Creatine Kinase and Progression Rate in Amyotrophic Lateral Sclerosis

 , , , , ,
, , , , ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Standard Protocol Approvals, Registrations, and Patient Consents
2.3. Aims of the Study
2.4. Statistical Analysis
3. Results
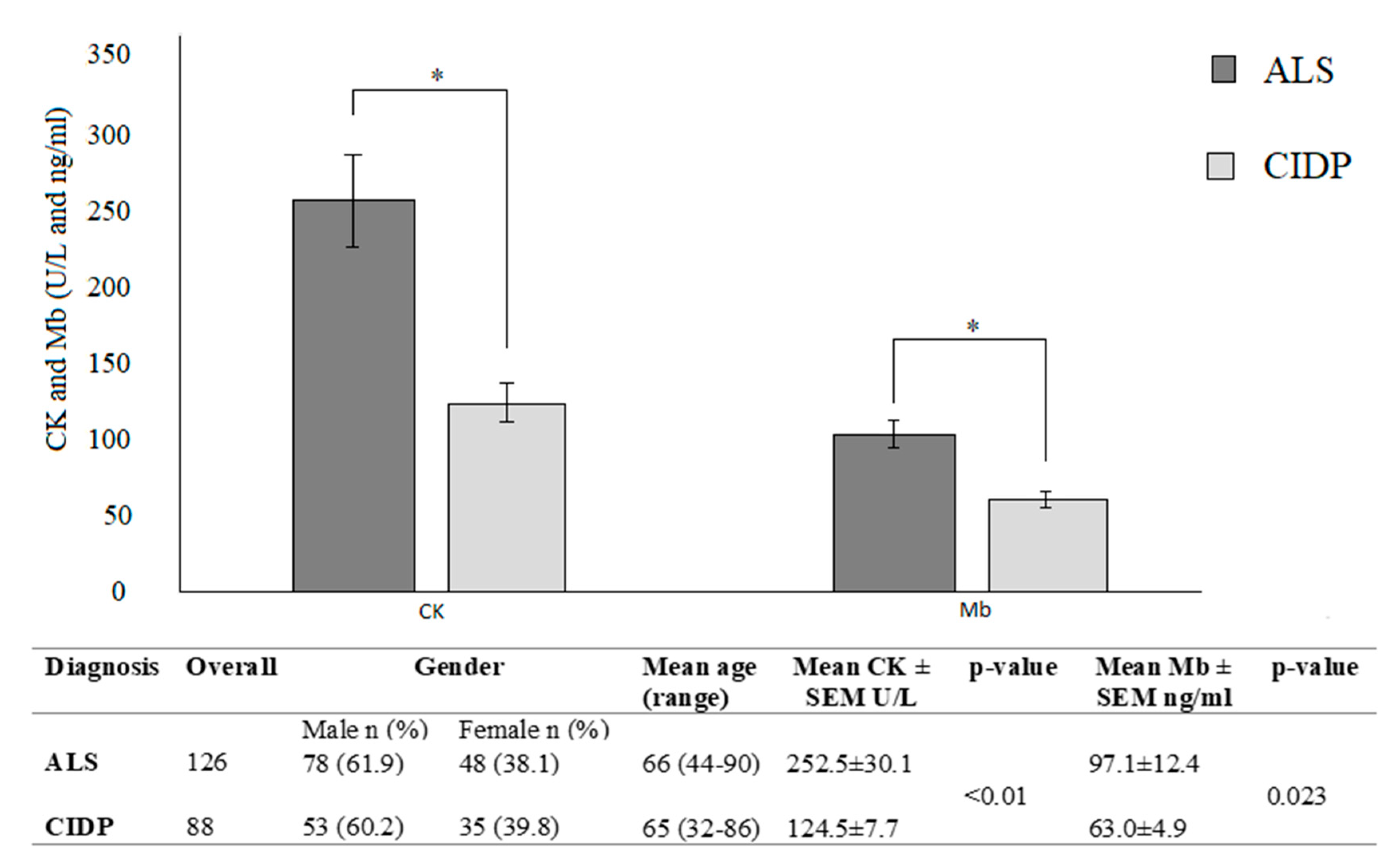
3.1. Baseline CK and Mb in ALS Patients
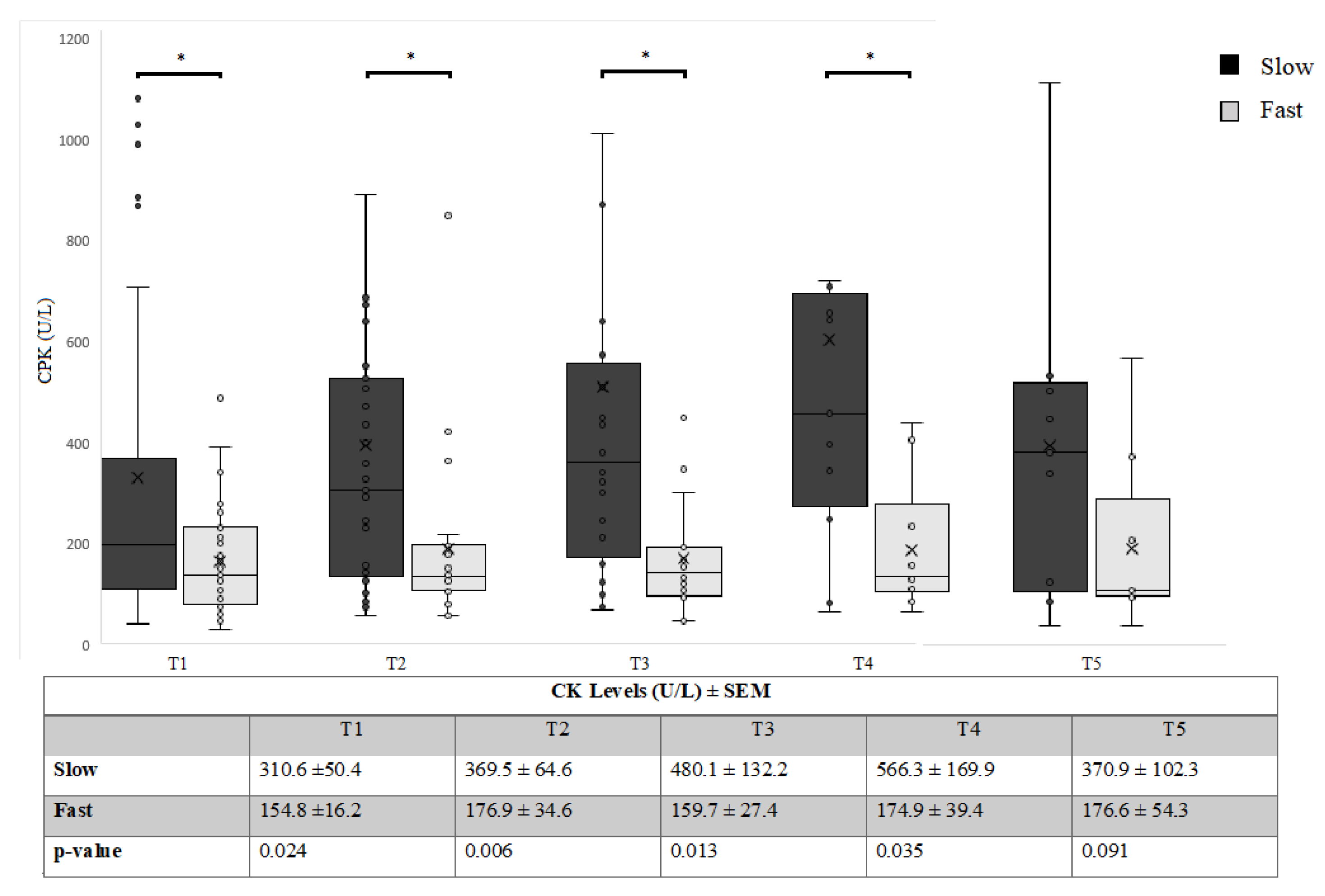
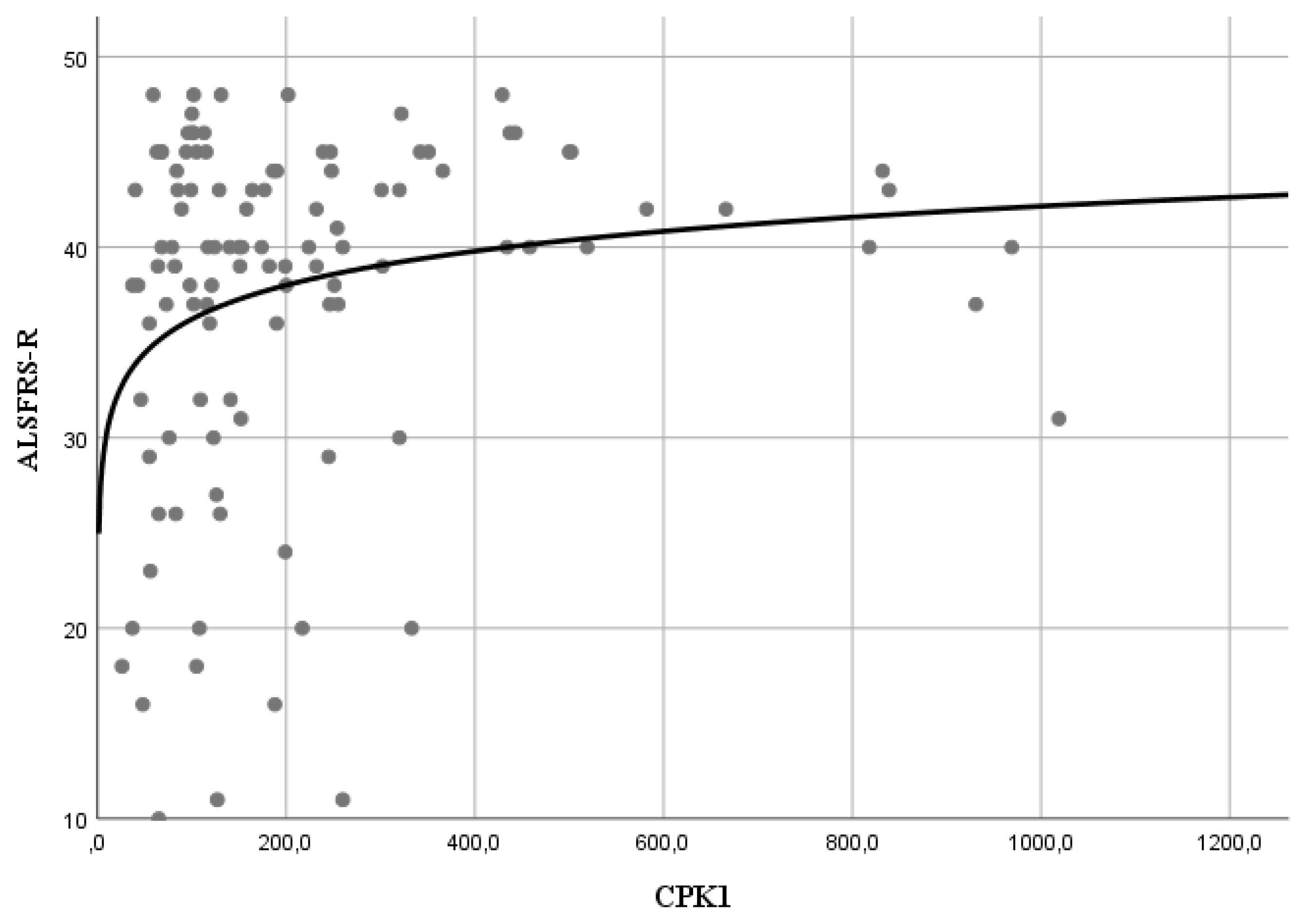
3.2. Relationship between CK and FAST/SLOW Progression over Time
3.3. CK and Mb in ALS and CIDP
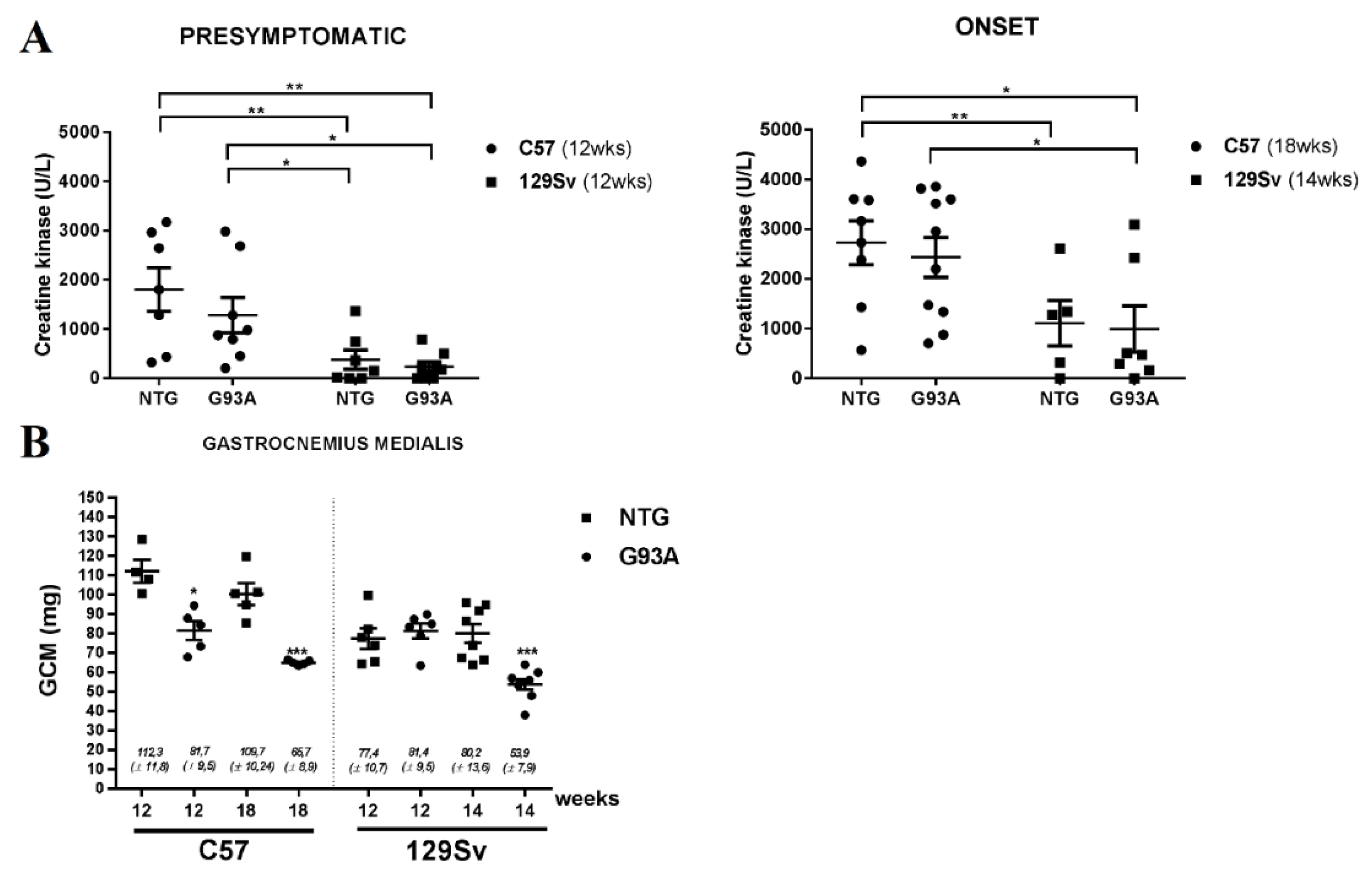
3.4. CK in Fast and Slow-Progressive ALS Mouse Models
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Brooks, B.R.; Miller, R.G.; Swash, M.; Munsat, T.L. El Escorial revisited: Revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Mot. Neuron Disord. 2000, 1, 293–299. [Google Scholar] [CrossRef]
- Traynor, B.J.; Codd, M.B.; Corr, B.; Forde, C.; Frost, E.; Hardiman, O.M. Clinical features of amyotrophic lateral sclerosis according to the El Escorial and Airlie House diagnostic criteria: A population-based study. Arch. Neurol. 2000, 57, 1171–1176. [Google Scholar] [CrossRef] [PubMed]
- Berardelli, A.; Inghilleri, M.; Formisano, R.; Accornero, N.; Manfredi, M. Stimulation of motor tracts in motor neuron disease. J. Neurol. Neurosurg. Psychiatry 1987, 50, 732–737. [Google Scholar] [CrossRef] [PubMed]
- Libonati, L.; Barone, T.F.; Ceccanti, M.; Cambieri, C.; Tartaglia, G.; Onesti, E.; Petrucci, A.; Frasca, V.; Inghilleri, M. Heteronymous H reflex in temporal muscle as sign of hyperexcitability in ALS patients. Clin. Neurophysiol. 2019, 130, 1455–1459. [Google Scholar] [CrossRef]
- Ruoppolo, G.; Schettino, I.; Frasca, V.; Giacomelli, E.; Prosperini, L.; Cambieri, C.; Roma, R.; Greco, A.; Mancini, P.; De Vincentiis, M.; et al. Dysphagia in amyotrophic lateral sclerosis: Prevalence and clinical findings. Acta Neurol. Scand. 2013, 128, 397–401. [Google Scholar] [CrossRef]
- Truini, A.; Biasiotta, A.; Onesti, E.; Di Stefano, G.; Ceccanti, M.; La Cesa, S.; Pepe, A.; Giordano, C.; Cruccu, G.; Inghilleri, M. Small-fibre neuropathy related to bulbar and spinal-onset in patients with ALS. J. Neurol. 2015, 262, 1014–1018. [Google Scholar] [CrossRef]
- Ceccanti, M.; Onesti, E.; Rubino, A.; Cambieri, C.; Tartaglia, G.; Miscioscia, A.; Frasca, V.; Inghilleri, M. Modulation of human corticospinal excitability by paired associative stimulation in patients with amyotrophic lateral sclerosis and effects of Riluzole. Brain Stimul. 2018, 11, 775–781. [Google Scholar] [CrossRef]
- Qiu, T.; Zhang, Y.; Tang, X.; Liu, X.; Wang, Y.; Zhou, C.; Luo, C.; Zhang, J. Precentral degeneration and cerebellar compensation in amyotrophic lateral sclerosis: A multimodal MRI analysis. Hum. Brain Mapp. 2019, 40, 3464–3474. [Google Scholar] [CrossRef]
- Turner, M.R.; Benatar, M. Ensuring continued progress in biomarkers for amyotrophic lateral sclerosis. Muscle Nerve 2015, 51, 14–18. [Google Scholar] [CrossRef]
- Said, G.; Krarup, C. Chronic inflammatory demyelinative polyneuropathy. Handb. Clin. Neurol. 2013, 115, 403–413. [Google Scholar] [CrossRef]
- Lima, A.F.; Evangelista, T.; de Carvalho, M. Increased creatine kinase and spontaneous activity on electromyography, in amyotrophic lateral sclerosis. Electromyogr. Clin. Neurophysiol. 2003, 43, 189–192. [Google Scholar]
- Panitch, H.S.; Franklin, G.M. Elevation of serum creatine phosphokinase in amyotrophic lateral sclerosis. Neurology 1972, 22, 964–966. [Google Scholar] [CrossRef]
- Amrit, A.N.; Anderson, M.S. Serum creatine phosphokinase in amyotrophic lateral sclerosis. Correlation with sex, duration, and skeletal muscle biopsy. Neurology 1974, 24, 834–837. [Google Scholar] [CrossRef]
- Edmonds, P.J.; Ziegler, D.K. Diagnostic value of serum creatine phosphokinase in motor neuron disease. South. Med. J. 1975, 68, 1388–1390. [Google Scholar] [CrossRef]
- Harrington, T.M.; Cohen, M.D.; Bartleson, J.D.; Ginsburg, W.W. Elevation of creatine kinase in amyotrophic lateral sclerosis. Potential confusion with polymyositis. Arthritis Rheum. 1983, 26, 201–205. [Google Scholar] [CrossRef]
- Sinaki, M.; Mulder, D.W. Amyotrophic lateral sclerosis: Relationship between serum creatine kinase level and patient survival. Arch. Phys. Med. Rehabil. 1986, 67, 169–171. [Google Scholar] [CrossRef]
- Felice, K.J.; North, W.A. Creatine kinase values in amyotrophic lateral sclerosis. J. Neurol. Sci. 1998, 160, S30–S32. [Google Scholar] [CrossRef]
- Süssmuth, S.D.; Tumani, H.; Ecker, D.; Ludolph, A.C. Amyotrophic lateral sclerosis: Disease stage related changes of tau protein and S100 beta in cerebrospinal fluid and creatine kinase in serum. Neurosci. Lett. 2003, 353, 57–60. [Google Scholar] [CrossRef]
- Chahin, N.; Sorenson, E.J. Serum creatine kinase levels in spinobulbar muscular atrophy and amyotrophic lateral sclerosis. Muscle Nerve 2009, 40, 126–129. [Google Scholar] [CrossRef]
- Gibson, S.B.; Kasarskis, E.J.; Hu, N.; Pulst, S.-M.; Mendiondo, M.S.; Matthews, D.E.; Mitsumoto, H.; Tandan, R.; Simmons, Z.; Kryscio, R.J.; et al. Relationship of creatine kinase to body composition, disease state, and longevity in ALS. Amyotroph. Lateral Scler. Front. Degener. 2015, 16, 473–477. [Google Scholar] [CrossRef]
- Rafiq, M.K.; Lee, E.; Bradburn, M.; McDermott, C.J.; Shaw, P.J. Creatine kinase enzyme level correlates positively with serum creatinine and lean body mass, and is a prognostic factor for survival in amyotrophic lateral sclerosis. Eur. J. Neurol. 2016, 23, 1071–1078. [Google Scholar] [CrossRef]
- Tai, H.; Cui, L.; Guan, Y.; Liu, M.; Li, X.; Shen, D.; Li, D.; Cui, B.; Fang, J.; Ding, Q.; et al. Correlation of Creatine Kinase Levels with Clinical Features and Survival in Amyotrophic Lateral Sclerosis. Front. Neurol. 2017, 8, 322. [Google Scholar] [CrossRef]
- Tai, H.; Cui, L.; Liu, M.; Guan, Y.; Li, X.; Shen, D.; Zhang, K.; Liu, S.; Wu, S.; Ding, Q.; et al. Creatine kinase level and its relationship with quantitative electromyographic characteristics in amyotrophic lateral sclerosis. Clin. Neurophysiol. 2018, 129, 926–930. [Google Scholar] [CrossRef]
- Wallimann, T.; Wyss, M.; Brdiczka, D.; Nicolay, K.; Eppenberger, H.M. Intracellular compartmentation, structure and function of creatine kinase isoenzymes in tissues with high and fluctuating energy demands: The “phosphocreatine circuit” for cellular energy homeostasis. Biochem. J. 1992, 281, 21–40. [Google Scholar] [CrossRef]
- Moghadam-Kia, S.; Oddis, C.V.; Aggarwal, R. Approach to asymptomatic creatine kinase elevation. Clevel. Clin. J. Med. 2016, 83, 37–42. [Google Scholar] [CrossRef]
- Wittenberg, J.B.; Wittenberg, B.A. Myoglobin function reassessed. J. Exp. Biol. 2003, 206, 2011–2020. [Google Scholar] [CrossRef]
- Ordway, G.A.; Garry, D.J. Myoglobin: An essential hemoprotein in striated muscle. J. Exp. Biol. 2004, 207, 3441–3446. [Google Scholar] [CrossRef]
- Kagawa, N.; Hizawa, K. Immunohistochemical study of myoglobin in neuromuscular diseases. Muscle Nerve 1985, 8, 9–16. [Google Scholar] [CrossRef]
- Liu, Y. Radioimmunoassay and immunohistochemical study of myoglobin in neuromuscular diseases. Zhonghua Yi Xue Za Zhi 1992, 72, 144–146. [Google Scholar]
- Khan, F.Y. Rhabdomyolysis: A review of the literature. Neth. J. Med. 2009, 67, 272–283. [Google Scholar]
- Marino, M.; Papa, S.; Crippa, V.; Nardo, G.; Peviani, M.; Cheroni, C.; Trolese, M.C.; Lauranzano, E.; Bonetto, V.; Poletti, A.; et al. Differences in protein quality control correlate with phenotype variability in 2 mouse models of familial amyotrophic lateral sclerosis. Neurobiol. Aging 2015, 36, 492–504. [Google Scholar] [CrossRef] [PubMed]
- Nardo, G.; Trolese, M.C.; Tortarolo, M.; Vallarola, A.; Freschi, M.; Pasetto, L.; Bonetto, V.; Bendotti, C. New Insights on the Mechanisms of Disease Course Variability in ALS from Mutant SOD1 Mouse Models. Brain Pathol. 2016, 26, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Nardo, G.; Trolese, M.C.; de Vito, G.; Cecchi, R.; Riva, N.; Dina, G.; Heath, P.R.; Quattrini, A.; Shaw, P.J.; Piazza, V.; et al. Immune response in peripheral axons delays disease progression in SOD1G93A mice. J. Neuroinflamm. 2016, 13, 261. [Google Scholar] [CrossRef]
- Joint Task Force of the EFNS and the PNS European Federation of Neurological Societies/Peripheral Nerve Society Guideline on management of paraproteinemic demyelinating neuropathies. Report of a Joint Task Force of the European Federation of Neurological Societies and the Peripheral Nerve Society—First revision. J. Peripher. Nerv. Syst. 2010, 15, 185–195. [Google Scholar] [CrossRef]
- Joyce, N.C.; Oskarsson, B.; Jin, L.-W. Muscle biopsy evaluation in neuromuscular disorders. Phys. Med. Rehabil. Clin. N. Am. 2012, 23, 609–631. [Google Scholar] [CrossRef]
- Al-Sarraj, S.; King, A.; Cleveland, M.; Pradat, P.-F.; Corse, A.; Rothstein, J.D.; Leigh, P.N.; Abila, B.; Bates, S.; Wurthner, J.; et al. Mitochondrial abnormalities and low grade inflammation are present in the skeletal muscle of a minority of patients with amyotrophic lateral sclerosis; an observational myopathology study. Acta Neuropathol. Commun. 2014, 2, 165. [Google Scholar] [CrossRef]
- Mazzaccara, C.; Labruna, G.; Cito, G.; Scarfò, M.; De Felice, M.; Pastore, L.; Sacchetti, L. Age-Related Reference Intervals of the Main Biochemical and Hematological Parameters in C57BL/6J, 129SV/EV and C3H/HeJ Mouse Strains. PLoS ONE 2008, 3, e3772. [Google Scholar] [CrossRef]
- Nardo, G.; Iennaco, R.; Fusi, N.; Heath, P.R.; Marino, M.; Trolese, M.C.; Ferraiuolo, L.; Lawrence, N.; Shaw, P.J.; Bendotti, C. Transcriptomic indices of fast and slow disease progression in two mouse models of amyotrophic lateral sclerosis. Brain 2013, 136, 3305–3332. [Google Scholar] [CrossRef]
- George, M.D.; McGill, N.-K.; Baker, J.F. Creatine kinase in the U.S. population: Impact of demographics, comorbidities, and body composition on the normal range. Medicine (Baltimore) 2016, 95, e4344. [Google Scholar] [CrossRef]
- Wong, E.T.; Cobb, C.; Umehara, M.K.; Wolff, G.A.; Haywood, L.J.; Greenberg, T.; Shaw, S.T. Heterogeneity of serum creatine kinase activity among racial and gender groups of the population. Am. J. Clin. Pathol. 1983, 79, 582–586. [Google Scholar] [CrossRef]
- Cronin, S.; Hardiman, O.; Traynor, B.J. Ethnic variation in the incidence of ALS: A systematic review. Neurology 2007, 68, 1002–1007. [Google Scholar] [CrossRef] [PubMed]
- Qadri, S.; Langefeld, C.D.; Milligan, C.; Caress, J.B.; Cartwright, M.S. Racial differences in intervention rates in individuals with ALS. Neurology 2019, 92, e1969–e1974. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.L.; Johnson, N.J.; Chen, J.T.; Cudkowicz, M.E.; Weisskopf, M.G. Race/ethnicity, socioeconomic status, and ALS mortality in the United States. Neurology 2016, 87, 2300–2308. [Google Scholar] [CrossRef] [PubMed]
- Csillik, B.; Nemcsók, J.; Chase, B.; Csillik, A.E.; Knyihár-Csillik, E. Infraterminal spreading and extrajunctional expression of nicotinic acetylcholine receptors in denervated rat skeletal muscle. Exp. Brain Res. 1999, 125, 426–434. [Google Scholar] [CrossRef] [PubMed]
- Ashley, Z.; Sutherland, H.; Lanmüller, H.; Russold, M.F.; Unger, E.; Bijak, M.; Mayr, W.; Boncompagni, S.; Protasi, F.; Salmons, S.; et al. Atrophy, but not necrosis, in rabbit skeletal muscle denervated for periods up to one year. Am. J. Physiol. Cell Physiol. 2007, 292, C440–C451. [Google Scholar] [CrossRef]
- Kiessling, W.R.; Ricker, K.; Pflughaupt, K.W.; Mertens, H.G.; Haubitz, I. Serum myoglobin in primary and secondary skeletal muscle disorders. J. Neurol. 1981, 224, 229–233. [Google Scholar] [CrossRef]
- Palma, E.; Inghilleri, M.; Conti, L.; Deflorio, C.; Frasca, V.; Manteca, A.; Pichiorri, F.; Roseti, C.; Torchia, G.; Limatola, C.; et al. Physiological characterization of human muscle acetylcholine receptors from ALS patients. Proc. Natl. Acad. Sci. USA 2011, 108, 20184–20188. [Google Scholar] [CrossRef]
- Palma, E.; Reyes-Ruiz, J.M.; Lopergolo, D.; Roseti, C.; Bertollini, C.; Ruffolo, G.; Cifelli, P.; Onesti, E.; Limatola, C.; Miledi, R.; et al. Acetylcholine receptors from human muscle as pharmacological targets for ALS therapy. Proc. Natl. Acad. Sci. USA 2016, 113, 3060–3065. [Google Scholar] [CrossRef]
- Musarò, A.; Dobrowolny, G.; Cambieri, C.; Onesti, E.; Ceccanti, M.; Frasca, V.; Pisano, A.; Cerbelli, B.; Lepore, E.; Ruffolo, G.; et al. Neuromuscular magnetic stimulation counteracts muscle decline in ALS patients: Results of a randomized, double-blind, controlled study. Sci. Rep. 2019, 9, 2837. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fast | Slow | p-Value | |
|---|---|---|---|
| Time since diagnosis (months ±SEM) | 15.4 ± 2.2 | 34.6 ± 4.8 | <0.05 |
| ALSFRS-R (±SEM) | 36.6 ± 1.7 | 38.26 ± 0.8 | >0.05 |
| Bulbar/spinal onset (n) | 19/27 | 20/60 | <0.05 |
| Male/female (n) | 26/22 | 47/31 | >0.05 |
| N | CK Levels (U/L) ± SEM | p-Value | Mb Levels (ng/mL) ± SEM | p-Value | ||
|---|---|---|---|---|---|---|
| Overall | 126 | 252.5 ± 30.1 | 97.1 ± 12.4 | |||
| Site of onset | Spinal | 91 | 287.2 ± 39.7 | <0.01 | 104.1 ± 15.9 | <0.05 |
| Bulbar | 35 | 161.4 ± 29.9 | 78.9 ± 18.7 | |||
| Rate of progression | Slow | 79 | 310.6 ± 50.4 | <0.05 | 116.9 ± 15.8 | <0.05 |
| Fast | 47 | 154.8 ± 16.2 | 63.7 ± 10.3 |
| T1 | T2 | T3 | T4 | T5 | |
|---|---|---|---|---|---|
| N | 126 | 115 | 102 | 96 | 61 |
| CPK ± SEM | 252.5 ± 30.1 | 297.7 ± 44.3 | 360.6 ± 76.8 | 420.3 ± 88.1 | 298.3 ± 69.5 |
| ALSFRS-R ± SEM | 37.6 ± 0.87 | 35.5 ± 1.3 | 33.6 ± 1.5 | 31.9 ± 2.1 | 27.8 ± 2.8 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ceccanti, M.; Pozzilli, V.; Cambieri, C.; Libonati, L.; Onesti, E.; Frasca, V.; Fiorini, I.; Petrucci, A.; Garibaldi, M.; Palma, E.; et al. Creatine Kinase and Progression Rate in Amyotrophic Lateral Sclerosis. Cells 2020, 9, 1174. https://doi.org/10.3390/cells9051174
Ceccanti M, Pozzilli V, Cambieri C, Libonati L, Onesti E, Frasca V, Fiorini I, Petrucci A, Garibaldi M, Palma E, et al. Creatine Kinase and Progression Rate in Amyotrophic Lateral Sclerosis. Cells. 2020; 9(5):1174. https://doi.org/10.3390/cells9051174
Chicago/Turabian StyleCeccanti, Marco, Valeria Pozzilli, Chiara Cambieri, Laura Libonati, Emanuela Onesti, Vittorio Frasca, Ilenia Fiorini, Antonio Petrucci, Matteo Garibaldi, Eleonora Palma, and et al. 2020. "Creatine Kinase and Progression Rate in Amyotrophic Lateral Sclerosis" Cells 9, no. 5: 1174. https://doi.org/10.3390/cells9051174
APA StyleCeccanti, M., Pozzilli, V., Cambieri, C., Libonati, L., Onesti, E., Frasca, V., Fiorini, I., Petrucci, A., Garibaldi, M., Palma, E., Bendotti, C., Fabbrizio, P., Trolese, M. C., Nardo, G., & Inghilleri, M. (2020). Creatine Kinase and Progression Rate in Amyotrophic Lateral Sclerosis. Cells, 9(5), 1174. https://doi.org/10.3390/cells9051174