CTRP9 Mediates Protective Effects in Cardiomyocytes via AMPK- and Adiponectin Receptor-Mediated Induction of Anti-Oxidant Response

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. PAB Model
2.2. Adult Human Tissue and Blood Samples
2.3. ELISA
2.4. Isolation of Recombinant CTRPs
2.5. Isolation of Recombinant CTRP9 from HEK293 Cells
2.6. Isolation of Adult Cardiomyocytes, Endothelial Cells and Fibroblasts
2.7. Isolation of Human Cardiomyocytes and HUVECs
2.8. Detection of ROS Formation
2.9. Measurement of Apoptosis
2.10. H9C2 Cardiomyoblast Culture and siRNA Transfection
2.11. RNA Isolation, RT-PCR and qPCR
2.12. Subcellular Fractionation and Western Blotting
2.13. Co-Immunoprecipitation
2.14. Pathway Reporter Array
2.15. Reporter Genes and Reporter Gene Assays
2.16. Statistical Analysis
3. Results
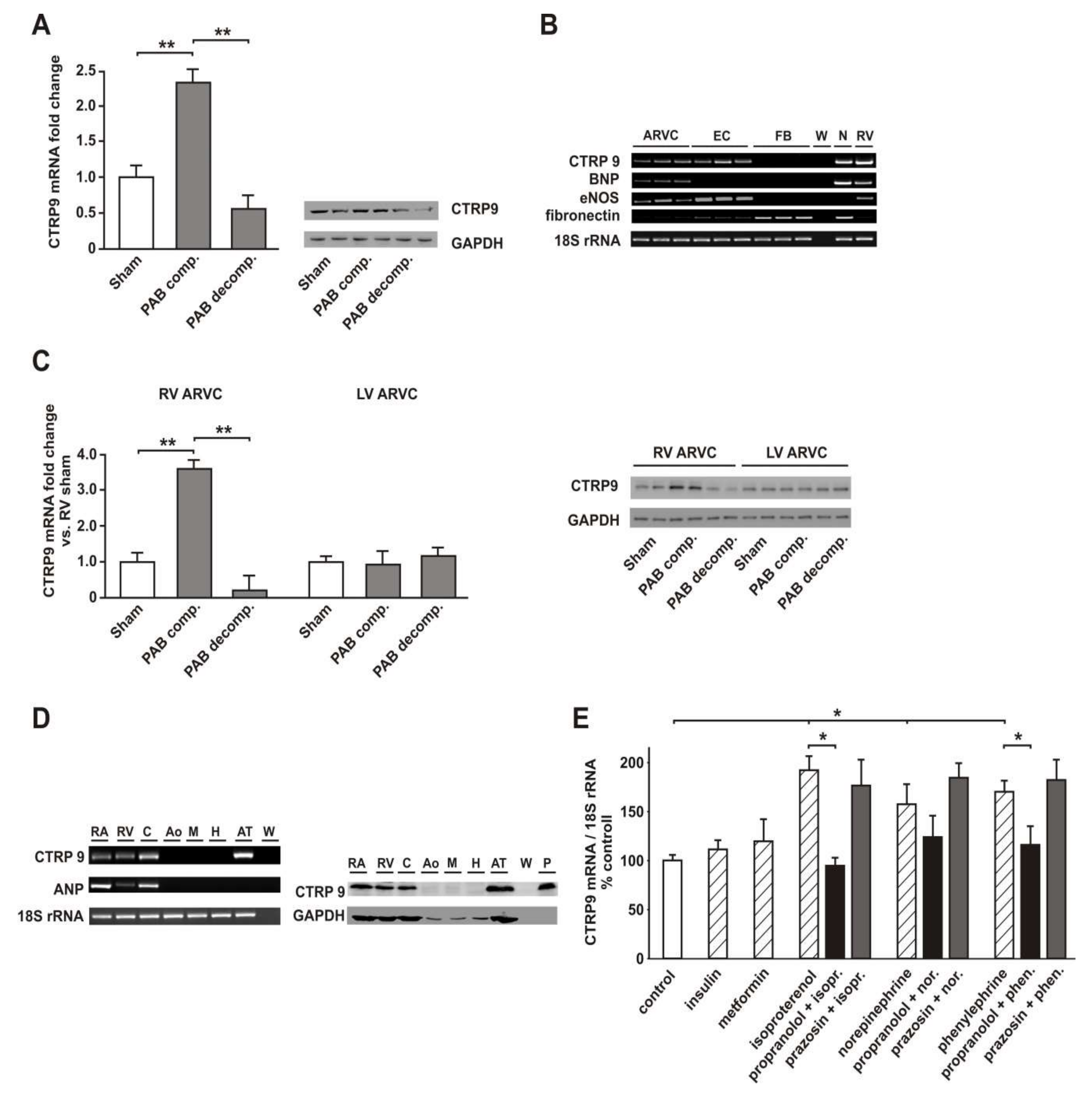
3.1. Two Clearly Distinct Stages of RV Disease are Reached
3.2. RV Failure is Associated with Altered Cardiomyocyte CTRP9 Expression
3.3. CTRP9 Mediates Anti-Oxidative Effects
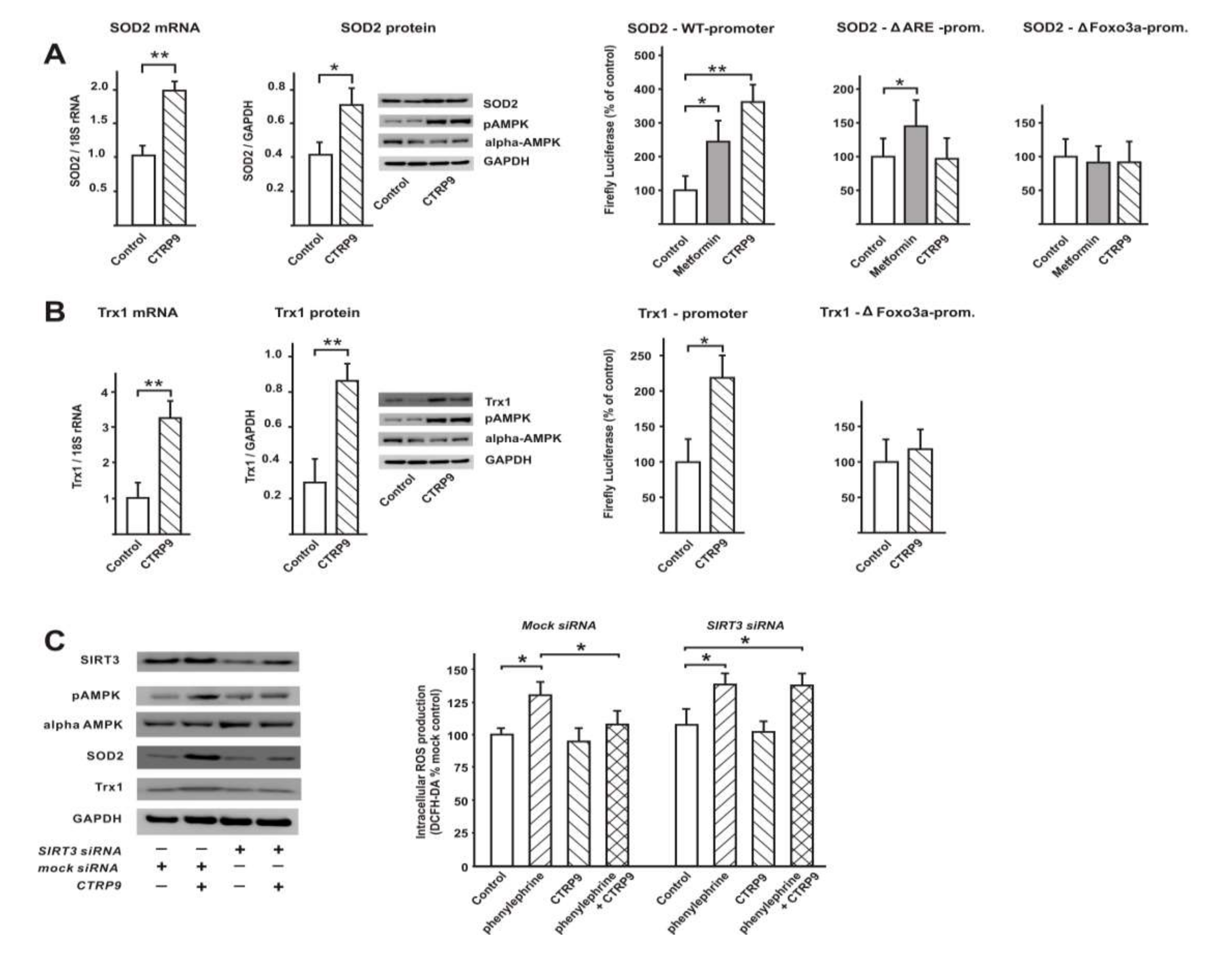
3.4. Transcriptional Regulation of Enzymes Involved in ROS Production or ROS Defense by CTRP9
3.4.1. Influence of CTRP9 on the Expression of Enzymes Involved in ROS Production or Defense in ARVCs
3.4.2. Influence of CTRP9 on the Expression of Anti-Oxidant Enzymes and Potentially Involved Transcription Factors in Cardiomyoblasts
3.5. Signaling Pathways Involved in Anti-Oxidant CTRP9 Effects
3.6. Role of Adiponectin Receptors in CTRP9-Mediated Signal Transduction
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACC | Acetyl-coenzyme A carboxylase |
| AdipoR | Adiponectin receptor |
| AMPK | AMP-activated protein kinase |
| Ara A | Adenine 9-β-D-arabinofuranoside |
| AP-1 | Activator protein 1 |
| ARE | Anti-oxidant response element |
| ARVC | Adult rat ventricular cardiomyocyte |
| AT | Adipose tissue |
| BNP | Brain natriuretic peptide |
| CsA | Cyclosporine A |
| CTRP | C1q/tumor necrosis factor-alpha-related protein |
| EC | Endothelial cell |
| eNOS | Endothelial NO synthase |
| ER | Endoplasmic reticulum |
| FCS | Fetal calf serum |
| Foxo3a | Forkhead box O3 |
| gC1qR | Globular C1q receptor |
| GPX | Glutathione peroxidase |
| HUVEC | Human umbilical vein endothelial cell |
| LV | Left ventricle |
| LV FS | LV fractional shortening |
| NFAT | Nuclear factor of activated T-cells |
| PAB | Pulmonary artery banding |
| PKA | Protein kinase A |
| RA | Right atrium |
| ROS | Reactive oxygen species |
| RV | Right ventricle |
| SOD | Superoxide dismutase |
| Trx | Thioredoxin |
References
- Fang, H.; Judd, R.L. Adiponectin Regulation and Function. Compr. Physiol. 2018, 8, 1031–1063. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.V.; Scherer, P.E. Adiponectin, the past two decades. J. Mol. Cell Boil. 2016, 8, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Wong, G.W.; Wang, J.; Hug, C.; Tsao, T.S.; Lodish, H.F. A family of Acrp30/adiponectin structural and functional paralogs. Proc. Natl. Acad. Sci. USA 2004, 101, 10302–10307. [Google Scholar] [CrossRef] [PubMed]
- Seldin, M.M.; Tan, S.Y.; Wong, G.W. Metabolic function of the CTRP family of hormones. Rev. Endocr. Metab. Disord. 2014, 15, 111–123. [Google Scholar] [CrossRef]
- Niemann, B.; Silber, R.E.; Rohrbach, S. Age-specific effects of short- and long-term caloric restriction on the expression of adiponectin and adiponectin receptors: Influence of intensity of food restriction. Exp. Gerontol. 2008, 43, 706–713. [Google Scholar] [CrossRef]
- Rohrbach, S.; Aurich, A.C.; Li, L.; Niemann, B. Age-associated loss in adiponectin-activation by caloric restriction: Lack of compensation by enhanced inducibility of adiponectin paralogs CTRP2 and CTRP7. Mol. Cell Endocrinol. 2007, 277, 26–34. [Google Scholar] [CrossRef][Green Version]
- Su, H.; Yuan, Y.; Wang, X.M.; Lau, W.B.; Wang, Y.; Wang, X.; Gao, E.; Koch, W.J.; Ma, X.L. Inhibition of CTRP9, a novel and cardiac-abundantly expressed cell survival molecule, by TNFalpha-initiated oxidative signaling contributes to exacerbated cardiac injury in diabetic mice. Basic. Res. Cardiol. 2013, 108, 315. [Google Scholar] [CrossRef]
- Appari, M.; Breitbart, A.; Brandes, F.; Szaroszyk, M.; Froese, N.; Korf-Klingebiel, M.; Mohammadi, M.M.; Grund, A.; Scharf, G.M.; Wang, H.; et al. C1q-TNF-Related Protein-9 Promotes Cardiac Hypertrophy and Failure. Circ. Res. 2017, 120, 66–77. [Google Scholar] [CrossRef]
- Yuan, Y.; Lau, W.B.; Su, H.; Sun, Y.; Yi, W.; Du, Y.; Christopher, T.; Lopez, B.; Wang, Y.; Ma, X.L. C1q-TNF-related protein-9, a novel cardioprotetcive cardiokine, requires proteolytic cleavage to generate a biologically active globular domain isoform. Am. J. Physiol. Endocrinol. Metab. 2015, 308, E891–E898. [Google Scholar] [CrossRef]
- Wong, G.W.; Krawczyk, S.A.; Kitidis-Mitrokostas, C.; Ge, G.; Spooner, E.; Hug, C.; Gimeno, R.; Lodish, H.F. Identification and characterization of CTRP9, a novel secreted glycoprotein, from adipose tissue that reduces serum glucose in mice and forms heterotrimers with adiponectin. FASEB J. 2009, 23, 241–258. [Google Scholar] [CrossRef]
- Wei, Z.; Lei, X.; Petersen, P.S.; Aja, S.; Wong, G.W. Targeted deletion of C1q/TNF-related protein 9 increases food intake, decreases insulin sensitivity, and promotes hepatic steatosis in mice. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E779–E790. [Google Scholar] [CrossRef] [PubMed]
- Kambara, T.; Ohashi, K.; Shibata, R.; Ogura, Y.; Maruyama, S.; Enomoto, T.; Uemura, Y.; Shimizu, Y.; Yuasa, D.; Matsuo, K.; et al. CTRP9 protein protects against myocardial injury following ischemia-reperfusion through AMP-activated protein kinase (AMPK)-dependent mechanism. J. Biol. Chem. 2012, 287, 18965–18973. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Yi, W.; Yuan, Y.; Lau, W.B.; Yi, D.; Wang, X.; Wang, Y.; Su, H.; Wang, X.; Gao, E.; et al. C1q/tumor necrosis factor-related protein-9, a novel adipocyte-derived cytokine, attenuates adverse remodeling in the ischemic mouse heart via protein kinase A activation. Circulation 2013, 128, S113–S120. [Google Scholar] [CrossRef]
- Zhao, D.; Feng, P.; Sun, Y.; Qin, Z.; Zhang, Z.; Tan, Y.; Gao, E.; Lau, W.B.; Ma, X.; Yang, J.; et al. Cardiac-derived CTRP9 protects against myocardial ischemia/reperfusion injury via calreticulin-dependent inhibition of apoptosis. Cell Death Dis. 2018, 9, 723. [Google Scholar] [CrossRef] [PubMed]
- Sardu, C.; Paolisso, G.; Marfella, R. Molecular mechanisms and therapeutic targets of inflammatory-related Cardiovascular diseases: From molecular mechanisms to therapeutic targets. Curr. Pharm. Des. 2020. [Google Scholar] [CrossRef] [PubMed]
- Sardu, C.; D’Onofrio, N.; Torella, M.; Portoghese, M.; Loreni, F.; Mureddu, S.; Signoriello, G.; Scisciola, L.; Barbieri, M.; Rizzo, M.R.; et al. Pericoronary fat inflammation and Major Adverse Cardiac Events (MACE) in prediabetic patients with acute myocardial infarction: Effects of metformin. Cardiovasc. Diabetol. 2019, 18, 126. [Google Scholar] [CrossRef]
- Gao, C.; Zhao, S.; Lian, K.; Mi, B.; Si, R.; Tan, Z.; Fu, F.; Wang, S.; Wang, R.; Ma, X.; et al. C1q/TNF-related protein 3 (CTRP3) and 9 (CTRP9) concentrations are decreased in patients with heart failure and are associated with increased morbidity and mortality. BMC Cardiovasc. Disord. 2019, 19, 139. [Google Scholar] [CrossRef]
- Wang, J.; Hang, T.; Cheng, X.M.; Li, D.M.; Zhang, Q.G.; Wang, L.J.; Peng, Y.P.; Gong, J.B. Associations of C1q/TNF-Related Protein-9 Levels in Serum and Epicardial Adipose Tissue with Coronary Atherosclerosis in Humans. BioMed Res. Int. 2015, 2015, 971683. [Google Scholar] [CrossRef]
- Wang, S.; Ling, Y.; Liang, W.; Shen, L. Association of serum C1q/TNF-related protein-3 (CTRP-3) in patients with coronary artery disease. BMC Cardiovasc. Disord. 2017, 17, 210. [Google Scholar] [CrossRef]
- Sasso, F.C.; Pafundi, P.C.; Marfella, R.; Calabro, P.; Piscione, F.; Furbatto, F.; Esposito, G.; Galiero, R.; Gragnano, F.; Rinaldi, L.; et al. Adiponectin and insulin resistance are related to restenosis and overall new PCI in subjects with normal glucose tolerance: The prospective AIRE Study. Cardiovasc. Diabetol. 2019, 18, 24. [Google Scholar] [CrossRef]
- Rana, M.S.; Christoffels, V.M.; Moorman, A.F. A molecular and genetic outline of cardiac morphogenesis. Acta Physiol. 2013, 207, 588–615. [Google Scholar] [CrossRef] [PubMed]
- Ryan, J.J.; Archer, S.L. Emerging concepts in the molecular basis of pulmonary arterial hypertension: Part I: Metabolic plasticity and mitochondrial dynamics in the pulmonary circulation and right ventricle in pulmonary arterial hypertension. Circulation 2015, 131, 1691–1702. [Google Scholar] [CrossRef] [PubMed]
- Dell’Italia, L.J. Anatomy and physiology of the right ventricle. Cardiol. Clin. 2012, 30, 167–187. [Google Scholar] [CrossRef] [PubMed]
- Schluter, K.D.; Schreiber, D. Adult ventricular cardiomyocytes: Isolation and culture. Methods Mol. Biol. 2005, 290, 305–314. [Google Scholar] [PubMed]
- Del Monte, F.; O’Gara, P.; Poole-Wilson, P.A.; Yacoub, M.; Harding, S.E. Cell geometry and contractile abnormalities of myocytes from failing human left ventricle. Cardiovasc. Res. 1995, 30, 281–290. [Google Scholar] [CrossRef]
- Aslam, M.; Troidl, C.; Tanislav, C.; Rohrbach, S.; Gunduz, D.; Hamm, C.W. Inhibition of Protein Prenylation of GTPases Alters Endothelial Barrier Function. Int. J. Mol. Sci. 2019, 21. [Google Scholar] [CrossRef]
- Watkins, S.J.; Borthwick, G.M.; Arthur, H.M. The H9C2 cell line and primary neonatal cardiomyocyte cells show similar hypertrophic responses in vitro. Vitr. Cell. Dev. Boil. Anim. 2011, 47, 125–131. [Google Scholar] [CrossRef]
- Niemann, B.; Chen, Y.; Teschner, M.; Li, L.; Silber, R.E.; Rohrbach, S. Obesity induces signs of premature cardiac aging in younger patients: The role of mitochondria. J. Am. Coll. Cardiol. 2011, 57, 577–585. [Google Scholar] [CrossRef]
- Kops, G.J.; Dansen, T.B.; Polderman, P.E.; Saarloos, I.; Wirtz, K.W.; Coffer, P.J.; Huang, T.T.; Bos, J.L.; Medema, R.H.; Burgering, B.M. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature 2002, 419, 316–321. [Google Scholar] [CrossRef]
- Jones, P.L.; Kucera, G.; Gordon, H.; Boss, J.M. Cloning and characterization of the murine manganous superoxide dismutase-encoding gene. Gene 1995, 153, 155–161. [Google Scholar] [CrossRef]
- Li, X.N.; Song, J.; Zhang, L.; LeMaire, S.A.; Hou, X.; Zhang, C.; Coselli, J.S.; Chen, L.; Wang, X.L.; Zhang, Y.; et al. Activation of the AMPK-FOXO3 pathway reduces fatty acid-induced increase in intracellular reactive oxygen species by upregulating thioredoxin. Diabetes 2009, 58, 2246–2257. [Google Scholar] [CrossRef] [PubMed]
- Asensio-Lopez, M.C.; Lax, A.; Pascual-Figal, D.A.; Valdes, M.; Sanchez-Mas, J. Metformin protects against doxorubicin-induced cardiotoxicity: Involvement of the adiponectin cardiac system. Free Radic. Biol. Med. 2011, 51, 1861–1871. [Google Scholar] [CrossRef] [PubMed]
- Hajri, T.; Tao, H.; Wattacheril, J.; Marks-Shulman, P.; Abumrad, N.N. Regulation of adiponectin production by insulin: Interactions with tumor necrosis factor-alpha and interleukin-6. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E350–E360. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Guo, Y.; Tao, L.; Lau, W.B.; Gan, L.; Yan, Z.; Guo, R.; Gao, E.; Wong, G.W.; Koch, W.L.; et al. C1q/Tumor Necrosis Factor-Related Protein-9 Regulates the Fate of Implanted Mesenchymal Stem Cells and Mobilizes Their Protective Effects Against Ischemic Heart Injury via Multiple Novel Signaling Pathways. Circulation 2017, 136, 2162–2177. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, P.; Rajesh, M.; Yoshihiro, K.; Hasko, G.; Pacher, P. Simple quantitative detection of mitochondrial superoxide production in live cells. Biochem. Biophys. Res. Commun. 2007, 358, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Redondo-Horcajo, M.; Romero, N.; Martinez-Acedo, P.; Martinez-Ruiz, A.; Quijano, C.; Lourenco, C.F.; Movilla, N.; Enriquez, J.A.; Rodriguez-Pascual, F.; Rial, E.; et al. Cyclosporine A-induced nitration of tyrosine 34 MnSOD in endothelial cells: Role of mitochondrial superoxide. Cardiovasc. Res. 2010, 87, 356–365. [Google Scholar] [CrossRef]
- Kukidome, D.; Nishikawa, T.; Sonoda, K.; Imoto, K.; Fujisawa, K.; Yano, M.; Motoshima, H.; Taguchi, T.; Matsumura, T.; Araki, E. Activation of AMP-activated protein kinase reduces hyperglycemia-induced mitochondrial reactive oxygen species production and promotes mitochondrial biogenesis in human umbilical vein endothelial cells. Diabetes 2006, 55, 120–127. [Google Scholar] [CrossRef]
- Sundaresan, N.R.; Gupta, M.; Kim, G.; Rajamohan, S.B.; Isbatan, A.; Gupta, M.P. Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J. Clin. Invest. 2009, 119, 2758–2771. [Google Scholar] [CrossRef]
- Pillai, V.B.; Sundaresan, N.R.; Kim, G.; Gupta, M.; Rajamohan, S.B.; Pillai, J.B.; Samant, S.; Ravindra, P.V.; Isbatan, A.; Gupta, M.P. Exogenous NAD blocks cardiac hypertrophic response via activation of the SIRT3-LKB1-AMP-activated kinase pathway. J. Biol. Chem. 2010, 285, 3133–3144. [Google Scholar] [CrossRef]
- Zheng, Q.; Yuan, Y.; Yi, W.; Lau, W.B.; Wang, Y.; Wang, X.; Sun, Y.; Lopez, B.L.; Christopher, T.A.; Peterson, J.M.; et al. C1q/TNF-related proteins, a family of novel adipokines, induce vascular relaxation through the adiponectin receptor-1/AMPK/eNOS/nitric oxide signaling pathway. Arter. Thromb. Vasc. Biol. 2011, 31, 2616–2623. [Google Scholar] [CrossRef]
- Schaffler, A.; Orso, E.; Palitzsch, K.D.; Buchler, C.; Drobnik, W.; Furst, A.; Scholmerich, J.; Schmitz, G. The human apM-1, an adipocyte-specific gene linked to the family of TNF’s and to genes expressed in activated T cells, is mapped to chromosome 1q21.3-q23, a susceptibility locus identified for familial combined hyperlipidaemia (FCH). Biochem. Biophys. Res. Commun. 1999, 260, 416–425. [Google Scholar] [CrossRef]
- Maruyama, S.; Shibata, R.; Ohashi, K.; Ohashi, T.; Daida, H.; Walsh, K.; Murohara, T.; Ouchi, N. Adiponectin ameliorates doxorubicin-induced cardiotoxicity through Akt protein-dependent mechanism. J. Biol. Chem. 2011, 286, 32790–32800. [Google Scholar] [CrossRef]
- Park, S.Y.; Choi, J.H.; Ryu, H.S.; Pak, Y.K.; Park, K.S.; Lee, H.K.; Lee, W. C1q tumor necrosis factor alpha-related protein isoform 5 is increased in mitochondrial DNA-depleted myocytes and activates AMP-activated protein kinase. J. Biol. Chem. 2009, 284, 27780–27789. [Google Scholar] [CrossRef]
- Yamauchi, T.; Nio, Y.; Maki, T.; Kobayashi, M.; Takazawa, T.; Iwabu, M.; Okada-Iwabu, M.; Kawamoto, S.; Kubota, N.; Kubota, T.; et al. Targeted disruption of AdipoR1 and AdipoR2 causes abrogation of adiponectin binding and metabolic actions. Nat. Med. 2007, 13, 332–339. [Google Scholar] [CrossRef]
- Schaffler, A.; Buechler, C. CTRP family: Linking immunity to metabolism. Trends Endocrinol. Metab. 2012, 23, 194–204. [Google Scholar] [CrossRef]
- Jorgensen, S.B.; Viollet, B.; Andreelli, F.; Frosig, C.; Birk, J.B.; Schjerling, P.; Vaulont, S.; Richter, E.A.; Wojtaszewski, J.F. Knockout of the alpha2 but not alpha1 5’-AMP-activated protein kinase isoform abolishes 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranosidebut not contraction-induced glucose uptake in skeletal muscle. J. Biol. Chem. 2004, 279, 1070–1079. [Google Scholar] [CrossRef]
- Pineiro, R.; Iglesias, M.J.; Gallego, R.; Raghay, K.; Eiras, S.; Rubio, J.; Dieguez, C.; Gualillo, O.; Gonzalez-Juanatey, J.R.; Lago, F. Adiponectin is synthesized and secreted by human and murine cardiomyocytes. FEBS Lett. 2005, 579, 5163–5169. [Google Scholar] [CrossRef]
- Gollmer, J.; Zirlik, A.; Bugger, H. Mitochondrial Mechanisms in Diabetic Cardiomyopathy. Diabetes Metab. J. 2020, 44, 33–53. [Google Scholar] [CrossRef]
- Niemann, B.; Rohrbach, S.; Miller, M.R.; Newby, D.E.; Fuster, V.; Kovacic, J.C. Oxidative Stress and Cardiovascular Risk: Obesity, Diabetes, Smoking, and Pollution: Part 3 of a 3-Part Series. J. Am. Coll. Cardiol. 2017, 70, 230–251. [Google Scholar] [CrossRef]
- Yu, X.H.; Zhang, D.W.; Zheng, X.L.; Tang, C.K. C1q tumor necrosis factor-related protein 9 in atherosclerosis: Mechanistic insights and therapeutic potential. Atherosclerosis 2018, 276, 109–116. [Google Scholar] [CrossRef]
- Arita, Y.; Kihara, S.; Ouchi, N.; Takahashi, M.; Maeda, K.; Miyagawa, J.; Hotta, K.; Shimomura, I.; Nakamura, T.; Miyaoka, K.; et al. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem. Biophys. Res. Commun. 1999, 257, 79–83. [Google Scholar] [CrossRef]
- Jung, C.H.; Lee, M.J.; Kang, Y.M.; Lee, Y.L.; Seol, S.M.; Yoon, H.K.; Kang, S.W.; Lee, W.J.; Park, J.Y. C1q/TNF-related protein-9 inhibits cytokine-induced vascular inflammation and leukocyte adhesiveness via AMP-activated protein kinase activation in endothelial cells. Mol. Cell Endocrinol. 2016, 419, 235–243. [Google Scholar] [CrossRef]
- Okamoto, Y.; Kihara, S.; Ouchi, N.; Nishida, M.; Arita, Y.; Kumada, M.; Ohashi, K.; Sakai, N.; Shimomura, I.; Kobayashi, H.; et al. Adiponectin reduces atherosclerosis in apolipoprotein E-deficient mice. Circulation 2002, 106, 2767–2770. [Google Scholar] [CrossRef]
- Colombo, S.L.; Moncada, S. AMPKalpha1 regulates the antioxidant status of vascular endothelial cells. Biochem. J. 2009, 421, 163–169. [Google Scholar] [CrossRef]
- Weng, C.F.; Wu, C.F.; Kao, S.H.; Chen, J.C.; Lin, H.H. Down-Regulation of miR-34a-5p Potentiates Protective Effect of Adipose-Derived Mesenchymal Stem Cells Against Ischemic Myocardial Infarction by Stimulating the Expression of C1q/Tumor Necrosis Factor-Related Protein-9. Front. Physiol. 2019, 10, 1445. [Google Scholar] [CrossRef]
- Branco, A.F.; Pereira, S.P.; Gonzalez, S.; Gusev, O.; Rizvanov, A.A.; Oliveira, P.J. Gene Expression Profiling of H9c2 Myoblast Differentiation towards a Cardiac-Like Phenotype. PLoS ONE 2015, 10, e0129303. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niemann, B.; Li, L.; Siegler, D.; Siegler, B.H.; Knapp, F.; Hanna, J.; Aslam, M.; Kracht, M.; Schulz, R.; Rohrbach, S. CTRP9 Mediates Protective Effects in Cardiomyocytes via AMPK- and Adiponectin Receptor-Mediated Induction of Anti-Oxidant Response. Cells 2020, 9, 1229. https://doi.org/10.3390/cells9051229
Niemann B, Li L, Siegler D, Siegler BH, Knapp F, Hanna J, Aslam M, Kracht M, Schulz R, Rohrbach S. CTRP9 Mediates Protective Effects in Cardiomyocytes via AMPK- and Adiponectin Receptor-Mediated Induction of Anti-Oxidant Response. Cells. 2020; 9(5):1229. https://doi.org/10.3390/cells9051229
Chicago/Turabian StyleNiemann, Bernd, Ling Li, Dorothee Siegler, Benedikt H. Siegler, Fabienne Knapp, Jakob Hanna, Muhammad Aslam, Michael Kracht, Rainer Schulz, and Susanne Rohrbach. 2020. "CTRP9 Mediates Protective Effects in Cardiomyocytes via AMPK- and Adiponectin Receptor-Mediated Induction of Anti-Oxidant Response" Cells 9, no. 5: 1229. https://doi.org/10.3390/cells9051229
APA StyleNiemann, B., Li, L., Siegler, D., Siegler, B. H., Knapp, F., Hanna, J., Aslam, M., Kracht, M., Schulz, R., & Rohrbach, S. (2020). CTRP9 Mediates Protective Effects in Cardiomyocytes via AMPK- and Adiponectin Receptor-Mediated Induction of Anti-Oxidant Response. Cells, 9(5), 1229. https://doi.org/10.3390/cells9051229