Liver Sinusoidal Endothelial Cells Contribute to Hepatic Antigen-Presenting Cell Function and Th17 Expansion in Cirrhosis


 , ,
, ,  ,
,
Abstract
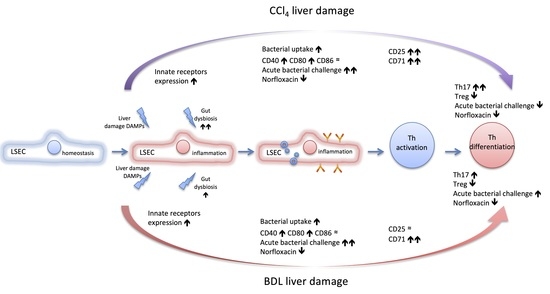
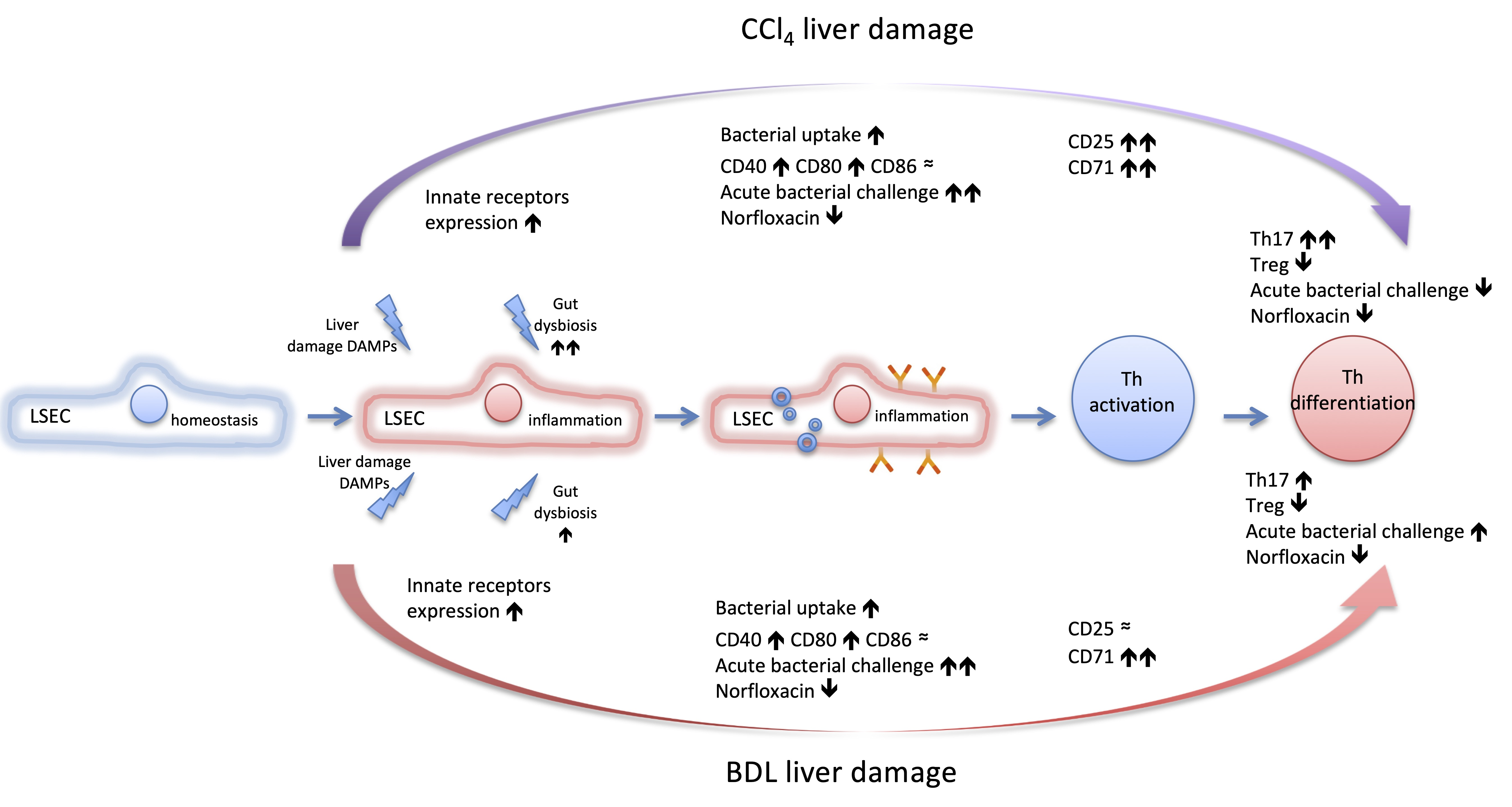{kind=link}
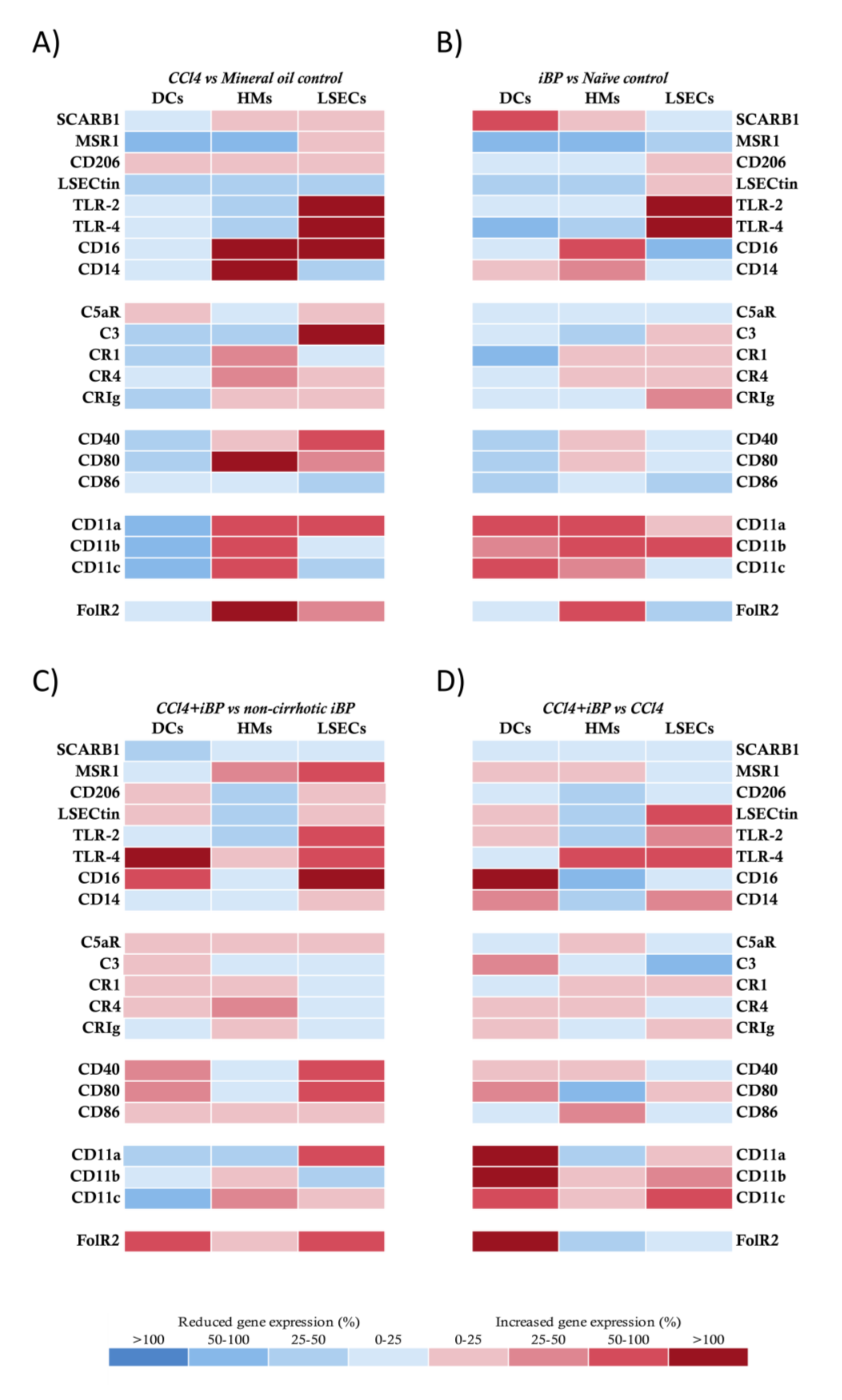{kind=link}
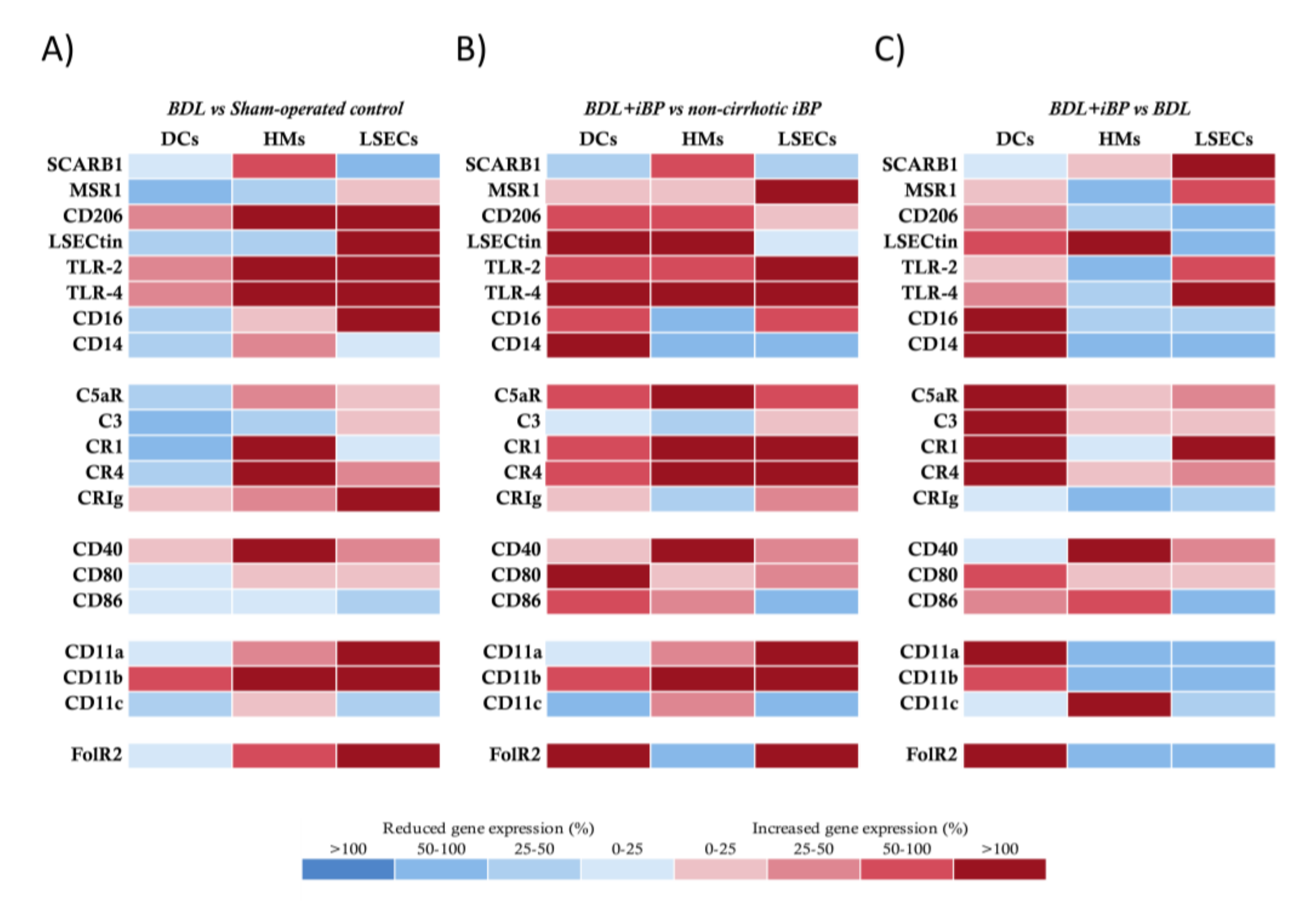{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals and Study Protocol
2.2. Liver APCs Isolation
2.3. Gene Expression Measurements
2.4. In Vitro Phagocytosis and Killing Assays and Liver APCs’ Co-Stimulatory Molecules Expression
2.5. Isolation of Splenic T Cells and Th Selection
2.6. Co-Culture of APCs with Th CD4+ Cells and Flow Cytometry Staining
2.7. Statistical Analysis
3. Results
3.1. Animals
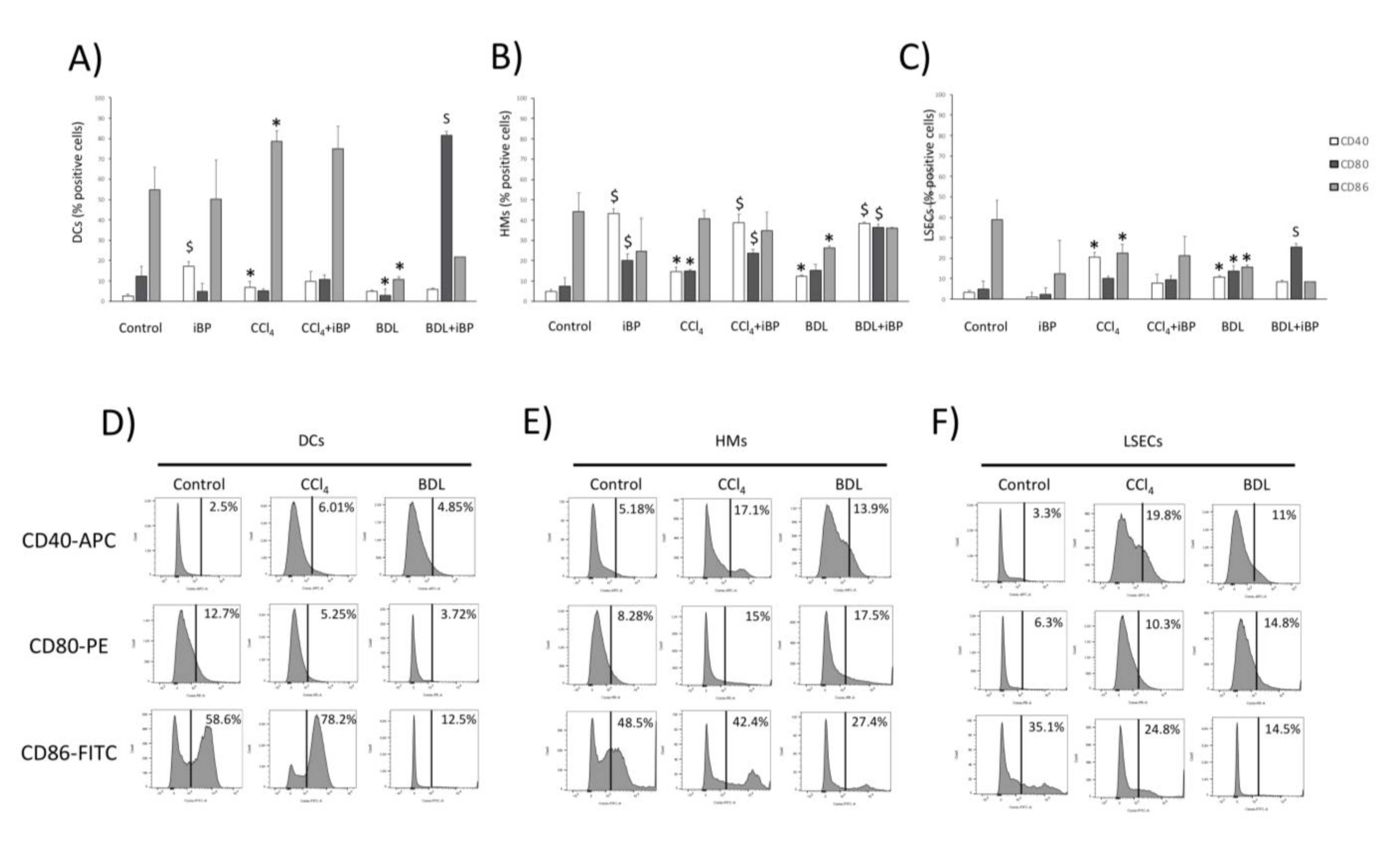
3.2. LSECs Displayed Immunosurveillance Markers during Experimental Liver Damage
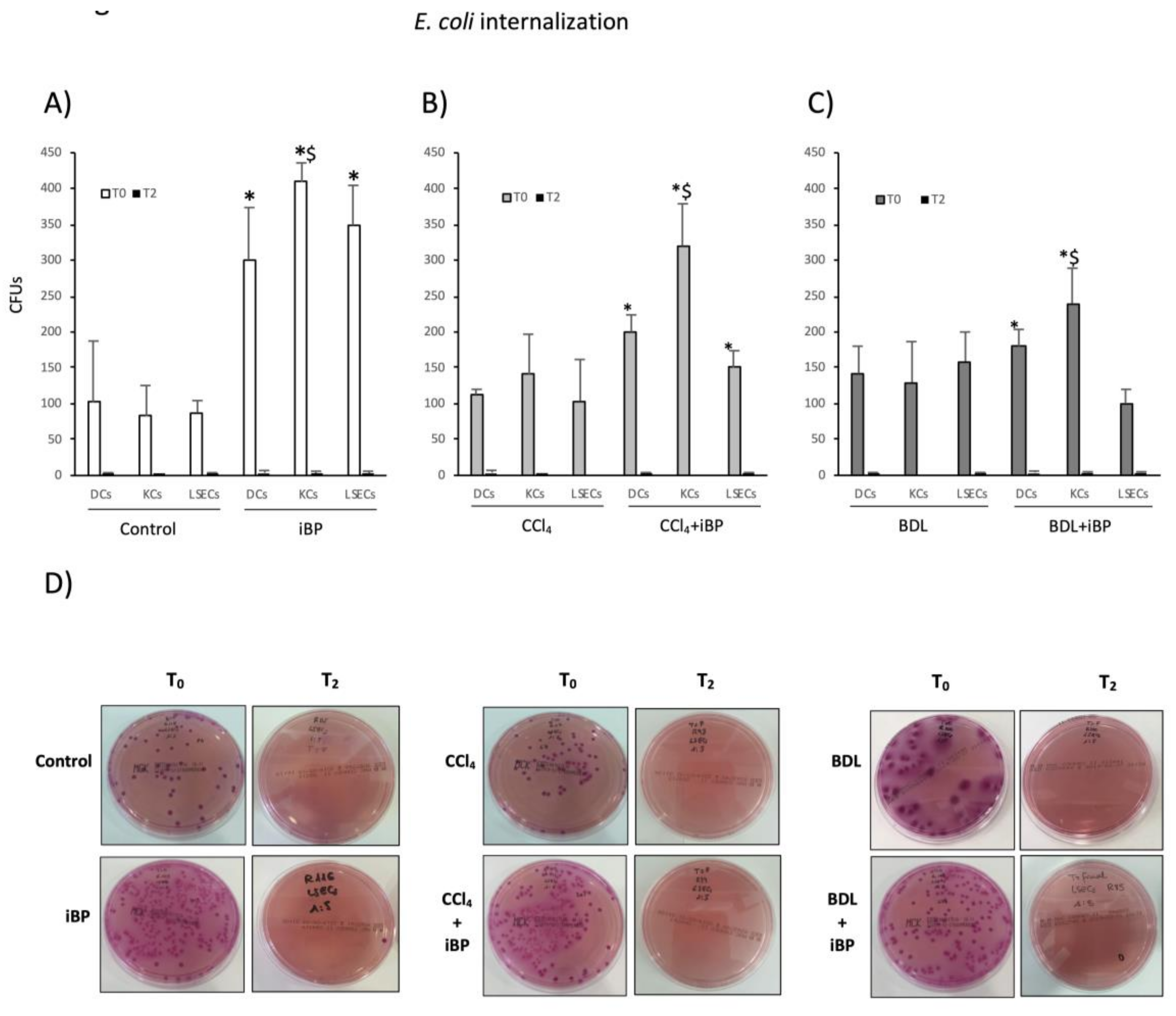
3.3. LSECs Significantly Cooperated in E. coli Internalization and Killing Activity in Experimental Cirrhosis
3.4. LSECs Participated in Bridging Innate and Adaptive Immunity during Experimental Cirrhosis
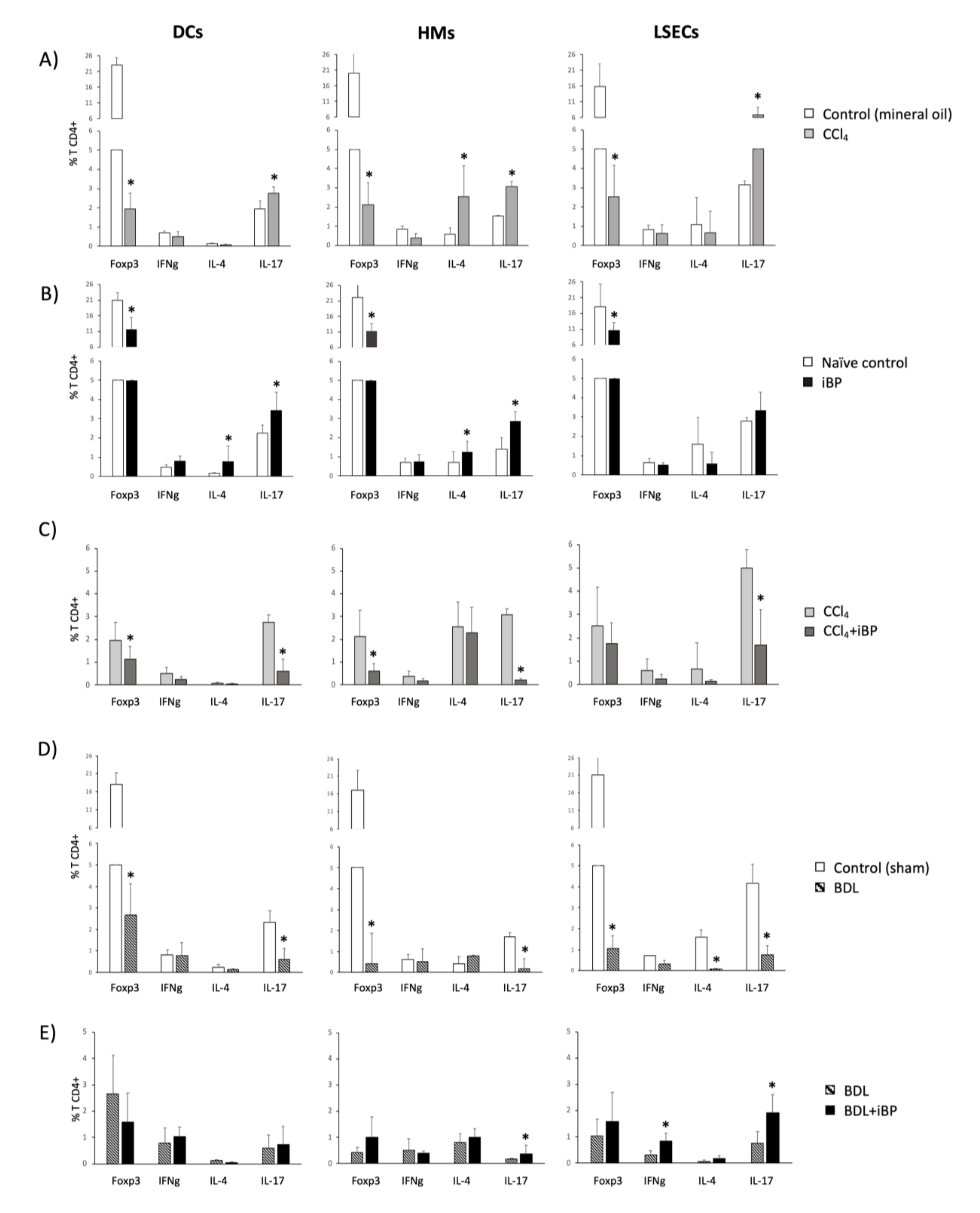
3.5. LSECs Stimulated the Activation and Differentiation of the Adaptive T CD4+ Cell Response in Rats with Cirrhosis
3.6. Norfloxacin Limited the Activation of Innate Immune Receptors and Preserved LSECs Functionality, Favoring LSEC-Dependent Treg Differentiation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Albillos, A.; Lario, M.; Alvarez-Mon, M. Cirrhosis-associated immune dysfunction: Distinctive features and clinical relevance. J. Hepatol. 2014, 61, 1385–1396. [Google Scholar] [CrossRef] [PubMed]
- Wiest, R.; Lawson, M.; Geuking, M. Pathological bacterial translocation in liver cirrhosis. J. Hepatol. 2014, 60, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Guillot, A.; Tacke, F. Liver Macrophages: Old Dogmas and New Insights. Hepatol. Commun. 2019, 3, 730–743. [Google Scholar] [CrossRef]
- Racanelli, V.; Rehermann, B. The liver as an immunological organ. Hepatology 2006, 43, S54–S62. [Google Scholar] [CrossRef] [PubMed]
- Thomson, A.W.; Knolle, P.A. Antigen-presenting cell function in the tolerogenic liver environment. Nat. Rev. Immunol. 2010, 10, 753–766. [Google Scholar] [CrossRef]
- Fouts, D.E.; Torralba, M.; Nelson, K.E.; Brenner, D.A.; Schnabl, B. Bacterial translocation and changes in the intestinal microbiome in mouse models of liver disease. J. Hepatol. 2012, 56, 1283–1292. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, F.; Lu, H.; Wang, B.; Chen, Y.; Lei, D.; Wang, Y.; Zhu, B.; Li, L. Characterization of fecal microbial communities in patients with liver cirrhosis. Hepatology 2011, 54, 562–572. [Google Scholar] [CrossRef]
- Albillos, A.; de la Hera, A.; Reyes, E.; Monserrat, J.; Munoz, L.; Nieto, M.; Prieto, A.; Sanz, E.; Alvarez-Mon, M. Tumour necrosis factor-alpha expression by activated monocytes and altered T-cell homeostasis in ascitic alcoholic cirrhosis: Amelioration with norfloxacin. J. Hepatol. 2004, 40, 624–631. [Google Scholar] [CrossRef]
- Muñoz, L.; Albillos, A.; Nieto, M.; Reyes, E.; Lledó, L.; Monserrat, J.; Sanz, E.; de la Hera, A.; Alvarez-Mon, M. Mesenteric Th1 polarization and monocyte TNF-alpha production: First steps to systemic inflammation in rats with cirrhosis. Hepatology 2005, 42, 411–419. [Google Scholar] [CrossRef]
- Lemmers, A.; Moreno, C.; Gustot, T.; Maréchal, R.; Degré, D.; Demetter, P.; De Nadai, P.; Geerts, A.; Quertinmont, E.; Vercruysse, V.; et al. The interleukin-17 pathway is involved in human alcoholic liver disease. Hepatology 2009, 49, 646–657. [Google Scholar] [CrossRef]
- Sun, H.Q.; Zhang, J.Y.; Zhang, H.; Zou, Z.S.; Wang, F.S.; Jia, J.H. Increased Th17 cells contribute to disease progression in patients with HBV-associated liver cirrhosis. J. Viral Hepat. 2012, 19, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Zapotoczny, B.; Szafranska, K.; Kus, E.; Braet, F.; Wisse, E.; Chlopicki, S.; Szymonski, M. Tracking Fenestrae Dynamics in Live Murine Liver Sinusoidal Endothelial Cells. Hepatology 2019, 69, 876–888. [Google Scholar] [CrossRef] [PubMed]
- Poisson, J.; Lemoinne, S.; Boulanger, C.; Durand, F.; Moreau, R.; Valla, D.; Rautou, P.E. Liver sinusoidal endothelial cells: Physiology and role in liver diseases. J. Hepatol. 2017, 66, 212–227. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, K.K.; McCourt, P.; Berg, T.; Crossley, C.; Couteur, D.L.; Wake, K.; Smedsrød, B. The scavenger endothelial cell: A new player in homeostasis and immunity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 303, R1217–R1230. [Google Scholar]
- Shetty, S.; Lalor, P.F.; Adams, D.H. Liver sinusoidal endothelial cells - gatekeepers of hepatic immunity. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 555–567. [Google Scholar] [CrossRef]
- Lalor, P.F.; Shields, P.; Grant, A.J.; Adams, D.H. Recruitment of lymphocytes to the human liver. Immunol. Cell Biol. 2002, 80, 52–64. [Google Scholar] [CrossRef]
- Runyon, B.A.; Sugano, S.; Kanel, G.; Mellencamp, M.A. A rodent model of cirrhosis, ascites, and bacterial peritonitis. Gastroenterology 1991, 100, 489–493. [Google Scholar] [CrossRef]
- Seki, E.; De Minicis, S.; Österreicher, C.H.; Kluwe, J.; Osawa, Y.; Brenner, D.A.; Schwabe, R.F. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat. Med. 2007, 13, 1324–1332. [Google Scholar] [CrossRef]
- Guarner, C.; González-Navajas, J.M.; Sánchez, E.; Soriando, G.; Francés, R.; Chiva, M.; Zapater, P.; Benlloch, S.; Muñoz, C.; Pascual, S.; et al. The detection of bacterial DNA in blood of rats with CCl(4)-induced cirrhosis with ascites represents episodes of bacterial translocation. Hepatology 2006, 44, 633–639. [Google Scholar] [CrossRef]
- Gracia-Sancho, J.; Laviña, B.; Rodríguez-Vilarrupla, A.; García-Calderó, H.; Bosch, J.; García-Pagán, J.C. Enhanced vasoconstrictor prostanoid production by sinusoidal endothelial cells increases portal perfusion pressure in cirrhotic rat livers. J. Hepatol. 2007, 47, 220–227. [Google Scholar] [CrossRef]
- Sørensen, K.K.; Simon-Santamaria, J.; McCuskey, R.S.; Smedsrød, B. Liver Sinusoidal Endothelial Cells. Compr. Physiol. 2015, 5, 1751–1774. [Google Scholar] [PubMed]
- Wiest, R.; Albillos, A.; Trauner, M.; Bajaj, J.S.; Jalan, R. Targeting the gut-liver axis in liver disease. J. Hepatol. 2017, 67, 1084–1103. [Google Scholar] [CrossRef] [PubMed]
- Irvine, K.M.; Ratnasekera, I.; Powell, E.E.; Hume, D.A. Causes and Consequences of Innate Immune Dysfunction in Cirrhosis. Front. Immunol. 2019, 10, 293. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.H.; Thomson, A.W. Dendritic cells and immune regulation in the liver. Gut 2003, 52, 307–314. [Google Scholar] [CrossRef]
- Pillarisetty, V.G.; Shah, A.B.; Miller, G.; Bleier, J.I.; DeMatteo, R.P. Liver dendritic cells are less immunogenic than spleen dendritic cells because of differences in subtype composition. J. Immunol. 2004, 172, 1009–1017. [Google Scholar] [CrossRef]
- Knolle, P.A.; Limmer, A. Neighborhood politics: The immunoregulatory function of organ-resident liver endothelial cells. Trends Immunol. 2001, 22, 432–437. [Google Scholar] [CrossRef]
- Crispe, I.N. The liver as a lymphoid organ. Annu. Rev. Immunol. 2009, 27, 147–163. [Google Scholar] [CrossRef]
- Yao, Z.; Mates, J.M.; Cheplowitz, A.M.; Hammer, L.P.; Maiseyeu, A.; Phillips, G.S.; Wewers, M.D.; Rajaram, M.V.; Robinson, J.M.; Anderson, C.L.; et al. Blood-Borne Lipopolysaccharide Is Rapidly Eliminated by Liver Sinusoidal Endothelial Cells via High-Density Lipoprotein. J. Immunol. 2016, 197, 2390–2399. [Google Scholar] [CrossRef]
- Géraud, C.; Schledzewski, K.; Demory, A.; Klein, D.; Kaus, M.; Peyre, F.; Sticht, C.; Evdokimov, K.; Lu, S.; Schmieder, A.; et al. Liver sinusoidal endothelium: A microenvironment-dependent differentiation program in rat including the novel junctional protein liver endothelial differentiation-associated protein-1. Hepatology 2010, 52, 313–326. [Google Scholar] [CrossRef]
- Limmer, A.; Ohl, J.; Kurts, C.; Ljunggren, H.G.; Reiss, Y.; Groettrup, M.; Momburg, F.; Arnold, B.; Knolle, P.A. Efficient presentation of exogenous antigen by liver endothelial cells to CD8+ T cells results in antigen-specific T-cell tolerance. Nat. Med. 2000, 6, 1348–1354. [Google Scholar] [CrossRef]
- Limmer, A.; Ohl, J.; Wingender, G.; Berg, M.; Jüngerkes, F.; Schumak, B.; Djandji, D.; Scholz, K.; Klevenz, A.; Hegenbarth, S.; et al. Cross-presentation of oral antigens by liver sinusoidal endothelial cells leads to CD8 T cell tolerance. Eur. J. Immunol. 2005, 35, 2970–2981. [Google Scholar] [CrossRef] [PubMed]
- Schurich, A.; Böttcher, J.P.; Burgdorf, S.; Penzler, P.; Hegenbarth, S.; Kern, M.; Dolf, A.; Endl, E.; Schultze, J.; Wiertz, E.; et al. Distinct kinetics and dynamics of cross-presentation in liver sinusoidal endothelial cells compared to dendritic cells. Hepatology 2009, 50, 909–919. [Google Scholar] [CrossRef] [PubMed]
- Von Oppen, N.; Schurich, A.; Hegenbarth, S.; Stabenow, D.; Tolba, R.; Weiskirchen, R.; Geerts, A.; Kolanus, W.; Knolle, P.; Diehl, L. Systemic antigen cross-presented by liver sinusoidal endothelial cells induces liver-specific CD8 T-cell retention and tolerization. Hepatology 2009, 49, 1664–1672. [Google Scholar] [CrossRef] [PubMed]
- Kruse, N.; Neumann, K.; Schrage, A.; Derkow, K.; Schott, E.; Erben, U.; Kühl, A.; Loddenkemper, C.; Zeitz, M.; Hamann, A.; et al. Priming of CD4+ T cells by liver sinusoidal endothelial cells induces CD25low forkhead box protein 3- regulatory T cells suppressing autoimmune hepatitis. Hepatology 2009, 50, 1904–1913. [Google Scholar] [CrossRef]
- Carambia, A.; Freund, B.; Schwinge, D. TGF-beta-dependent induction of CD4(+)CD25(+)Foxp3(+) Tregs by liver sinusoidal endothelial cells. J. Hepatol. 2014, 61, 594–599. [Google Scholar] [CrossRef]
- Schurich, A.; Berg, M.; Stabenow, D.; Böttcher, J.; Kern, M.; Schild, H.J.; Kurts, C.; Schuette, V.; Burgdorf, S.; Diehl, L.; et al. Dynamic regulation of CD8 T cell tolerance induction by liver sinusoidal endothelial cells. J. Immunol. 2010, 184, 4107–4114. [Google Scholar] [CrossRef]
- Connolly, M.K.; Bedrosian, A.S.; Malhotra, A.; Henning, J.R.; Ibrahim, J.; Vera, V.; Cieza-Rubio, N.E.; Hassan, B.U.; Pachter, H.L.; Cohen, S.; et al. In hepatic fibrosis, liver sinusoidal endothelial cells acquire enhanced immunogenicity. J. Immunol. 2010, 185, 2200–2208. [Google Scholar] [CrossRef]
- Hammerich, L.; Heymann, F.; Tacke, F. Role of IL-17 and Th17 cells in liver diseases. Clin. Dev. Immunol. 2011, 2011, 345803. [Google Scholar] [CrossRef]
- Francés, R.; Zapater, P.; González-Navajas, J.M.; Muñoz, C.; Caño, R.; Moreu, R.; Pascual, S.; Bellot, P.; Pérez-Mateo, M.; Such, J. Bacterial DNA in patients with cirrhosis and noninfected ascites mimics the soluble immune response established in patients with spontaneous bacterial peritonitis. Hepatology 2008, 47, 78–985. [Google Scholar] [CrossRef]
- Yanguas, S.C.; da Silva, T.C.; Pereira, I.V.; Maes, M.; Willebrords, J.; Shestopalov, V.I.; Goes, B.M.; Nogueira, M.S.; de Castro, I.A.; Romualdo, G.R.; et al. Genetic ablation of pannexin1 counteracts liver fibrosis in a chemical, but not in a surgical mouse model. Arch. Toxicol. 2018, 92, 2607–2627. [Google Scholar] [CrossRef]
- Martin-Mateos, R.; Alvarez-Mon, M.; Albillos, A. Dysfunctional Immune Response in Acute-on-Chronic Liver Failure: It Takes Two to Tango. Front. Immunol. 2019, 10, 973. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Yang, J.; Liu, W.; Tang, X.; Chen, J.; Zhao, D.; Wang, M.; Xu, F.; Lu, Y.; Liu, B.; et al. Liver sinusoidal endothelial cell lectin, LSECtin, negatively regulates hepatic T-cell immune response. Gastroenterology 2009, 137, 1498–1508. [Google Scholar] [CrossRef] [PubMed]
- Cabrera-Rubio, R.; Patterson, A.M.; Cotter, P.D.; Beraza, N. Cholestasis induced by bile duct ligation promotes changes in the intestinal microbiome in mice. Sci. Rep. 2019, 9, 12324. [Google Scholar] [CrossRef]
- Nevzorova, Y.A.; Boyer-Diaz, Z.; Cubero, F.J.; Gracia-Sancho, J. Animal Models for Liver Disease - A Practical Approach for Translational Research. J. Hepatol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Dalhoff, A.; Shalit, I. Immunomodulatory effects of quinolones. Lancet Infect. Dis. 2003, 3, 359–371. [Google Scholar] [CrossRef]
- Zapater, P.; Caño, R.; Llanos, L.; Ruiz–Alcaraz, A.J.; Pascual, S.; Barquero, C.; Moreu, R.; Bellot, P.; Horga, J.F.; Muñoz, C.; et al. Norfloxacin modulates the inflammatory response and directly affects neutrophils in patients with decompensated cirrhosis. Gastroenterology 2009, 137, 1669–1679. [Google Scholar] [CrossRef]
- Juanola, O.; Gómez-Hurtado, I.; Zapater, P.; Moratalla, A.; Caparrós, E.; Piñero, P.; González-Navajas, J.M.; Giménez, P.; Such, J.; Francés, R. Selective intestinal decontamination with norfloxacin enhances a regulatory T cell-mediated inflammatory control mechanism in cirrhosis. Liver Int. 2016, 36, 1811–1820. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caparrós, E.; Juanola, O.; Gómez-Hurtado, I.; Puig-Kroger, A.; Piñero, P.; Zapater, P.; Linares, R.; Tarín, F.; Martínez-López, S.; Gracia-Sancho, J.; et al. Liver Sinusoidal Endothelial Cells Contribute to Hepatic Antigen-Presenting Cell Function and Th17 Expansion in Cirrhosis. Cells 2020, 9, 1227. https://doi.org/10.3390/cells9051227
Caparrós E, Juanola O, Gómez-Hurtado I, Puig-Kroger A, Piñero P, Zapater P, Linares R, Tarín F, Martínez-López S, Gracia-Sancho J, et al. Liver Sinusoidal Endothelial Cells Contribute to Hepatic Antigen-Presenting Cell Function and Th17 Expansion in Cirrhosis. Cells. 2020; 9(5):1227. https://doi.org/10.3390/cells9051227
Chicago/Turabian StyleCaparrós, Esther, Oriol Juanola, Isabel Gómez-Hurtado, Amaya Puig-Kroger, Paula Piñero, Pedro Zapater, Raquel Linares, Fabián Tarín, Sebastián Martínez-López, Jordi Gracia-Sancho, and et al. 2020. "Liver Sinusoidal Endothelial Cells Contribute to Hepatic Antigen-Presenting Cell Function and Th17 Expansion in Cirrhosis" Cells 9, no. 5: 1227. https://doi.org/10.3390/cells9051227
APA StyleCaparrós, E., Juanola, O., Gómez-Hurtado, I., Puig-Kroger, A., Piñero, P., Zapater, P., Linares, R., Tarín, F., Martínez-López, S., Gracia-Sancho, J., González-Navajas, J. M., & Francés, R. (2020). Liver Sinusoidal Endothelial Cells Contribute to Hepatic Antigen-Presenting Cell Function and Th17 Expansion in Cirrhosis. Cells, 9(5), 1227. https://doi.org/10.3390/cells9051227