrossRef]
- Goessl, C.; Krause, H. Fluorescent methylation-specific polymerase chain reaction for DNA-based detection of prostate cancer in bodily fluids. Cancer Res. 2000, 60, 5941–5945. [Google Scholar]
- Goessl, C.; Muller, M. DNA-based detection of prostate cancer in blood, urine, and ejaculates. Ann. N. Y. Acad. Sci. 2001, 945, 51–58. [Google Scholar] [CrossRef]
- Jeronimo, C.; Usadel, H. Quantitative GSTP1 hypermethylation in bodily fluids of patients with prostate cancer. Urology 2002, 60, 1131–1135. [Google Scholar] [CrossRef]
- Payne, S.R.; Serth, J. DNA methylation biomarkers of prostate cancer: Confirmation of candidates and evidence urine is the most sensitive body fluid for non-invasive detection. Prostate 2009, 69, 1257–1269. [Google Scholar] [CrossRef]
- Goering, W.; Kloth, M. DNA methylation changes in prostate cancer. Methods Mol. Biol. 2012, 863, 47–66. [Google Scholar] [CrossRef]
- Lowe, R.; Rakyan, V.K. Marmal-aid—A database for Infinium HumanMethylation450. BMC Bioinform. 2013, 14, 359. [Google Scholar] [CrossRef] [PubMed]
- Reinert, T.; Scholer, L.V. Analysis of circulating tumour DNA to monitor disease burden following colorectal cancer surgery. Gut 2016, 65, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, K.S. Locked nucleic acids can enhance the analytical performance of quantitative methylation-specific polymerase chain reaction. J. Mol. Diagn. 2008, 10, 33–42. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Huggett, J.F.; Foy, C.A. The digital MIQE guidelines: Minimum Information for Publication of Quantitative Digital PCR Experiments. Clin. Chem. 2013, 59, 892–902. [Google Scholar] [CrossRef] [PubMed]
- Jensen, S.Ø.; Øgaard, N. Novel DNA methylation biomarkers show high sensitivity and specificity for blood-based detection of colorectal cancer—A clinical biomarker discovery and validation study. Clin. Epigenetics 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Hochberg, Y.; Benjamini, Y. More powerful procedures for multiple significance testing. Stat. Med. 1990, 9, 811–818. [Google Scholar] [CrossRef]
- Yanagita, M.; Redig, A.J. A Prospective Evaluation of Circulating Tumor Cells and Cell-Free DNA in EGFR-Mutant Non-Small Cell Lung Cancer Patients Treated with Erlotinib on a Phase II Trial. Clin. Cancer Res. 2016, 22, 6010–6020. [Google Scholar] [CrossRef]
- Lapin, M.; Oltedal, S. Fragment size and level of cell-free DNA provide prognostic information in patients with advanced pancreatic cancer. J. Transl. Med. 2018, 16, 300. [Google Scholar] [CrossRef]
- Gautschi, O.; Bigosch, C. Circulating deoxyribonucleic Acid as prognostic marker in non-small-cell lung cancer patients undergoing chemotherapy. J. Clin. Oncol. 2004, 22, 4157–4164. [Google Scholar] [CrossRef]
- Feng, J.; Gang, F. Plasma cell-free DNA and its DNA integrity as biomarker to distinguish prostate cancer from benign prostatic hyperplasia in patients with increased serum prostate-specific antigen. Int. Urol. Nephrol. 2013, 45, 1023–1028. [Google Scholar] [CrossRef]
- Altimari, A.; Grigioni, A.D. Diagnostic role of circulating free plasma DNA detection in patients with localized prostate cancer. Am. J. Clin. Pathol. 2008, 129, 756–762. [Google Scholar] [CrossRef] [PubMed]
- Allen, D.; Butt, A. Role of cell-free plasma DNA as a diagnostic marker for prostate cancer. Ann. N. Y. Acad. Sci. 2004, 1022, 76–80. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.; Stephan, C. Increased cell-free DNA in plasma of patients with metastatic spread in prostate cancer. Cancer Lett. 2004, 205, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Boddy, J.L.; Gal, S. Prospective study of quantitation of plasma DNA levels in the diagnosis of malignant versus benign prostate disease. Clin. Cancer Res. 2005, 11, 1394–1399. [Google Scholar] [CrossRef] [PubMed]
- Schwarzenbach, H.; Alix-Panabieres, C. Cell-free tumor DNA in blood plasma as a marker for circulating tumor cells in prostate cancer. Clin. Cancer Res. 2009, 15, 1032–1038. [Google Scholar] [CrossRef]
- Hennigan, S.T.; Trostel, S.Y. Low Abundance of Circulating Tumor DNA in Localized Prostate Cancer. JCO Precis. Oncol. 2019, 1–13. [Google Scholar] [CrossRef]
- Shen, S.Y.; Singhania, R. Sensitive tumour detection and classification using plasma cell-free DNA methylomes. Nature 2018, 563, 579–583. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metastatic PCa (n = 65) | Localized PCa (n = 102) | Benign Prostatic Hyperplasia (n = 61) | Healthy Controls (n = 36) | |
|---|---|---|---|---|
| Age (years): median (min-max) | 72 (47–86) | 66 (43–81) | 71 (49–88) | > 45 |
| PSA (ng/mL): median | 42.0 | 9.4 | 4.3 | NA |
| (min-max) | (1.9–1171) | (1.2–812) | (0.5–42.8) | |
| cT-stage: n (%) | ||||
| cT1–2 | 15 (23) | 82 (80) | NA | NA |
| cT3–4 | 48 (74) | 17 (17) | NA | NA |
| Unknown | 2 (3) | 3 (3) | NA | NA |
| Gleason Grade Group: n (%) | ||||
| I | 3 (5) | 2 (2) | NA | NA |
| II | 6 (9) | 69 (67) | NA | NA |
| III | 10 (15) | 20 (20) | NA | NA |
| IV | 13 (20) | 2 (2) | NA | NA |
| V | 31 (48) | 6 (6) | NA | NA |
| Unknown | 2 (3) | 3 (3) | NA | NA |
| Regional lymph node metastasis status: n (%) | ||||
| N1 | 15 (23) | 4 (4) | NA | NA |
| N0 | 6 (9) | 27 (26) | NA | NA |
| NX | 1 (2) | 53 (52) | NA | NA |
| Unknown | 43 (66) | 18 (18) | NA | NA |
| Metastasis status: n (%) | ||||
| M0 | 0 (0) | 102 (100) | NA | NA |
| M1 | 65 (100) | 0 (0) | NA | NA |
| Metastasis location/volume: n (%) | ||||
| M1–lymph node only (non regional) | 4 (6) | NA | NA | NA |
| M1–Low tumor volume | 31 (48) | NA | NA | NA |
| M1–High tumor volume | 28 (43) | NA | NA | NA |
| Unknown | 2 (3) | NA | NA | NA |
| Treatment after diagnosis: n (%) | ||||
| RP | 0 (0) | 90 (88) | NA | NA |
| RT | 0 (0) | 3 (3) | NA | NA |
| Active surveillance | 0 (0) | 1 (1) | NA | NA |
| Antiandrogen blockage (Bicalutamide) | 0 (0) | 2 (2) | NA | NA |
| Androgen deprivation therapy | 65 (100) | 6 (6) | NA | NA |
| Biochemical recurrence (BCR) after RP: n (%) | ||||
| BCR | NA | 19 (21) | NA | NA |
| No BCR | NA | 67 (75) | NA | NA |
| Unknown | NA | 4 (4) | ||
| Plasma volume (mL): Mean (min-max) | 6.8 (1.4–19.9) | 8.9 (2.6–20.0) | 10.2 (6.5–19.7) | 6.7 (4.7–7.7) |
| cfDNA concentration (ng/mL plasma): Median (min-max) | 6.5 | 5.6 | 6.0 | 6.0 |
| (2.0–196.4) | (2.0–47.3) | (2.7–35.7) | (2.1–10.3) |
| Variable | Characteristics | Univariate | Multivariate | ||||
|---|---|---|---|---|---|---|---|
| HR (95% CI) | p-val | C-index | HR (95% CI) | p-val | C-index | ||
| Endpoint: mCRPC | |||||||
| Methylated ctDNA | No ctDNA vs. ctDNA | 3.1 (1.7–6.6) | 0.001 | 0.654 | 3.0 (1.3–6.9) | 0.012 | 0.715 |
| Tumor volume | Low vs. high | 2.9 (1.5–5.6) | 0.001 | 0.657 | 1.8 (0.8–3.8) | 0.202 | |
| Endpoint: PCa-Specific Survival | |||||||
| Methylated ctDNA | No ctDNA vs. ctDNA | 3.3 (1.1–9.8) | 0.034 | 0.639 | - | - | - |
| Tumor volume | Low vs. high | 2.4 (0.94–6.2) | 0.068 | 0.636 | - | - | - |
| Endpoint: Overall Survival | |||||||
| Methylated ctDNA | No ctDNA vs. ctDNA | 2.5 (1.1–5.5) | 0.029 | 0.597 | - | - | - |
| Tumor volume | Low vs. high | 2.1 (0.97–4.3) | 0.060 | 0.605 | - | - | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bjerre, M.T.; Nørgaard, M.; Larsen, O.H.; Jensen, S.Ø.; Strand, S.H.; Østergren, P.; Fode, M.; Fredsøe, J.; Ulhøi, B.P.; Mortensen, M.M.; et al. Epigenetic Analysis of Circulating Tumor DNA in Localized and Metastatic Prostate Cancer: Evaluation of Clinical Biomarker Potential. Cells 2020, 9, 1362. https://doi.org/10.3390/cells9061362
Bjerre MT, Nørgaard M, Larsen OH, Jensen SØ, Strand SH, Østergren P, Fode M, Fredsøe J, Ulhøi BP, Mortensen MM, et al. Epigenetic Analysis of Circulating Tumor DNA in Localized and Metastatic Prostate Cancer: Evaluation of Clinical Biomarker Potential. Cells. 2020; 9(6):1362. https://doi.org/10.3390/cells9061362
Chicago/Turabian StyleBjerre, Marianne Trier, Maibritt Nørgaard, Ole Halfdan Larsen, Sarah Østrup Jensen, Siri H. Strand, Peter Østergren, Mikkel Fode, Jacob Fredsøe, Benedicte Parm Ulhøi, Martin Mørck Mortensen, and et al. 2020. "Epigenetic Analysis of Circulating Tumor DNA in Localized and Metastatic Prostate Cancer: Evaluation of Clinical Biomarker Potential" Cells 9, no. 6: 1362. https://doi.org/10.3390/cells9061362
APA StyleBjerre, M. T., Nørgaard, M., Larsen, O. H., Jensen, S. Ø., Strand, S. H., Østergren, P., Fode, M., Fredsøe, J., Ulhøi, B. P., Mortensen, M. M., Jensen, J. B., Borre, M., & Sørensen, K. D. (2020). Epigenetic Analysis of Circulating Tumor DNA in Localized and Metastatic Prostate Cancer: Evaluation of Clinical Biomarker Potential. Cells, 9(6), 1362. https://doi.org/10.3390/cells9061362