Two Birds with One Stone: NFAT1-MDM2 Dual Inhibitors for Cancer Therapy †


Abstract
1. Introduction
2. MDM2 as a Molecule Target for Cancer Therapy
3. NFAT1 as a Target for Cancer Therapy
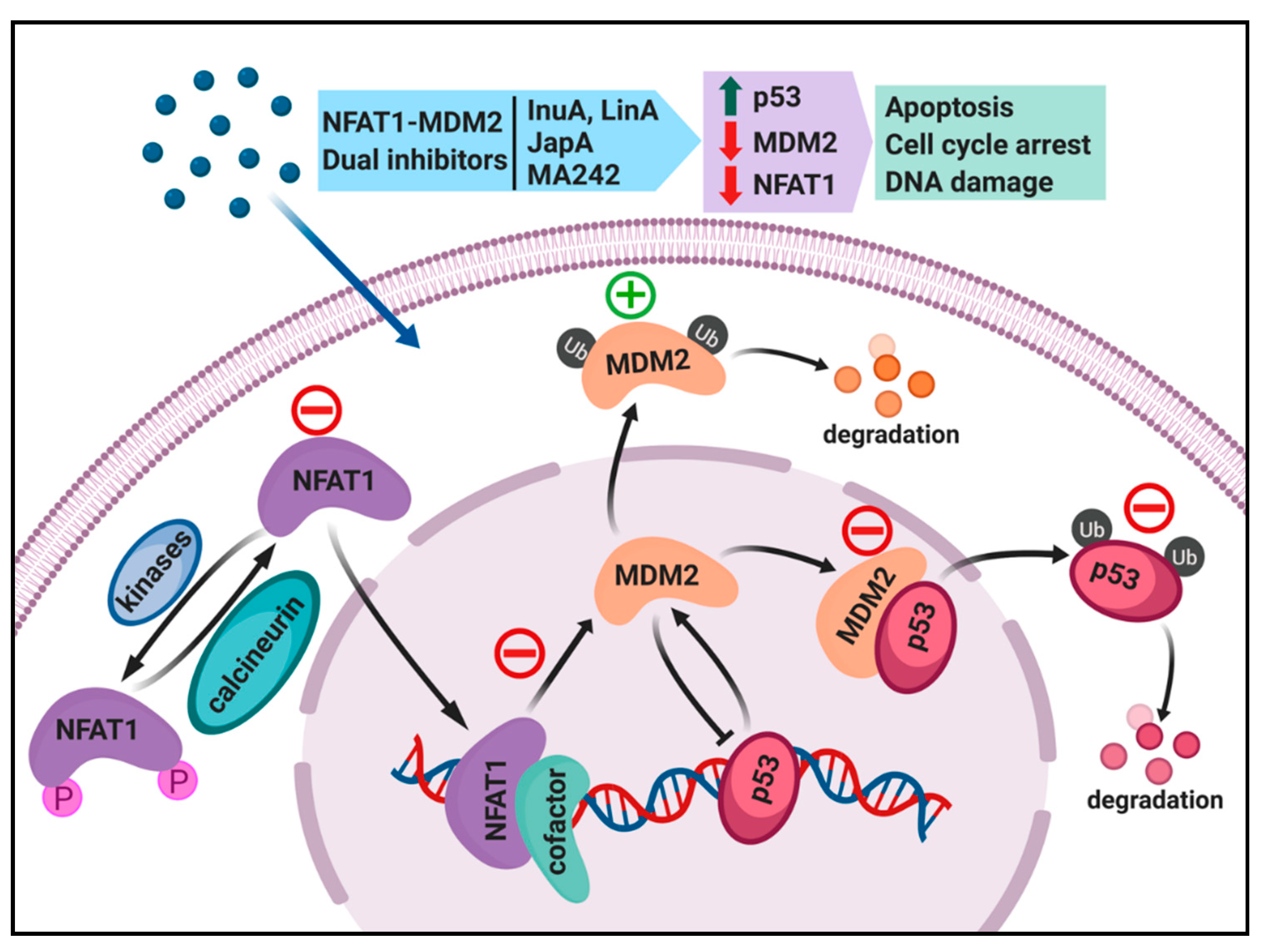
4. Dual Inhibition of the NFAT1 and MDM2 Pathways for Cancer Therapy
4.1. Regulation of MDM2 Expression by NFAT1 and Clinical Relevance
4.2. Search for NFAT1 and MDM2 Inhibitors
4.3. Discovery and Evaluation of Dual NFAT1 and MDM2 Inhibitors
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cahilly-Snyder, L.; Yang-Feng, T.; Francke, U.; George, D.L. Molecular analysis and chro- mosomal mapping of amplified genes isolated from a transformed mouse 3T3 cell line. Somat. Cell Mol. Genet. 1987, 13, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Ware, P.L.; Snow, A.N.; Gvalani, M.; Pettenati, M.J.; Qasem, S.A. MDM2 copy numbers in well-differentiated and dedifferentiated liposarcoma: Characterizing progression to high-grade tumours. Am. J. Clin. Pathol. 2014, 141, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Momand, J.; Jung, D.; Wilczynski, S.; Niland, J. The MDM2 gene amplification database. Nucleic Acids Res. 1998, 26, 3453–3459. [Google Scholar] [CrossRef]
- Onel, K.; Cordon-Cardo, C. MDM2 and prognosis. Mol. Cancer Res. 2004, 2, 1–8. [Google Scholar] [PubMed]
- Nag, S.; Qin, J.; Srivenugopal, K.S.; Wang, M.; Zhang, R. The MDM2-p53 pathway revisited. J. Biomed. Res. 2013, 27, 254–271. [Google Scholar]
- Karni-Schmidt, O.; Lokshin, M.; Prives, C. The roles of MDM2 and MDMX in cancer. Annu. Rev. Pathol. 2016, 11, 617–644. [Google Scholar] [CrossRef]
- Wade, M.; Li, Y.C.; Wahl, G.M. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat. Rev. Cancer 2013, 13, 83–96. [Google Scholar] [CrossRef]
- Oliner, J.D.; Kinzler, K.W.; Meltzer, P.S.; George, D.L.; Vogelstein, B. Amplification of a gene encoding a p53-associated protein in human sarcomas. Nature 1992, 358, 80–83. [Google Scholar] [CrossRef]
- Momand, J.; Zambetti, G.P.; Olson, D.C.; George, D.; Levine, A.J. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 1992, 69, 1237–1245. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, M.; Wang, H.; Agrawal, S.; Zhang, R. Antisense therapy targeting MDM2 oncogene in prostate cancer: Effects on proliferation, apoptosis, multiple gene expression, and chemotherapy. Proc. Natl. Acad. Sci. USA 2003, 100, 11636–11641. [Google Scholar] [CrossRef]
- Gu, L.; Zhu, N.; Zhang, H.; Durden, D.L.; Feng, Y.; Zhou, M. Regulation of XIAP translation and induction by MDM2 following irradiation. Cancer Cell 2009, 15, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, Z.; Cheng, J.; Li, M.; Wang, W.; Xu, W.; Wang, H.; Zhang, R. Transcription factor NFAT1 activates the mdm2 oncogene independent of p53. J. Biol. Chem. 2012, 287, 30468–30476. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.J.; Wang, W.; Zhang, R. Experimental therapy of advanced breast cancer: Targeting NFAT1–MDM2–p53 pathway. Prog. Mol. Biol. Transl. Sci. 2017, 151, 195–216. [Google Scholar] [PubMed]
- Prives, C.; Hall, P.A. The p53 pathway. J. Pathol. 1999, 187, 112–126. [Google Scholar] [CrossRef]
- Sionov, R.V.; Haupt, Y. The cellular response to p53: The decision between life and death. Oncogene 1999, 18, 6145–6157. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Lu, X. Live or let die: The cell’s response to p53. Nat. Rev. Cancer 2002, 2, 594–604. [Google Scholar] [CrossRef]
- Abbas, T.; Dutta, A. p21 in cancer: Intricate networks and multiple activities. Nat. Rev. Cancer 2009, 9, 400–414. [Google Scholar] [CrossRef]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef]
- Kubbutat, M.H.; Jones, S.N.; Vousden, K.H. Regulation of p53 stability by Mdm2. Nature 1997, 387, 299–303. [Google Scholar] [CrossRef]
- Honda, R.; Tanaka, H.; Yasuda, H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997, 420, 25–27. [Google Scholar] [CrossRef]
- Iwakuma, T.; Lozano, G. MDM2, an introduction. Mol. Cancer Res. 2003, 1, 993–1000. [Google Scholar] [PubMed]
- Shaikh, M.F.; Morano, F.; Lee, J.; Gleeson, E.; Babcock, B.D.; Michl, J.; Sarafraz-Yazdim, E.; Pincus, M.R.; Bowne, W.B. Emerging Role of MDM2 as target for anti-cancer therapy: A review. Ann. Clin. Lab. Sci. 2016, 46, 627–634. [Google Scholar] [PubMed]
- Fakharzadeh, S.S.; Trusko, S.P.; George, D.L. Tumorigenic potential associated with enhanced expression of a gene that is amplified in a mouse tumor cell line. EMBO J. 1991, 10, 1565–1569. [Google Scholar] [CrossRef] [PubMed]
- Moll, U.M.; Petrenko, O. The MDM2-p53 interaction. Mol. Cancer Res. 2003, 1, 1001–1008. [Google Scholar] [PubMed]
- Ding, K.; Lu, Y.; Nikolovska-Coleska, Z.; Wang, G.; Qiu, S.; Shangary, S.; Gao, W.; Qin, D.; Stuckey, J.; Krajewski, K.; et al. Structure-based design of spiro-oxindoles as potent, specific small-molecule inhibitors of the MDM2-p53 interaction. J. Med. Chem. 2006, 49, 3432–3435. [Google Scholar] [CrossRef] [PubMed]
- Shangary, S.; Qin, D.; McEachern, D.; Liu, M.; Miller, R.S.; Qiu, S.; Nikolovska-Coleska, Z.; Ding, K.; Wang, G.; Chen, J.; et al. Temporal activation of p53 by a specific MDM2 inhibitor is selectively toxic to tumours and leads to complete tumor growth inhibition. Proc. Natl. Acad. Sci. USA 2008, 105, 3933–3938. [Google Scholar] [CrossRef]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef]
- Beloglazkina, A.; Zyk, N.; Majouga, A.; Beloglazkina, E. Recent small-molecule inhibitors of the p53-MDM2 protein-protein interaction. Molecules 2020, 25, 1211. [Google Scholar] [CrossRef]
- Klein, C.; Vassilev, L.T. Targeting the p53-MDM2 interaction to treat cancer. Br. J. Cancer 2004, 91, 1415–1419. [Google Scholar] [CrossRef]
- Buolamwini, J.K.; Addo, J.; Kamath, S.; Patil, S.; Mason, D.; Ores, M. Small molecule antagonists of the MDM2 oncoprotein as anticancer agents. Curr. Cancer Drug Targets 2005, 5, 57–68. [Google Scholar] [CrossRef]
- Burgess, A.; Chia, K.M.; Haupt, S.; Thomas, D.; Haupt, Y.; Lim, E. Clinical Overview of MDM2/X-Targeted Therapies. Front. Oncol. 2016, 6, 7. [Google Scholar] [CrossRef] [PubMed]
- Furet, P.; Masuya, K.; Kallen, J.; Stachyra-Valat, T.; Ruetz, S.; Guagnano, V.; Holzer, P.; Mah, R.; Stutz, S.; Vaupel, A.; et al. Discovery of a novel class of highly potent inhibitors of the p53-MDM2 interaction by structure-based design starting from a conformational argument. Bioorg. Med. Chem. Lett. 2016, 26, 4837–4841. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhang, Z.; Hill, D.L.; Chen, X.; Wang, H.; Zhang, R. Genistein, a dietary isoflavone, down-regulates the MDM2 oncogene at both transcriptional and posttranslational levels. Cancer Res. 2005, 65, 8200–8208. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhang, Z.; Hill, D.L.; Wang, H.; Zhang, R. Curcumin, a dietary component, has anticancer, chemosensitization, and radiosensitization effects by down-regulating the MDM2 oncogene through the PI3K/mTOR/ETS2 pathway. Cancer Res. 2007, 67, 1988–1996. [Google Scholar] [CrossRef]
- Wang, W.; Rayburn, E.R.; Velu, S.E.; Nadkarni, D.H.; Murugesan, S.; Zhang, R. In vitro and in vivo anticancer activity of novel synthetic makaluvamine analogues. Clin. Cancer Res. 2009, 15, 3511–3518. [Google Scholar] [CrossRef]
- Wang, W.; Qin, J.J.; Voruganti, S.; Nijampatnam, B.; Velu, S.E.; Ruan, K.H.; Hu, M.; Zhou, J.; Zhang, R. Discovery and characterization of dual inhibitors of MDM2 and NFAT1 for Pancreatic cancer therapy. Cancer Res. 2018, 78, 5656–5667. [Google Scholar] [CrossRef]
- Wang, W.; Cheng, J.W.; Qin, J.J.; Hu, B.; Li, X.; Nijampatnam, B.; Velu, S.E.; Fan, J.; Yang, X.R.; Zhang, R. MDM2-NFAT1 dual inhibitor, MA242: Effective against hepatocellular carcinoma, independent of p53. Cancer Lett. 2019, 459, 156–167. [Google Scholar] [CrossRef]
- Wang, W.; Zhao, Y.; Rayburn, E.R.; Hill, D.L.; Wang, H.; Zhang, R. In vitro anti-cancer activity and structure-activity relationships of natural products isolated from fruits of Panax ginseng. Cancer Chemother. Pharmacol. 2007, 59, 589–601. [Google Scholar] [CrossRef]
- Nag, S.A.; Qin, J.J.; Wang, W.; Wang, M.H.; Wang, H.; Zhang, R. Ginsenosides as anticancer agents: In vitro and in vivo activities, structure-activity relationships, and molecular mechanisms of action. Front. Pharmacol. 2012, 3, 25. [Google Scholar] [CrossRef]
- Wang, W.; Qin, J.J.; Voruganti, S.; Wang, M.H.; Sharma, H.; Patil, S.; Zhou, J.; Wang, H.; Mukhopadhyay, D.; Buolamwini, J.K.; et al. Identification of a new class of MDM2 inhibitor that inhibits growth of orthotopic pancreatic tumors in mice. Gastroenterology 2014, 147, 893–902. [Google Scholar] [CrossRef]
- Wang, W.; Qin, J.J.; Voruganti, S.; Srivenugopal, K.S.; Nag, S.; Patil, S.; Sharma, H.; Wang, M.H.; Wang, H.; Buolamwini, J.K.; et al. The pyrido[b]indole MDM2 inhibitor SP-141 exerts potent therapeutic effects in breast cancer models. Nat. Commun. 2014, 5, 5086. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.J.; Wang, W.; Li, X.; Deokar, H.; Buolamwini, J.K.; Zhang, R. Inhibiting β-Catenin by β-Carboline-Type MDM2 Inhibitor for Pancreatic Cancer Therapy. Front. Pharmacol. 2018, 9, 5. [Google Scholar] [CrossRef]
- Qin, J.J.; Nag, S.; Wang, W.; Zhou, J.; Zhang, W.D.; Wang, H.; Zhang, R. NFAT as cancer target: Mission possible? Biochim. Biophys. Acta. 2014, 1846, 297–311. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Shaw, K.T.; Raghavan, A.; Aramburu, J.; Garcia-Cozar, F.; Perrino, B.A.; Hogan, P.G.; Rao, A. Interaction of calcineurin with a domain of the transcription factor NFAT1 that controls nuclear import. Proc. Natl. Acad. Sci. USA 1996, 93, 8907–8912. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Rodriguez, C.; Aramburu, J.; Rakeman, A.S.; Rao, A. NFAT5, a constitutively nuclear NFAT protein that does not cooperate with Fos and Jun. Proc. Natl. Acad. Sci. USA 1999, 96, 7214–7219. [Google Scholar] [CrossRef]
- Mancini, M.; Toker, A. NFAT proteins: Emerging roles in cancer progression. Nat. Rev. Cancer 2009, 9, 810–820. [Google Scholar] [CrossRef] [PubMed]
- Yoeli-Lerner, M.; Yiu, G.K.; Rabinovitz, I.; Erhardt, P.; Jauliac, S.; Toker, A. Akt blocks breast cancer cell motility and invasion through the transcription factor NFAT. Mol. Cell 2005, 20, 539–550. [Google Scholar] [CrossRef]
- Yoeli-Lerner, M.; Chin, Y.R.; Hansen, C.K.; Toker, A. Akt/protein kinase b and glycogen synthase kinase-3beta signaling pathway regulates cell migration through the NFAT1 transcription factor. Mol. Cancer Res. 2009, 7, 425–432. [Google Scholar] [CrossRef]
- Baumgart, S.; Glesel, E.; Singh, G.; Chen, N.M.; Reutlinger, K.; Zhang, J.; Billadeau, D.D.; Fernandez–Zapico, M.E.; Gress, T.M.; Singh, S.K.; et al. Restricted heterochromatin formation links NFATc2 repressor activity with growth promotion in pancreatic cancer. Gastroenterology 2012, 142, 388–398. [Google Scholar] [CrossRef]
- Marafioti, T.; Pozzobon, M.; Hansmann, M.L.; Ventura, R.; Pileri, S.A.; Roberton, H.; Gesk, S.; Gaulard, P.; Barth, T.F.; Du, M.Q.; et al. The NFATc1 transcription factor is widely expressed in white cells and translocates from the cytoplasm to the nucleus in a subset of human lymphomas. Br. J. Haematol. 2005, 128, 333–342. [Google Scholar] [CrossRef]
- Pham, L.V.; Tamayo, A.T.; Yoshimura, L.C.; Lin-Lee, Y.C.; Ford, R.J. Constitutive NFkappaB and NFAT activation in aggressive B-cell lymphomas synergistically activates the CD154 gene and maintains lymphoma cell survival. Blood 2005, 106, 3940–3947. [Google Scholar] [CrossRef]
- Szuhai, K.; Ijszenga, M.; de Jong, D.; Karseladze, A.; Tanke, H.J.; Hogendoorn, P.C. The NFATc2 gene is involved in a novel cloned translocation in a Ewing sarcoma variant that couples its function in immunology to oncology. Clin. Cancer Res. 2009, 15, 2259–2268. [Google Scholar] [CrossRef] [PubMed]
- Arbajian, E.; Magnusson, L.; Brosjo, O.; Wejde, J.; Folpe, A.L.; Nord, K.H.; Mertens, F. A benign vascular tumor with a new fusion gene: EWSR1-NFATC1 in hemangioma of the bone. Am. J. Surg. Pathol. 2013, 37, 613–616. [Google Scholar] [CrossRef] [PubMed]
- Sankar, S.; Lessnick, S.L. Promiscuous partnerships in Ewing’s sarcoma. Cancer Genet. 2011, 204, 351–365. [Google Scholar] [CrossRef] [PubMed]
- Buchholz, M.; Schatz, A.; Wagner, M.; Michl, P.; Linhart, T.; Adler, G.; Gress, T.M.; Ellenrieder, V. Overexpression of c-myc in pancreatic cancer caused by ectopic activation of NFATc1 and the Ca2+/calcineurin signaling pathway. EMBO J. 2006, 25, 3714–3724. [Google Scholar] [CrossRef]
- Singh, G.; Singh, S.K.; König, A.; Reutlinger, K.; Nye, M.D.; Adhikary, T.; Eilers, M.; Gress, T.M.; Fernandez-Zapico, M.E.; Ellenrieder, V. Sequential activation of NFAT and c-Myc transcription factors mediates the TGF-beta switch from a suppressor to a promoter of cancer cell proliferation. J. Biol. Chem. 2010, 285, 27241–27250. [Google Scholar] [CrossRef]
- Horsley, V.; Aliprantis, A.O.; Polak, L.; Glimcher, L.H.; Fuchs, E. NFATc1 balances quiescence and proliferation of skin stem cells. Cell 2008, 132, 299–310. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial–mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- Yiu, G.K.; Kaunisto, A.; Chin, Y.R.; Toker, A. NFAT promotes carcinoma invasive migration through glypican-6. Biochem. J. 2011, 440, 157–166. [Google Scholar] [CrossRef]
- Yiu, G.K.; Toker, A. NFAT induces breast cancer cell invasion by promoting the induction of cyclooxygenase-2. J. Biol. Chem. 2006, 281, 12210–12217. [Google Scholar] [CrossRef]
- Chen, M.; O’Connor, K.L. Integrin alpha6beta4 promotes expression of autotaxin/ENPP2 autocrine motility factor in breast carcinoma cells. Oncogene 2005, 24, 5125–5130. [Google Scholar] [CrossRef] [PubMed]
- Jauliac, S.; Lopez-Rodriguez, C.; Shaw, L.M.; Brown, L.F.; Rao, A.; Toker, A. The role of NFAT transcription factors in integrin-mediated carcinoma invasion. Nat. Cell Biol. 2002, 4, 540–544. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Jana, S.; Biswas, S.; Mandal, P.K.; Bhattacharyya, A. Cooperative involvement of NFAT and SnoN mediates transforming growth factor-beta (TGF-beta) induced EMT in metastatic breast cancer (MDA-MB 231) cells. Clin. Exp. Metastasis 2013, 30, 1019–1031. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.Y.; Zong, C.S.; Xia, W.; Wei, Y.; Ali-Seyed, M.; Li, Z.; Broglio, K.; Berry, D.A.; Hung, M.C. MDM2 promotes cell motility and invasiveness by regulating E-cadherin degradation. Mol. Cell Biol. 2006, 26, 7269–7282. [Google Scholar] [CrossRef]
- Baggott, R.R.; Alfranca, A.; Lopez-Maderuelo, D.; Mohamed, T.M.; Escolano, A.; Oller, J.; Ornes, B.C.; Kurusamy, S.; Rowther, F.B.; Brown, J.E.; et al. Plasma membrane calcium ATPase isoform 4 inhibits vascular endothelial growth factor-mediated angiogenesis through interaction with calcineurin. Arterioscler Thromb Vasc Biol. 2014, 34, 2310–2320. [Google Scholar] [CrossRef]
- Medyouf, H.; Alcalde, H.; Berthier, C.; Guillemin, M.C.; dos Santos, N.R.; Janin, A.; Decaudin, D.; de Thé, H.; Ghysdael, J. Targeting calcineurin activation as a therapeutic strategy for T-cell acute lymphoblastic leukemia. Nat. Med. 2007, 13, 736–741. [Google Scholar] [CrossRef]
- Lee, C.R.; Chun, J.N.; Kim, S.Y.; Park, S.; Kim, S.H.; Park, E.J.; Kim, I.S.; Cho, N.H.; Kim, I.G.; So, I.; et al. Cyclosporin A suppresses prostate cancer cell growth through CaMKKβ/AMPK-mediated inhibition of mTORC1 signaling. Biochem. Pharmacol. 2012, 84, 425–431. [Google Scholar] [CrossRef]
- Garrido, W.; Munoz, M.; San Martin, R.; Quezada, C. FK506 confers chemosensitivity to anticancer drugs in glioblastoma multiforme cells by decreasing the expression of the multiple resistance-associated protein-1. Biochem. Biophys. Res. Commun. 2011, 411, 62–68. [Google Scholar] [CrossRef]
- Romano, S.; Di Pace, A.; Sorrentino, A.; Bisogni, R.; Sivero, L.; Romano, M.F. FK506 binding proteins as targets in anticancer therapy. Anticancer Agents Med. Chem. 2010, 10, 651–656. [Google Scholar] [CrossRef]
- Liu, J.; Farmer, J.D., Jr.; Lane, W.S.; Friedman, J.; Weissman, I.; Schreiber, S.L. Calcineurin is a common target of cyclophilin–cyclosporin A and FKBP–FK506 complexes. Cell 1991, 66, 807–815. [Google Scholar] [CrossRef]
- Siamakpour-Reihani, S.; Caster, J.; Bandhu Nepal, D.; Courtwright, A.; Hilliard, E.; Usary, J.; Ketelsen, D.; Darr, D.; Shen, X.J.; Patterson, C.; et al. The role of calcineurin/NFAT in SFRP2 induced angiogenesis-a rationale for breast cancer treatment with the calcineurin inhibitor tacrolimus. PLoS ONE 2011, 6, e20412. [Google Scholar] [CrossRef] [PubMed]
- Courtwright, A.; Siamakpour-Reihani, S.; Arbiser, J.L.; Banet, N.; Hilliard, E.; Fried, L.; Livasy, C.; Ketelsen, D.; Nepal, D.B.; Perou, C.M.; et al. Secreted frizzle-related protein 2 stimulates angiogenesis via a calcineurin/NFAT signaling pathway. Cancer Res. 2009, 69, 4621–4628. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Wang, Q.; Yang, S.; Chen, C.; Li, X.; Liu, J.; Zou, Z.; Cai, D. Quercetin inhibits angiogenesis by targeting calcineurin in the xenograft model of human breast cancer. Eur. J. Pharmacol. 2016, 781, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Le Roy, C.; Deglesne, P.A.; Chevallier, N.; Beitar, T.; Eclache, V.; Quettier, M.; Boubaya, M.; Letestu, R.; Lévy, V.; Ajchenbaum-Cymbalista, F.; et al. The degree of BCR and NFAT activation predicts clinical outcomes in chronic lymphocytic leukemia. Blood 2012, 120, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; van Berkel, T.J.; Biessen, E.A. Therapeutic potential of VIVIT, a selective peptide inhibitor of nuclear factor of activated T cells, in cardiovascular disorders. Cardiovasc. Drug Rev. 2007, 25, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Hogan, P.G. Calcium-NFAT transcriptional signalling in T cell activation and T cell exhaustion. Cell Calcium. 2017, 63, 66–69. [Google Scholar] [CrossRef]
- Muller, M.R.; Rao, A. NFAT, immunity and cancer: A transcription factor comes of age. Nat. Rev. Immunol. 2010, 10, 645–656. [Google Scholar] [CrossRef]
- Tie, X.; Han, S.; Meng, L.; Wang, Y.; Wu, A. NFAT1 is highly expressed in, and regulates the invasion of, glioblastoma multiforme cells. PLoS ONE 2013, 8, e66008. [Google Scholar] [CrossRef]
- Shou, J.; Jing, J.; Xie, J.; You, L.; Jing, Z.; Yao, J.; Han, W.; Pan, H. Nuclear factor of activated T cells in cancer development and treatment. Cancer Lett. 2015, 361, 174–184. [Google Scholar] [CrossRef]
- Oikawa, T.; Nakamura, A.; Onishi, N.; Yamada, T.; Matsuo, K.; Saya, H. Acquired expression of NFATc1 downregulates E-cadherin and promotes cancer cell invasion. Cancer Res. 2013, 73, 5100–5109. [Google Scholar] [CrossRef]
- Fougere, M.; Gaudineau, B.; Barbier, J.; Guaddachi, F.; Feugeas, J.P.; Auboeuf, D.; Jauliac, S. NFAT3 transcription factor inhibits breast cancer cell motility by targeting the Lipocalin 2 gene. Oncogene 2010, 29, 2292–2301. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.J.; Wang, W.; Voruganti, S.; Wang, H.; Zhang, W.D.; Zhang, R. Identification of a new class of natural product MDM2 inhibitor: In vitro and in vivo anti-breast cancer activities and target validation. Oncotarget 2015, 6, 2623–2640. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.J.; Wang, W.; Voruganti, S.; Wang, H.; Zhang, W.D.; Zhang, R. Inhibiting NFAT1 for breast cancer therapy: New insights into the mechanism of action of MDM2 inhibitor JapA. Oncotarget 2015, 6, 33106–33119. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.J.; Wang, W.; Sarkar, S.; Voruganti, S.; Agarwal, R.; Zhang, R.; Inulanolide, A. as a new dual inhibitor of NFAT1-MDM2 pathway for breast cancer therapy. Oncotarget 2016, 7, 32566–32578. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.J.; Sarkar, S.; Voruganti, S.; Agarwal, R.; Wang, W.; Zhang, R. Identification of linear-iifolianoid A as a novel dual NFAT1 and MDM2 inhibitor for human cancer therapy. J. Biomed. Res. 2016, 30, 322–333. [Google Scholar] [PubMed]
- Baksh, S.; Widlund, H.R.; Frazer-Abel, A.A.; Du, J.; Fosmire, S.; Fisher, D.E.; DeCaprio, J.A.; Modiano, J.F.; Burakoff, S.J. NFATc2-mediated repression of cyclin-dependent kinase 4 expression. Mol. Cell 2002, 10, 1071–1081. [Google Scholar] [CrossRef]
- Hodge, M.R.; Ranger, A.M.; Charles de la Brousse, F.; Hoey, T.; Grusby, M.J.; Glimcher, L.H. Hyperproliferation and dysregulation of IL-4 expression in NF-ATp-deficient mice. Immunity 1996, 4, 397–405. [Google Scholar] [CrossRef]
- Carvalho, L.D.; Teixeira, L.K.; Carrossini, N.; Caldeira, A.T.; Ansel, K.M.; Rao, A.; Viola, J.P. The NFAT1 transcription factor is a repressor of cyclin A2 gene expression. Cell Cycle 2007, 6, 1789–1795. [Google Scholar] [CrossRef]
- Chebel, A.; Rouault, J.P.; Urbanowicz, I.; Baseggio, L.; Chien, W.W.; Salles, G.; Ffrench, M. Transcriptional activation of hTERT, the human telomerase reverse transcriptase, by nuclear factor of activated T cells. J. Biol. Chem. 2009, 284, 35725–35734. [Google Scholar] [CrossRef]
- Otto, T.; Sicinski, P. Cell cycle proteins as promising targets in cancer therapy. Nat. Rev. Cancer 2017, 17, 93–115. [Google Scholar] [CrossRef]
- Harris, A.L. Development of cancer metabolism as a therapeutic target: New pathways, patient studies, stratification and combination therapy. Br. J. Cancer 2020, 122, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Zaal, E.A.; Berkers, C.R. The Influence of Metabolism on Drug Response in Cancer. Front. Oncol. 2018, 8, 500. [Google Scholar] [CrossRef] [PubMed]
- Simabuco, F.M.; Morale, M.G.; Pavan, I.C.B.; Morelli, A.P.; Silva, F.R.; Tamura, R.E. p53 and metabolism: From mechanism to therapeutics. Oncotarget 2018, 9, 23780–23823. [Google Scholar] [CrossRef] [PubMed]
- Riscal, R.; Le Cam, L.; Linares, L.K. Chromatin-bound MDM2, a new player in metabolism. Mol. Cell Oncol. 2016, 3, e1210560. [Google Scholar] [CrossRef]
- Riscal, R.; Schrepfer, E.; Arena, G.; Cissé, M.Y.; Bellvert, F.; Heuillet, M.; Rambow, F.; Bonneil, E.; Sabourdy, F.; Vincent, C.; et al. Chromatin-Bound MDM2 Regulates Serine Metabolism and Redox Homeostasis Independently of p53. Mol. Cell 2016, 62, 890–902. [Google Scholar] [CrossRef]
- Arena, G.; Cissé, M.Y.; Pyrdziak, S.; Chatre, L.; Riscal, R.; Fuentes, M.; Arnold, J.J.; Kastner, M.; Gayte, L.; Bertrand-Gaday, C.; et al. Mitochondrial MDM2 regulates respiratory complex I activity independently of p53. Mol. Cell 2018, 69, 594–609. [Google Scholar] [CrossRef]
- Maguire, M.; Nield, P.C.; Devling, T.; Jenkins, R.E.; Park, B.K.; Polański, R.; Vlatković, N.; Boyd, M.T. MDM2 regulates dihydrofolate reductase activity through monoubiquitination. Cancer Res. 2008, 68, 3232–3242. [Google Scholar] [CrossRef]
- Shi, L.; Tu, B.P. Acetyl-CoA and the regulation of metabolism: Mechanisms and consequences. Curr. Opin. Cell Biol. 2015, 33, 125–131. [Google Scholar] [CrossRef]
- Lee, J.V.; Berry, C.T.; Kim, K.; Sen, P.; Kim, T.; Carrer, A.; Trefely, S.; Zhao, S.; Fernandez, S.; Barney, L.E.; et al. Acetyl-CoA promotes glioblastoma cell adhesion and migration through Ca2+-NFAT signaling. Genes Dev. 2018, 32, 497–511. [Google Scholar] [CrossRef]
- Liu, P.; Wang, Y.; Li, X. Targeting the untargetable KRAS in cancer therapy. Acta Pharm. Sin. B 2019, 9, 871–879. [Google Scholar] [CrossRef]
- Waters, A.M.; Der, C.J. KRAS: The critical driver and therapeutic target for pancreatic cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a031435. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.; Menon, D.; Bernfeld, E.; Mroz, V.; Kalan, S.; Loayza, D.; Foster, D.A. Aspartate Rescues S-phase Arrest Caused by Suppression of Glutamine Utilization in KRas-driven Cancer Cells. J. Biol. Chem. 2016, 291, 9322–9329. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Saqcena, M.; Foster, D.A. Synthetic lethality in KRas-driven cancer cells created by glutamine deprivation. Oncoscience 2015, 2, 807–808. [Google Scholar]
- Tisato, V.; Voltan, R.; Gonelli, A.; Secchiero, P.; Zauli, G. MDM2/X inhibitors under clinical evaluation: Perspectives for the management of hematological malignancies and pediatric cancer. J. Hematol. Oncol. 2017, 10, 133. [Google Scholar] [CrossRef] [PubMed]
- Canon, J.; Osgood, T.; Olson, S.H.; Saiki, A.Y.; Robertson, R.; Yu, D.; Eksterowicz, J.; Ye, Q.; Jin, L.; Chen, A.; et al. The MDM2 inhibitor AMG 232 demonstrates robust antitumor efficacy and potentiates the activity of p53-inducing cytotoxic agents. Mol. Cancer Ther. 2015, 14, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Hata, A.N.; Rowley, S.; Archibald, H.L.; Gomez-Caraballo, M.; Siddiqui, F.M.; Ji, F.; Jung, J.; Light, M.; Lee, J.S.; Debussche, L.; et al. Synergistic activity and heterogeneous acquired resistance of combined MDM2 and MEK inhibition in KRAS mutant cancers. Oncogene 2017, 36, 6581–6591. [Google Scholar] [CrossRef] [PubMed]
- Conradt, L.; Henrich, A.; Wirth, M.; Reichert, M.; Lesina, M.; Algül, H.; Schmid, R.M.; Krämer, O.H.; Saur, D.; Schneider, G. Mdm2 inhibitors synergize with topoisomerase II inhibitors to induce p53-independent pancreatic cancer cell death. Int. J. Cancer 2013, 132, 2248–2257. [Google Scholar] [CrossRef]
- Baumgart, S.; Chen, N.M.; Siveke, J.T.; König, A.; Zhang, J.S.; Singh, S.K.; Wolf, E.; Bartkuhn, M.; Esposito, I.; Heßmann, E.; et al. Inflammation-induced NFATc1-STAT3 transcription complex promotes pancreatic cancer initiation by KrasG12D. Cancer Discov. 2014, 4, 688–701. [Google Scholar] [CrossRef]
- Ramsay, R.R.; Popovic-Nikolic, M.R.; Nikolic, K.; Uliassi, E.; Bolognesi, M.L. A perspective on multi-target drug discovery and design for complex diseases. Clin. Transl. Med. 2018, 7, 3. [Google Scholar] [CrossRef]


{kind=link}
| Compound | Structure | In Vitro and In Vivo Activities |
|---|---|---|
| Japonicone A (JapA) [82,83] |  | Cell growth inhibition Decreased cell proliferation Inhibition of colony formation G2/M phase arrest Increased apoptosis Inhibition of tumor growth and lung metastasis in breast cancer xenograft models |
| Inulanolide A (InuA) [84] |  | Cell growth inhibition Decreased cell proliferation Inhibition of colony formation G2/M phase arrest Increased apoptosis Inhibition of cell migration and invasion Inhibition of tumor growth and lung metastasis in breast cancer orthotopic models |
| Lineariifolianoid A (LinA) [85] |  | Cell growth inhibition Decreased cell proliferation Inhibition of colony formation G2/M phase arrest Increased apoptosis Inhibition of cell migration and invasion |
| MA242 [36,37] |  | Cell growth inhibition Decreased cell proliferation Inhibition of colony formation G2/M phase arrest Increased apoptosis Inhibition of cell migration and invasion Inhibition of pancreatic tumor growth and metastasis |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, W.; Zafar, A.; Rajaei, M.; Zhang, R. Two Birds with One Stone: NFAT1-MDM2 Dual Inhibitors for Cancer Therapy. Cells 2020, 9, 1176. https://doi.org/10.3390/cells9051176
Wang W, Zafar A, Rajaei M, Zhang R. Two Birds with One Stone: NFAT1-MDM2 Dual Inhibitors for Cancer Therapy. Cells. 2020; 9(5):1176. https://doi.org/10.3390/cells9051176
Chicago/Turabian StyleWang, Wei, Atif Zafar, Mehrdad Rajaei, and Ruiwen Zhang. 2020. "Two Birds with One Stone: NFAT1-MDM2 Dual Inhibitors for Cancer Therapy" Cells 9, no. 5: 1176. https://doi.org/10.3390/cells9051176
APA StyleWang, W., Zafar, A., Rajaei, M., & Zhang, R. (2020). Two Birds with One Stone: NFAT1-MDM2 Dual Inhibitors for Cancer Therapy. Cells, 9(5), 1176. https://doi.org/10.3390/cells9051176