Generation of Multipotent Stem Cells from Adult Human Peripheral Blood Following the Treatment with Platelet-Derived Mitochondria

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. PB-IPC Cell Culture
2.2. Isolation of Mitochondria from Platelets
2.3. Flow Cytometry
2.4. Retinal Pigment Epithelium (RPE) Cell Differentiation of Mitochondrion-Induced PB-IPC (miPB-IPC)
2.5. Neuronal Differentiation of Mitochondrion-Induced PB-IPC (miPB-IPC)
2.6. Colony Analysis
2.7. Tumor Formation Assay
2.8. Tracking RFP-Labeled Mitochondria in PB-IPC
2.9. Transmission Electron Microscopy (TEM)
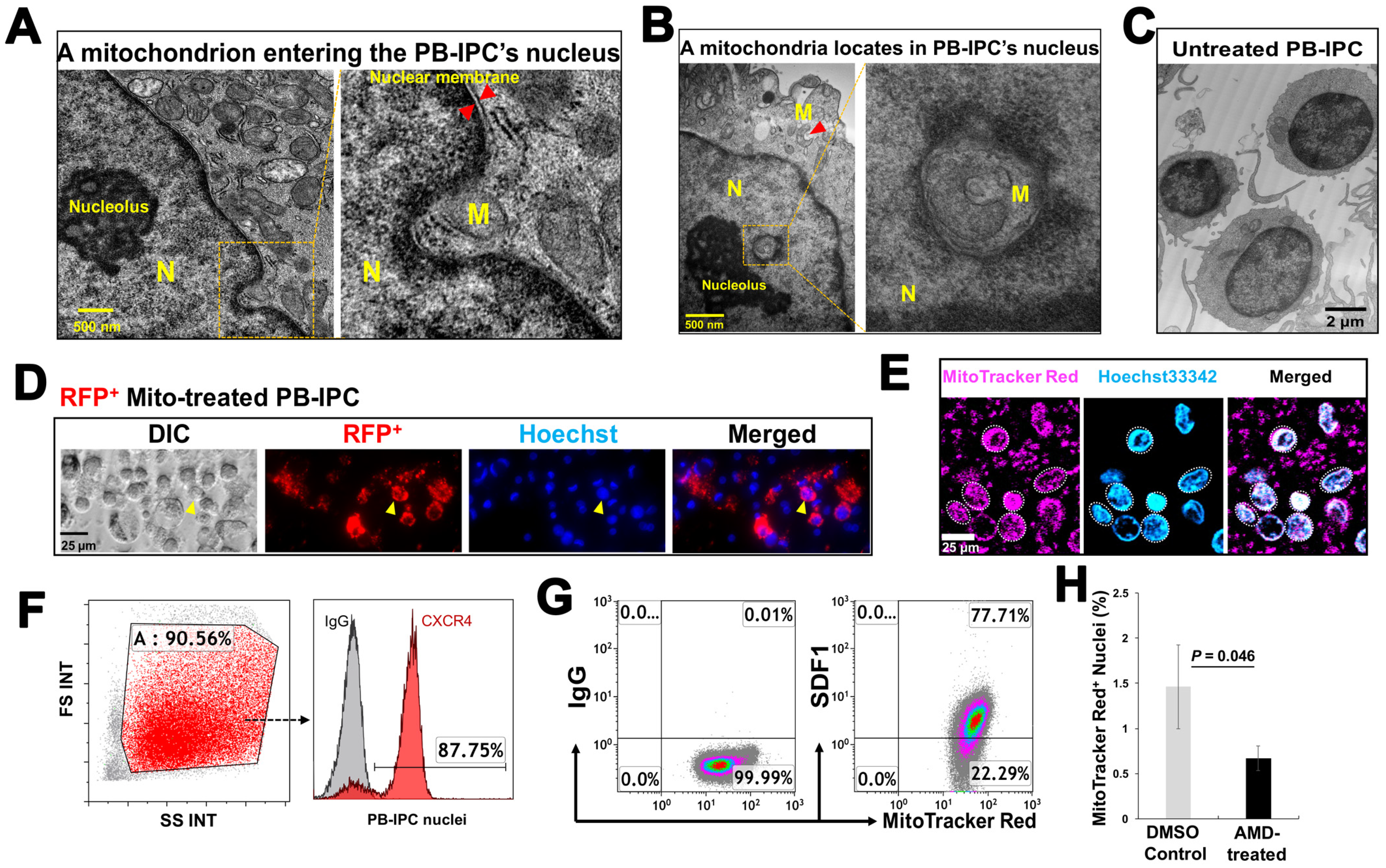
2.10. Blocking Experiment with CXCR4 Receptor Antagonist AMD 3100
2.11. Quantitative Real Time PCR
2.12. RNA-seq
2.13. Statistics
3. Results
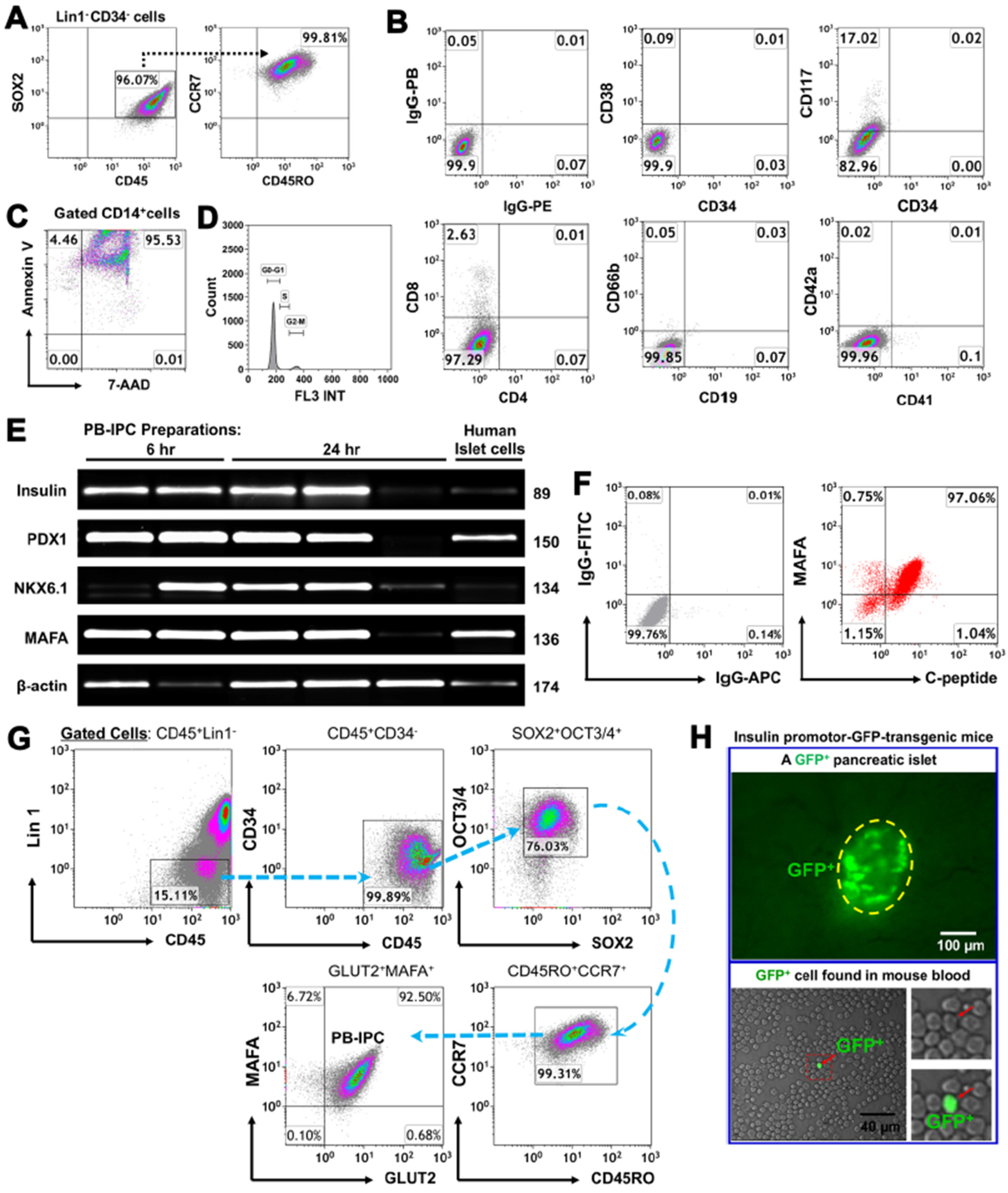
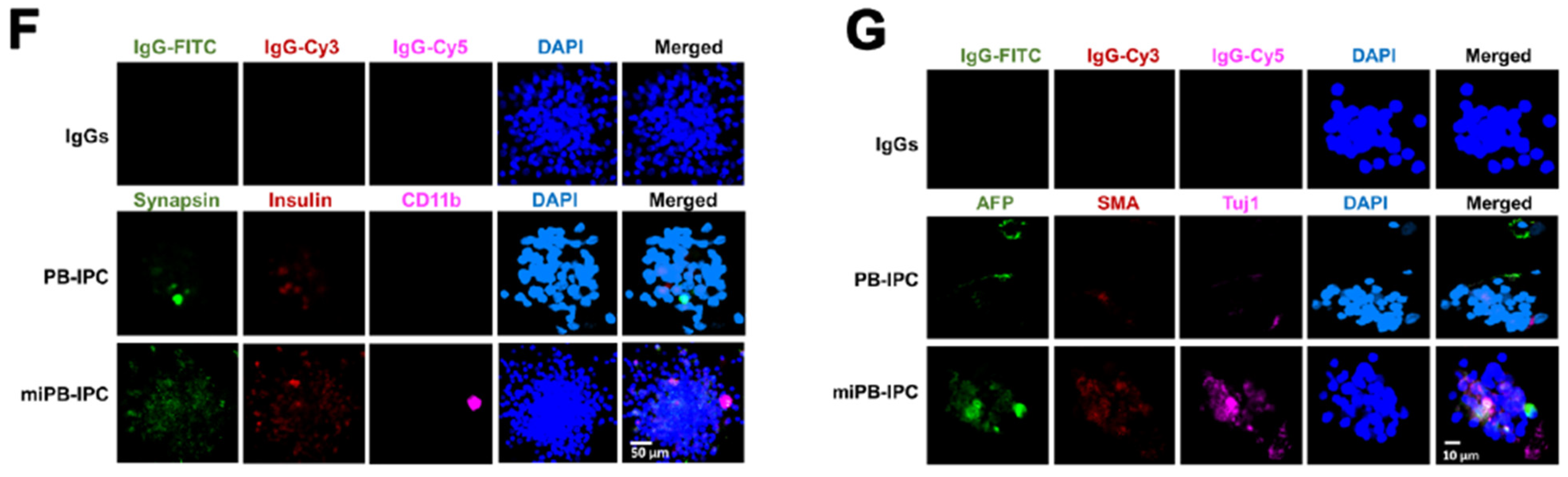
3.1. Adult Peripheral Blood-Derived PB-IPC Display Human Islet β Cell-Specific Markers
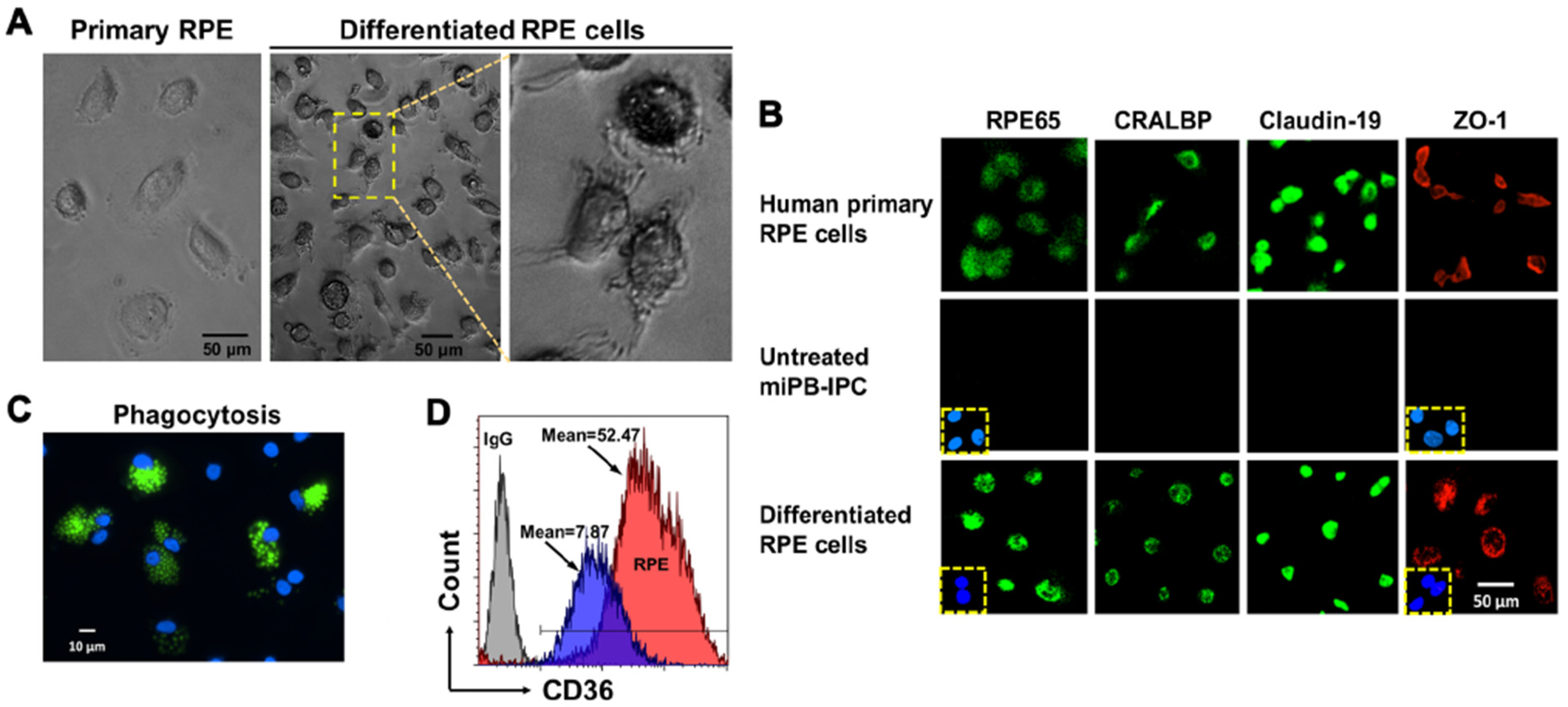
3.2. Ex Vivo Differentiation of Mitochondrion-Induced PB-IPC (miPB-IPC) into Retinal Pigment Epithelium (RPE) Cells
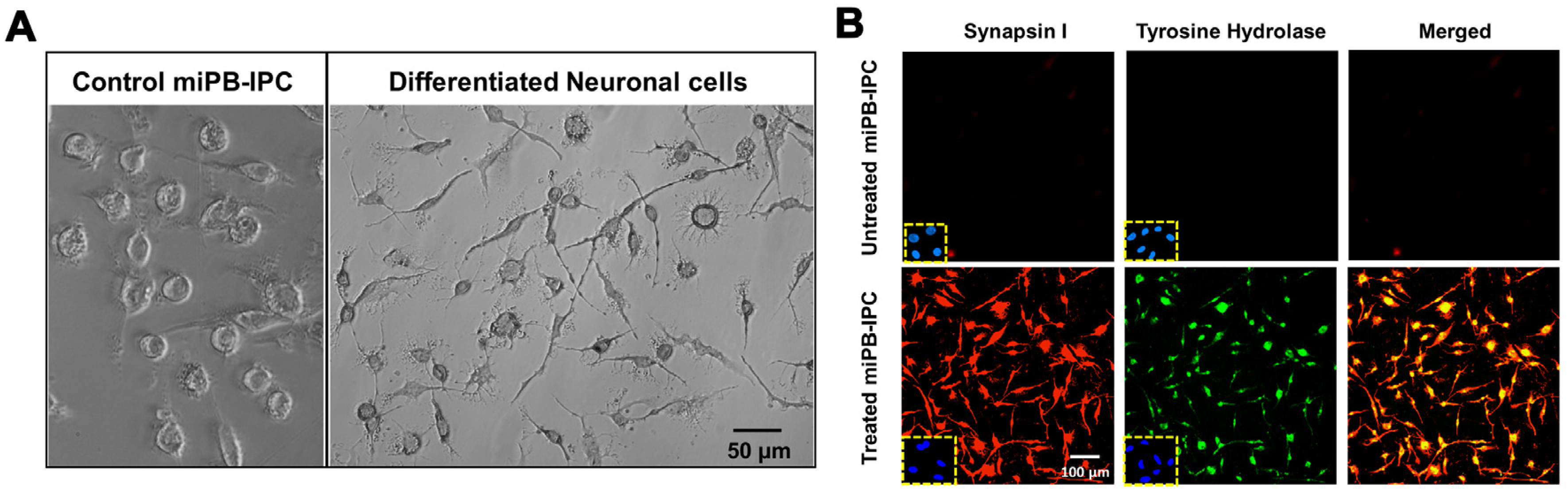
3.3. Ex Vivo Differentiation of Mitochondrion-Induced PB-IPC (miPB-IPC) into Neuronal Cells
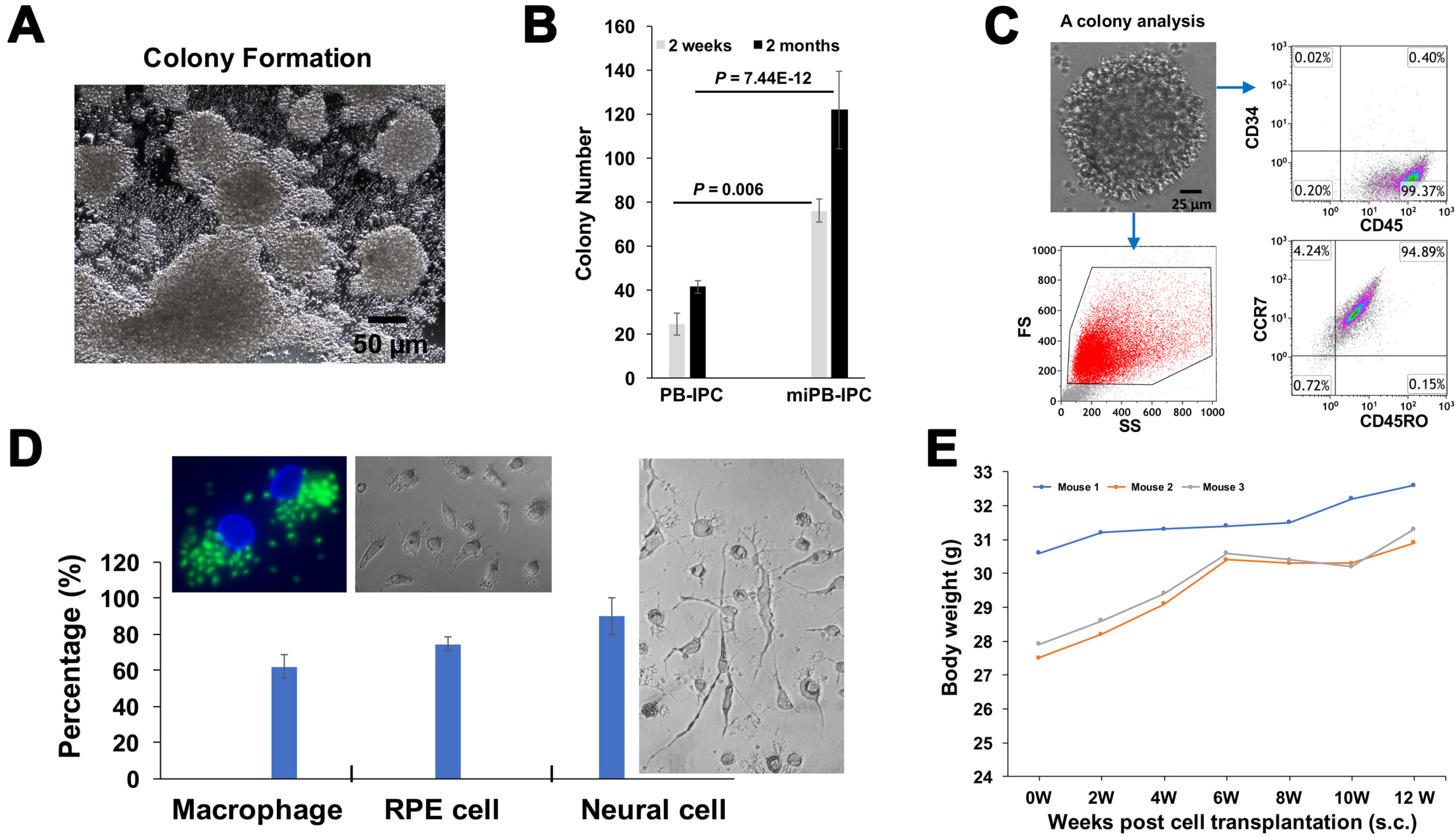
3.4. Clonal Analysis of miPB-IPC
3.5. Penetration of Mitochondria into Nuclei of PB-IPC
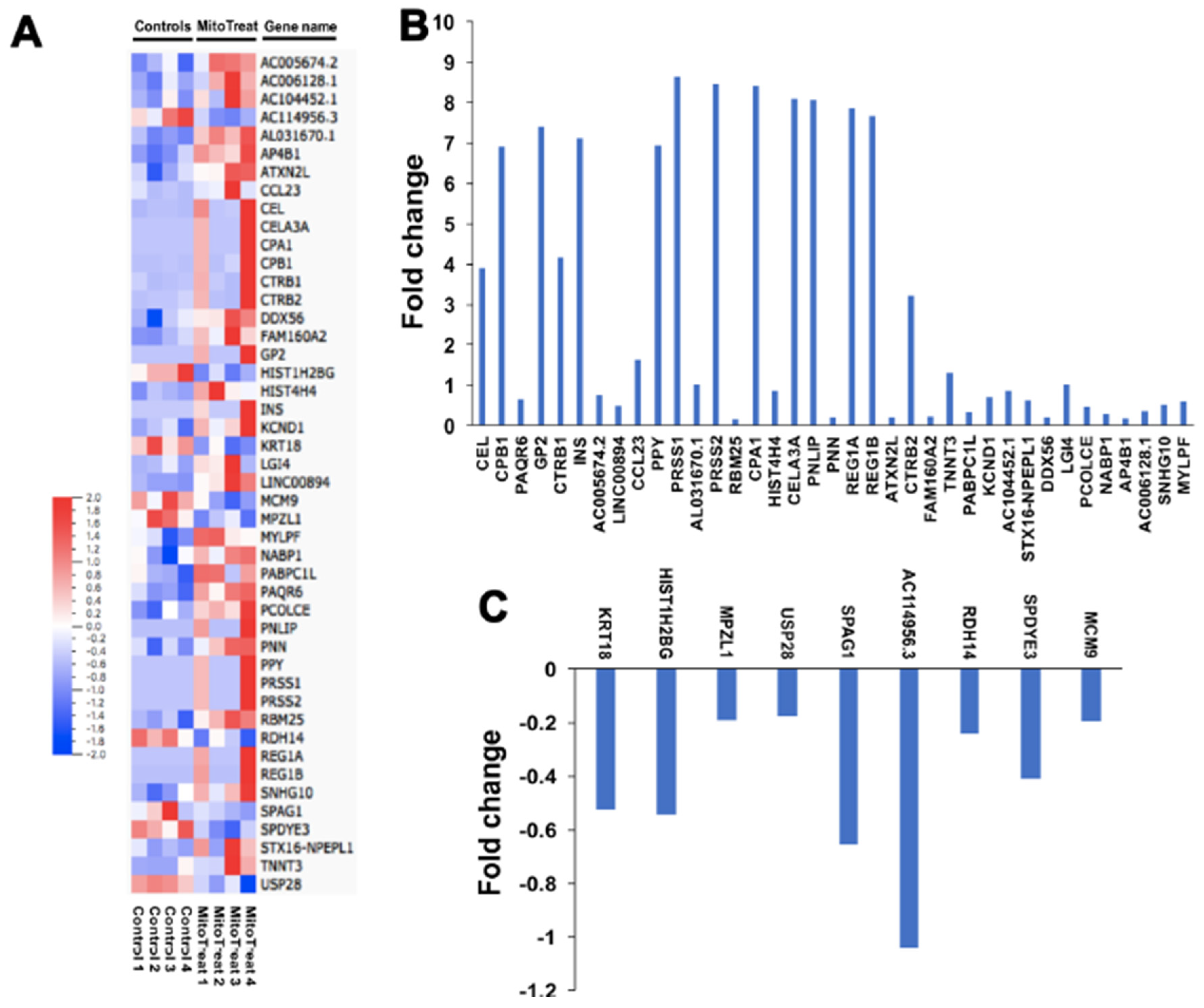
3.6. Genetic and Epigenetic Changes in PB-IPC after the Treatment with Mitochondria
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nanditha, A.; Ma, R.C.; Ramachandran, A.; Snehalatha, C.; Chan, J.C.; Chia, K.S.; Shaw, J.E.; Zimmet, P.Z. Diabetes in Asia and the Pacific: Implications for the Global Epidemic. Diabetes Care 2016, 39, 472–485. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Wang, L.; He, J.; Bi, Y.; Li, M.; Wang, T.; Wang, L.; Jiang, Y.; Dai, M.; Lu, J.; et al. Prevalence and control of diabetes in Chinese adults. JAMA 2013, 310, 948–959. [Google Scholar] [CrossRef] [PubMed]
- Lotfy, M.; Adeghate, J.; Kalasz, H.; Singh, J.; Adeghate, E. Chronic Complications of Diabetes Mellitus: A Mini Review. Curr. Diabetes Rev. 2017, 13, 3–10. [Google Scholar] [PubMed]
- DeFronzo, R.A.; Ferrannini, E.; Groop, L.; Henry, R.R.; Herman, W.H.; Holst, J.J.; Hu, F.B.; Kahn, C.R.; Raz, I.; Shulman, G.I.; et al. Type 2 diabetes mellitus. Nat. Rev. Dis. Primers 2015, 1, 15019. [Google Scholar] [CrossRef]
- Wong, E.; Backholer, K.; Gearon, E.; Harding, J.; Freak-Poli, R.; Stevenson, C.; Peeters, A. Diabetes and risk of physical disability in adults: A systematic review and meta-analysis. Lancet Diabetes Endocrinol. 2013, 1, 106–114. [Google Scholar] [CrossRef]
- Defuria, J.; Belkina, A.C.; Jagannathan-Bogdan, M.; Snyder-Cappione, J.; Carr, J.D.; Nersesova, Y.R.; Markham, D.; Strissel, K.J.; Watkins, A.A.; Zhu, M.; et al. B cells promote inflammation in obesity and type 2 diabetes through regulation of T-cell function and an inflammatory cytokine profile. Proc. Natl. Acad. Sci. USA 2013, 110, 5133–5138. [Google Scholar] [CrossRef]
- Winer, D.A.; Winer, S.; Shen, L.; Wadia, P.P.; Yantha, J.; Paltser, G.; Tsui, H.; Wu, P.; Davidson, M.G.; Alonso, M.N.; et al. B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat. Med. 2011, 17, 610–617. [Google Scholar] [CrossRef]
- Wu, D.; Molofsky, A.B.; Liang, H.E.; Ricardo-Gonzalez, R.R.; Jouihan, H.A.; Bando, J.K.; Chawla, A.; Locksley, R.M. Eosinophils sustain adipose alternatively activated macrophages associated with glucose homeostasis. Science 2011, 332, 243–247. [Google Scholar] [CrossRef]
- Bapat, S.P.; Myoung, S.J.; Fang, S.; Liu, S.; Zhang, Y.; Cheng, A.; Zhou, C.; Liang, Y.; LeBlanc, M.; Liddle, C.; et al. Depletion of fat-resident Treg cells prevents age-associated insulin resistance. Nature 2015, 528, 137–141. [Google Scholar] [CrossRef]
- Tsai, S.; Clemente-Casares, X.; Revelo, X.S.; Winer, S.; Winer, D.A. Are obesity-related insulin resistance and type 2 diabetes autoimmune diseases? Diabetes 2015, 64, 1886–1897. [Google Scholar] [CrossRef]
- Bluestone, J.A.; Herold, K.; Eisenbarth, G. Genetics, pathogenesis and clinical interventions in type 1 diabetes. Nature 2010, 464, 1293–1300. [Google Scholar] [PubMed]
- Winer, S.; Chan, Y.; Paltser, G.; Truong, D.; Tsui, H.; Bahrami, J.; Dorfman, R.; Wang, Y.; Zielenski, J.; Mastronardi, F.; et al. Normalization of obesity-associated insulin resistance through immunotherapy. Nat. Med. 2009, 15, 921–929. [Google Scholar] [CrossRef] [PubMed]
- Winer, S.; Winer, D.A. The adaptive immune system as a fundamental regulator of adipose tissue inflammation and insulin resistance. Immunol. Cell Biol. 2012, 90, 755–762. [Google Scholar] [PubMed]
- Liu, J.; Divoux, A.; Sun, J.; Zhang, J.; Clement, K.; Glickman, J.N.; Sukhova, G.K.; Wolters, P.J.; Du, J.; Gorgun, C.Z.; et al. Genetic deficiency and pharmacological stabilization of mast cells reduce diet-induced obesity and diabetes in mice. Nat. Med. 2009, 15, 940–945. [Google Scholar] [CrossRef]
- Olefsky, J.M.; Glass, C.K. Macrophages, inflammation, and insulin resistance. Annu. Rev. Physiol. 2010, 72, 219–246. [Google Scholar] [CrossRef]
- Talukdar, S.; Oh, d.Y.; Bandyopadhyay, G.; Li, D.; Xu, J.; McNelis, J.; Lu, M.; Li, P.; Yan, Q.; Zhu, Y.; et al. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat. Med. 2012, 18, 1407–1412. [Google Scholar]
- Bernal-Mizrachi, E.; Kulkarni, R.N.; Scott, D.K.; Mauvais-Jarvis, F.; Stewart, A.F.; Garcia-Ocana, A. Human beta-cell proliferation and intracellular signaling part 2: Still driving in the dark without a road map. Diabetes 2014, 63, 819–831. [Google Scholar] [CrossRef]
- Stewart, A.F.; Hussain, M.A.; Garcia-Ocana, A.; Vasavada, R.C.; Bhushan, A.; Bernal-Mizrachi, E.; Kulkarni, R.N. Human beta-cell proliferation and intracellular signaling: Part 3. Diabetes 2015, 64, 1872–1885. [Google Scholar] [CrossRef]
- Odorico, J.; Markmann, J.; Melton, D.; Greenstein, J.; Hwa, A.; Nostro, C.; Rezania, A.; Oberholzer, J.; Pipeleers, D.; Yang, L.; et al. Report of the Key Opinion Leaders Meeting on Stem Cell-derived Beta Cells. Transplantation 2018, 102, 1223–1229. [Google Scholar] [CrossRef]
- Pagliuca, F.W.; Millman, J.R.; Gurtler, M.; Segel, M.; Van, D.A.; Ryu, J.H.; Peterson, Q.P.; Greiner, D.; Melton, D.A. Generation of functional human pancreatic beta cells in vitro. Cell 2014, 159, 428–439. [Google Scholar] [CrossRef]
- Rezania, A.; Bruin, J.E.; Arora, P.; Rubin, A.; Batushansky, I.; Asadi, A.; O’Dwyer, S.; Quiskamp, N.; Mojibian, M.; Albrecht, T.; et al. Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat. Biotechnol. 2014, 32, 1121–1133. [Google Scholar] [CrossRef] [PubMed]
- Kelly, O.G.; Chan, M.Y.; Martinson, L.A.; Kadoya, K.; Ostertag, T.M.; Ross, K.G.; Richardson, M.; Carpenter, M.K.; D’Amour, K.A.; Kroon, E.; et al. Cell-surface markers for the isolation of pancreatic cell types derived from human embryonic stem cells. Nat. Biotechnol. 2011, 29, 750–756. [Google Scholar] [CrossRef] [PubMed]
- Chabannon, C.; Kuball, J.; Bondanza, A.; Dazzi, F.; Pedrazzoli, P.; Toubert, A.; Ruggeri, A.; Fleischhauer, K.; Bonini, C. Hematopoietic stem cell transplantation in its 60s: A platform for cellular therapies. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef]
- Staal, F.J.; Baum, C.; Cowan, C.; Dzierzak, E.; Hacein-Bey-Abina, S.; Karlsson, S.; Lapidot, T.; Lemischka, I.; Mendez-Ferrer, S.; Mikkers, H.; et al. Stem cell self-renewal: Lessons from bone marrow, gut and iPS toward clinical applications. Leukemia 2011, 25, 1095–1102. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Jiang, Z.; Zhao, T.; Ye, M.; Hu, C.; Yin, Z.; Li, H.; Zhang, Y.; Diao, Y.; Li, Y.; et al. Reversal of type 1 diabetes via islet beta cell regeneration following immune modulation by cord blood-derived multipotent stem cells. Bmc Med. 2012, 10, 3. [Google Scholar] [CrossRef] [PubMed]
- Delgado, E.; Perez-Basterrechea, M.; Suarez-Alvarez, B.; Zhou, H.; Revuelta, E.M.; Garcia-Gala, J.M.; Perez, S.; Alvarez-Viejo, M.; Menendez, E.; Lopez-Larrea, C.; et al. Modulation of Autoimmune T-Cell Memory by Stem Cell Educator Therapy: Phase 1/2 Clinical Trial. EBioMedicine 2015, 2, 2024–2036. [Google Scholar] [PubMed]
- Zhao, Y.; Jiang, Z.; Zhao, T.; Ye, M.; Hu, C.; Zhou, H.; Yin, Z.; Chen, Y.; Zhang, Y.; Wang, S.; et al. Targeting insulin resistance in type 2 diabetes via immune modulation of cord blood-derived multipotent stem cells (CB-SCs) in stem cell educator therapy: Phase I/II clinical trial. Bmc Med. 2013, 11, 160. [Google Scholar] [CrossRef]
- Li, Y.; Yan, B.; Wang, H.; Li, H.; Li, Q.; Zhao, D.; Chen, Y.; Zhang, Y.; Li, W.; Zhang, J.; et al. Hair regrowth in alopecia areata patients following Stem Cell Educator therapy. Bmc Med. 2015, 13, 87. [Google Scholar]
- Zhao, Y.; Jiang, Z.; Delgado, E.; Li, H.; Zhou, H.; Hu, W.; Perez-Basterrechea, M.; Janostakova, A.; Tan, Q.; Wang, J.; et al. Platelet-Derived Mitochondria Display Embryonic Stem Cell Markers and Improve Pancreatic Islet beta-cell Function in Humans. Stem Cells Transl. Med. 2017, 6, 1684–1697. [Google Scholar] [CrossRef]
- Zhao, Y.; Huang, Z.; Lazzarini, P.; Wang, Y.; Di, A.; Chen, M. A unique human blood-derived cell population displays high potential for producing insulin. Biochem. Biophys. Res. Commun. 2007, 360, 205–211. [Google Scholar] [CrossRef]
- Asmussen, N.; Lin, Z.; McClure, M.J.; Schwartz, Z.; Boyan, B.D. Regulation of extracellular matrix vesicles via rapid responses to steroid hormones during endochondral bone formation. Steroids 2019, 142, 43–47. [Google Scholar] [CrossRef]
- Zhao, Y.; Glesne, D.; Huberman, E. A human peripheral blood monocyte-derived subset acts as pluripotent stem cells. Proc. Natl. Acad. Sci. USA 2003, 100, 2426–2431. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, H.; Mazzone, T. Identification of stem cells from human umbilical cord blood with embryonic and hematopoietic characteristics. Exp. Cell Res. 2006, 312, 2454–2464. [Google Scholar] [CrossRef]
- Andrzejewska, A.; Lukomska, B.; Janowski, M. Concise Review: Mesenchymal Stem Cells: From Roots to Boost. Stem Cells 2019, 37, 855–864. [Google Scholar] [CrossRef]
- Matsuoka, T.A.; Artner, I.; Henderson, E.; Means, A.; Sander, M.; Stein, R. The MafA transcription factor appears to be responsible for tissue-specific expression of insulin. Proc. Natl. Acad. Sci. USA 2004, 101, 2930–2933. [Google Scholar] [CrossRef]
- Ao, J.; Wood, J.P.; Chidlow, G.; Gillies, M.C.; Casson, R.J. Retinal pigment epithelium in the pathogenesis of age-related macular degeneration and photobiomodulation as a potential therapy? Clin. Exp. Ophthalmol. 2018, 46, 670–686. [Google Scholar] [CrossRef]
- Chichagova, V.; Hallam, D.; Collin, J.; Zerti, D.; Dorgau, B.; Felemban, M.; Lako, M.; Steel, D.H. Cellular regeneration strategies for macular degeneration: Past, present and future. Eye (Lond) 2018, 32, 946–971. [Google Scholar] [CrossRef]
- Finnemann, S.C.; Silverstein, R.L. Differential roles of CD36 and alphavbeta5 integrin in photoreceptor phagocytosis by the retinal pigment epithelium. J. Exp. Med. 2001, 194, 1289–1298. [Google Scholar] [CrossRef]
- Daubner, S.C.; Le, T.; Wang, S. Tyrosine hydroxylase and regulation of dopamine synthesis. Arch Biochem. Biophys. 2011, 508, 1–12. [Google Scholar] [CrossRef]
- Agulnick, A.D.; Ambruzs, D.M.; Moorman, M.A.; Bhoumik, A.; Cesario, R.M.; Payne, J.K.; Kelly, J.R.; Haakmeester, C.; Srijemac, R.; Wilson, A.Z.; et al. Insulin-Producing Endocrine Cells Differentiated In Vitro From Human Embryonic Stem Cells Function in Macroencapsulation Devices In Vivo. Stem Cells Transl. Med. 2015, 4, 1214–1222. [Google Scholar] [CrossRef]
- Veres, A.; Faust, A.L.; Bushnell, H.L.; Engquist, E.N.; Kenty, J.H.; Harb, G.; Poh, Y.C.; Sintov, E.; Gurtler, M.; Pagliuca, F.W.; et al. Charting cellular identity during human in vitro beta-cell differentiation. Nature 2019, 569, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Kroon, E.; Martinson, L.A.; Kadoya, K.; Bang, A.G.; Kelly, O.G.; Eliazer, S.; Young, H.; Richardson, M.; Smart, N.G.; Cunningham, J.; et al. Pancreatic endoderm derived from human embryonic stem cells generates glucose-responsive insulin-secreting cells in vivo. Nat. Biotechnol. 2008, 26, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Boyd, A.S.; Rodrigues, N.P.; Lui, K.O.; Fu, X.; Xu, Y. Concise review: Immune recognition of induced pluripotent stem cells. Stem Cells 2012, 30, 797–803. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Zhang, Z.N.; Rong, Z.; Xu, Y. Immunogenicity of induced pluripotent stem cells. Nature 2011, 474, 212–215. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Zhang, Z.N.; Westenskow, P.D.; Todorova, D.; Hu, Z.; Lin, T.; Rong, Z.; Kim, J.; He, J.; Wang, M.; et al. Humanized Mice Reveal Differential Immunogenicity of Cells Derived from Autologous Induced Pluripotent Stem Cells. Cell Stem Cell 2015, 17, 353–359. [Google Scholar] [CrossRef]
- Sneddon, J.B.; Tang, Q.; Stock, P.; Bluestone, J.A.; Roy, S.; Desai, T.; Hebrok, M. Stem Cell Therapies for Treating Diabetes: Progress and Remaining Challenges. Cell Stem Cell 2018, 22, 810–823. [Google Scholar] [CrossRef]
- Brandes, D.; Schofield, B.H.; Anton, E. Nuclear mitochondria? Science 1965, 149, 1373–1374. [Google Scholar] [CrossRef]
- Bloom, G.D. A nucleus with cytoplasmic features. J. Cell Biol. 1967, 35, 266–268. [Google Scholar] [CrossRef]
- Jensen, H.; Engedal, H.; Saetersdal, T.S. Ultrastructure of mitochondria-containining nuclei in human myocardial cells. Virchows Arch. B Cell Pathol. 1976, 1–12. [Google Scholar]
- Takemura, G.; Takatsu, Y.; Sakaguchi, H.; Fujiwara, H. Intranuclear mitochondria in human myocardial cells. Pathol. Res. Pr. 1997, 193, 305–311. [Google Scholar] [CrossRef]
- Nishimura, K.; Fukuda, A.; Hisatake, K. Mechanisms of the Metabolic Shift during Somatic Cell Reprogramming. Int. J. Mol. Sci. 2019, 20, 2254. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, H.; Hu, W.; Song, X.; Zhao, Y. Generation of Multipotent Stem Cells from Adult Human Peripheral Blood Following the Treatment with Platelet-Derived Mitochondria. Cells 2020, 9, 1350. https://doi.org/10.3390/cells9061350
Yu H, Hu W, Song X, Zhao Y. Generation of Multipotent Stem Cells from Adult Human Peripheral Blood Following the Treatment with Platelet-Derived Mitochondria. Cells. 2020; 9(6):1350. https://doi.org/10.3390/cells9061350
Chicago/Turabian StyleYu, Haibo, Wei Hu, Xiang Song, and Yong Zhao. 2020. "Generation of Multipotent Stem Cells from Adult Human Peripheral Blood Following the Treatment with Platelet-Derived Mitochondria" Cells 9, no. 6: 1350. https://doi.org/10.3390/cells9061350
APA StyleYu, H., Hu, W., Song, X., & Zhao, Y. (2020). Generation of Multipotent Stem Cells from Adult Human Peripheral Blood Following the Treatment with Platelet-Derived Mitochondria. Cells, 9(6), 1350. https://doi.org/10.3390/cells9061350