Single-Cell RNA Sequencing and Its Combination with Protein and DNA Analyses


Abstract
1. Introduction
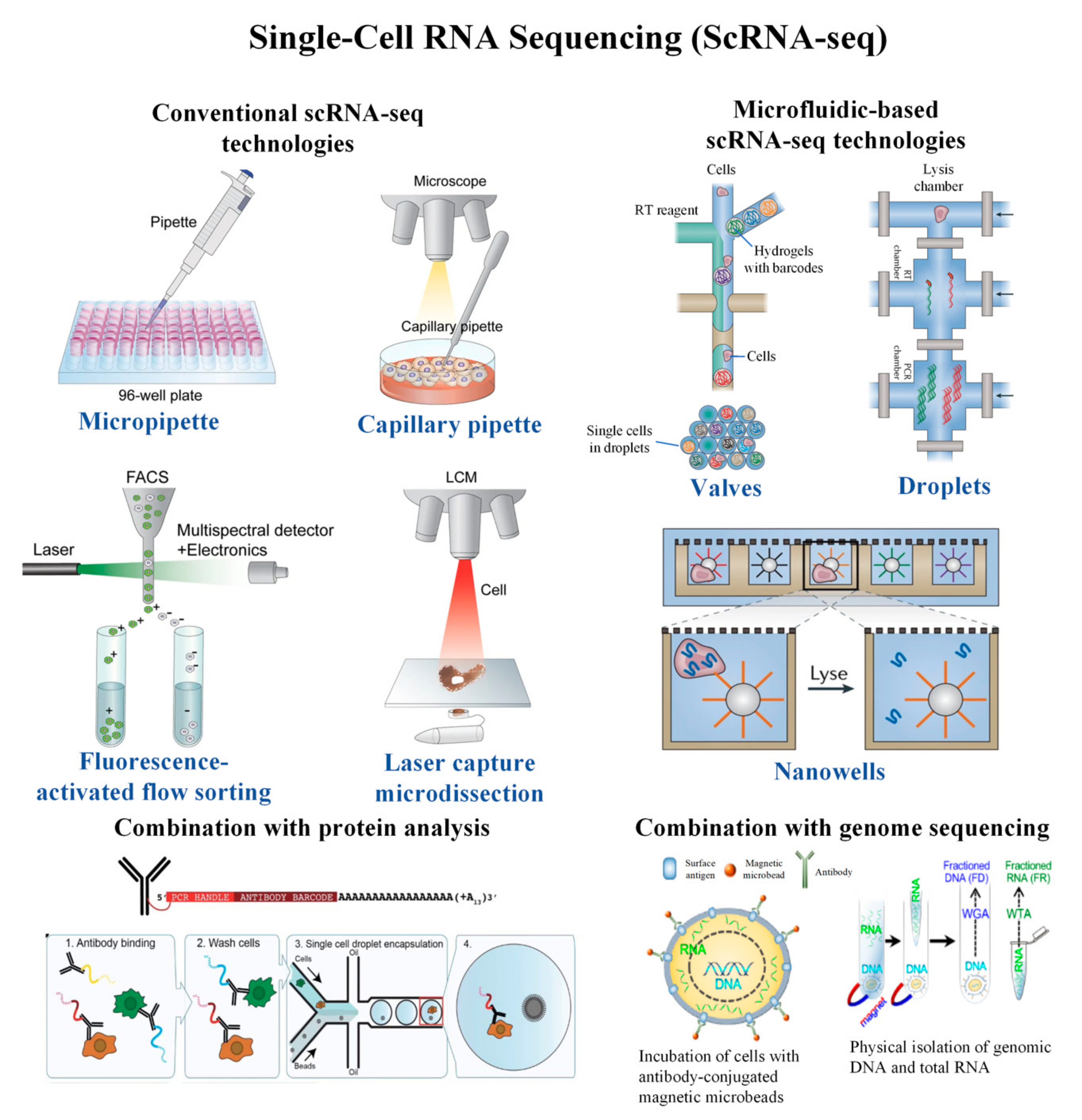
2. Conventional scRNA-seq Technologies
2.1. Smart-seq 1 and 2
2.2. SCRB-seq
2.3. CEL-seq 1 and 2
2.4. MARS-seq 1 and 2
2.5. Quartz-seq 1 and 2
2.6. SUPeR-seq
2.7. MATQ-seq
3. Microfluidic-Based scRNA-seq Technologies
3.1. Valve-Based scRNA-seq Technologies
3.1.1. Multilayer Microfluidic Device for scRNA-seq
3.1.2. Microfluidic Hydrodynamic Trap Array for scRNA-seq
3.1.3. MID-RNA-seq
3.1.4. Hydro-seq
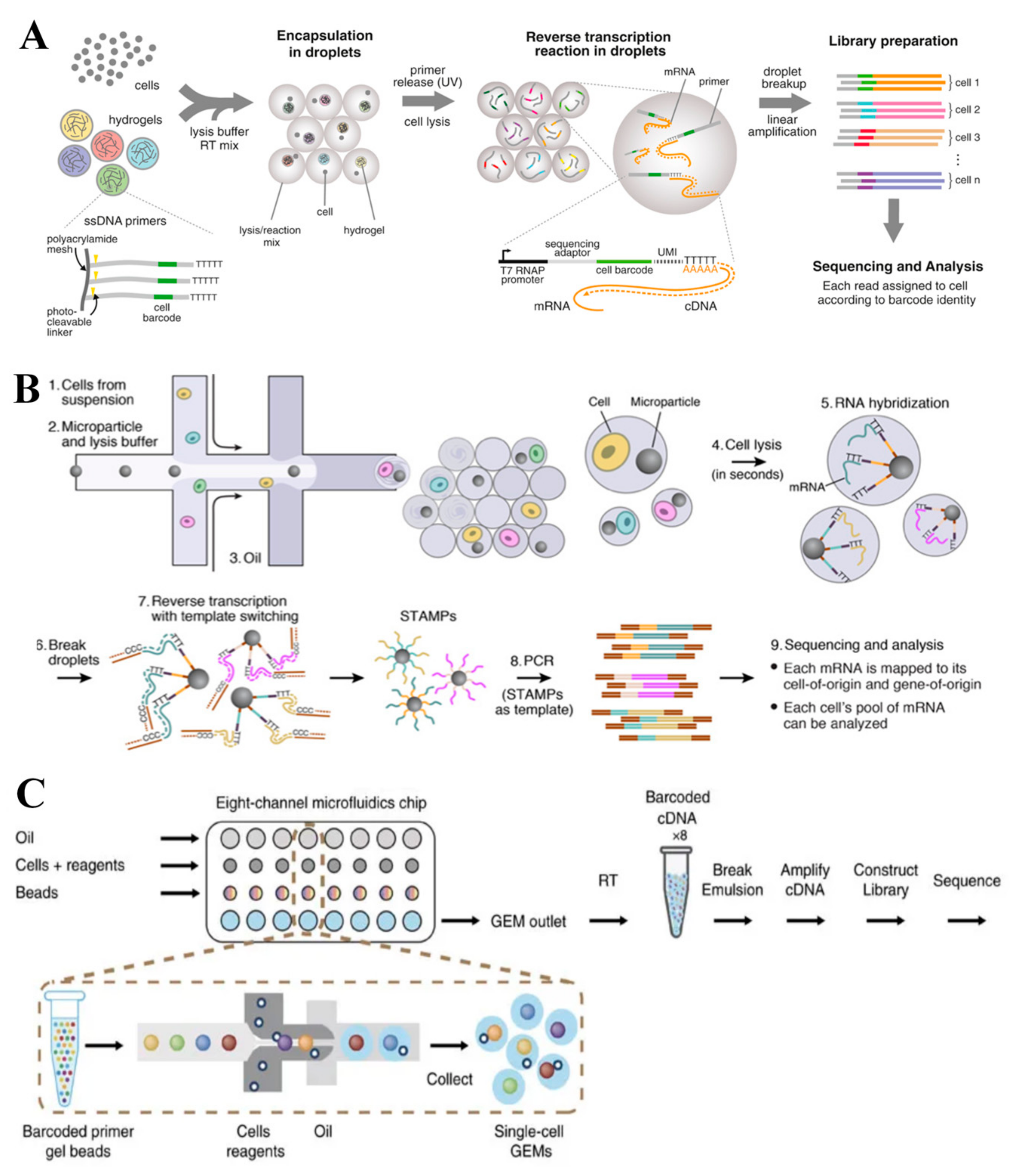
3.2. Droplet-Based scRNA-seq Technologies
3.2.1. Hi-SCL
3.2.2. In-Drop
3.2.3. Drop-seq
3.2.4. 10x Genomics
3.2.5. MULTI-seq
3.3. Nanowell-Based scRNA-seq Technologies
3.3.1. Cytoseq
3.3.2. Microwell-seq
3.3.3. Seq-Well
3.3.4. SCOPE-seq
3.3.5. scFTD-seq
4. Combination of scRNA-seq with Proteomic Analysis
4.1. Cite-seq
4.2. Reap-seq
4.3. PDMS Nanowell and seq
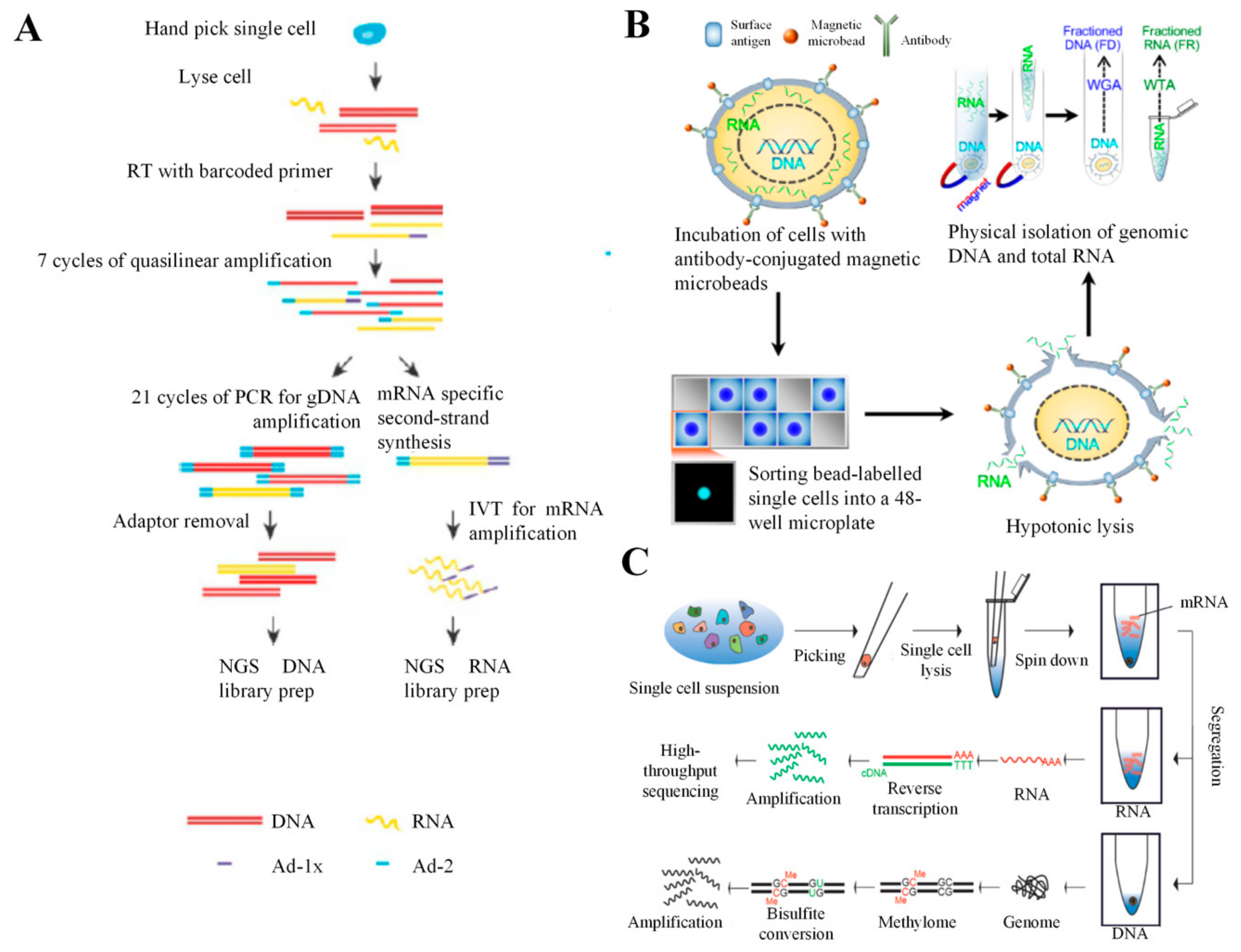
5. Combination of scRNA-seq with DNA Analysis
5.1. DR-seq
5.2. G and T-seq
5.3. SIDR-seq
5.4. CORTAD-seq
5.5. scTrio-seq
6. Commercial scRNA-seq Technologies
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Olsson:, A.; Venkatasubramanian, M.; Chaudhri, V.K.; Aronow, B.J.; Salomonis, N.; Singh, H.; Grimes, H.L. Single-cell analysis of mixed-lineage states leading to a binary cell fate choice. Nature 2016, 537, 698–702. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018, 36, 411–420. [Google Scholar] [CrossRef]
- Papalexi, E.; Satija, R. Single-cell RNA sequencing to explore immune cell heterogeneity. Nat. Rev. Immunol. 2018, 18, 35. [Google Scholar] [CrossRef] [PubMed]
- Fischer, D.S.; Fiedler, A.K.; Kernfeld, E.M.; Genga, R.M.; Bastidas-Ponce, A.; Bakhti, M.; Lickert, H.; Hasenauer, J.; Maehr, R.; Theis, F.J. Inferring population dynamics from single-cell RNA-sequencing time series data. Nat. Biotechnol. 2019, 37, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Dong, X.; Freytag, S.; Lê Cao, K.-A.; Su, S.; JalalAbadi, A.; Amann-Zalcenstein, D.; Weber, T.S.; Seidi, A.; Jabbari, J.S. Benchmarking single cell RNA-sequencing analysis pipelines using mixture control experiments. Nat. Methods 2019, 16, 479–487. [Google Scholar] [CrossRef]
- Stegle, O.; Teichmann, S.A.; Marioni, J.C. Computational and analytical challenges in single-cell transcriptomics. Nat. Rev. Genet. 2015, 16, 133–145. [Google Scholar] [CrossRef]
- Crow, M.; Paul, A.; Ballouz, S.; Huang, Z.J.; Gillis, J. Characterizing the replicability of cell types defined by single cell RNA-sequencing data using MetaNeighbor. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef]
- Usoskin, D.; Furlan, A.; Islam, S.; Abdo, H.; Lönnerberg, P.; Lou, D.; Hjerling-Leffler, J.; Haeggström, J.; Kharchenko, O.; Kharchenko, P.V. Unbiased classification of sensory neuron types by large-scale single-cell RNA sequencing. Nat. Neurosci. 2015, 18, 145. [Google Scholar] [CrossRef]
- Farrell, J.A.; Wang, Y.; Riesenfeld, S.J.; Shekhar, K.; Regev, A.; Schier, A.F. Single-cell reconstruction of developmental trajectories during zebrafish embryogenesis. Science 2018, 360, eaar3131. [Google Scholar] [CrossRef]
- Packer, J.; Trapnell, C. Single-cell multi-omics: An engine for new quantitative models of gene regulation. Trends Genet. 2018, 34, 653–665. [Google Scholar] [CrossRef]
- Grün, D.; Lyubimova, A.; Kester, L.; Wiebrands, K.; Basak, O.; Sasaki, N.; Clevers, H.; Van Oudenaarden, A. Single-cell messenger RNA sequencing reveals rare intestinal cell types. Nature 2015, 525, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Xue, Z.; Huang, K.; Cai, C.; Cai, L.; Jiang, C.-y.; Feng, Y.; Liu, Z.; Zeng, Q.; Cheng, L.; Sun, Y.E. Genetic programs in human and mouse early embryos revealed by single-cell RNA sequencing. Nature 2013, 500, 593–597. [Google Scholar] [CrossRef] [PubMed]
- Hashimshony, T.; Wagner, F.; Sher, N.; Yanai, I. CEL-Seq: Single-cell RNA-Seq by multiplexed linear amplification. Cell Rep. 2012, 2, 666–673. [Google Scholar] [CrossRef] [PubMed]
- Hashimshony, T.; Senderovich, N.; Avital, G.; Klochendler, A.; de Leeuw, Y.; Anavy, L.; Gennert, D.; Li, S.; Livak, K.J.; Rozenblatt-Rosen, O. CEL-Seq2: Sensitive highly-multiplexed single-cell RNA-Seq. Genome Biol. 2016, 17, 77. [Google Scholar] [CrossRef]
- Soumillon, M.; Cacchiarelli, D.; Semrau, S.; van Oudenaarden, A.; Mikkelsen, T.S. Characterization of directed differentiation by high-throughput single-cell RNA-Seq. BioRxiv 2014, 003236. [Google Scholar]
- Stephenson, W.; Donlin, L.T.; Butler, A.; Rozo, C.; Bracken, B.; Rashidfarrokhi, A.; Goodman, S.M.; Ivashkiv, L.B.; Bykerk, V.P.; Orange, D.E. Single-cell RNA-seq of rheumatoid arthritis synovial tissue using low-cost microfluidic instrumentation. Nat. Commun. 2018, 9, 1–10. [Google Scholar] [CrossRef]
- Bose, S.; Wan, Z.; Carr, A.; Rizvi, A.H.; Vieira, G.; Pe’er, D.; Sims, P.A. Scalable microfluidics for single-cell RNA printing and sequencing. Genome Biol. 2015, 16, 120. [Google Scholar] [CrossRef]
- Moon, H.-S.; Je, K.; Min, J.-W.; Park, D.; Han, K.-Y.; Shin, S.-H.; Park, W.-Y.; Yoo, C.E.; Kim, S.-H. Inertial-ordering-assisted droplet microfluidics for high-throughput single-cell RNA-sequencing. Lab Chip 2018, 18, 775–784. [Google Scholar] [CrossRef]
- Streets, A.M.; Zhang, X.; Cao, C.; Pang, Y.; Wu, X.; Xiong, L.; Yang, L.; Fu, Y.; Zhao, L.; Tang, F. Microfluidic single-cell whole-transcriptome sequencing. Proc. Natl. Acad. Sci. USA 2014, 111, 7048–7053. [Google Scholar] [CrossRef]
- Picelli, S. Single-cell RNA-sequencing: The future of genome biology is now. Rna Biol. 2017, 14, 637–650. [Google Scholar] [CrossRef]
- Saliba, A.-E.; Westermann, A.J.; Gorski, S.A.; Vogel, J. Single-cell RNA-seq: Advances and future challenges. Nucleic Acids Res. 2014, 42, 8845–8860. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.; Khoo, W.H.; Moran, I.; Croucher, P.I.; Phan, T.G. Single cell RNA sequencing of rare immune cell populations. Front. Immunol. 2018, 9, 1553. [Google Scholar] [CrossRef] [PubMed]
- Macosko, E.Z.; Basu, A.; Satija, R.; Nemesh, J.; Shekhar, K.; Goldman, M.; Tirosh, I.; Bialas, A.R.; Kamitaki, N.; Martersteck, E.M. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 2015, 161, 1202–1214. [Google Scholar] [CrossRef] [PubMed]
- Klein, A.M.; Mazutis, L.; Akartuna, I.; Tallapragada, N.; Veres, A.; Li, V.; Peshkin, L.; Weitz, D.A.; Kirschner, M.W. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 2015, 161, 1187–1201. [Google Scholar] [CrossRef] [PubMed]
- Gierahn, T.M.; Wadsworth II, M.H.; Hughes, T.K.; Bryson, B.D.; Butler, A.; Satija, R.; Fortune, S.; Love, J.C.; Shalek, A.K. Seq-Well: Portable, low-cost RNA sequencing of single cells at high throughput. Nat. Methods 2017, 14, 395–398. [Google Scholar] [CrossRef]
- Han, X.; Wang, R.; Zhou, Y.; Fei, L.; Sun, H.; Lai, S.; Saadatpour, A.; Zhou, Z.; Chen, H.; Ye, F. Mapping the mouse cell atlas by microwell-seq. Cell 2018, 172, 1091–1107.e17. [Google Scholar] [CrossRef]
- Zhang, H.; Cui, N.; Cai, Y.; Lei, F.; Weitz, D.A. Single-cell sequencing leads a new era of profiling transcriptomic landscape. J. Bio-X Res. 2018, 1, 2–6. [Google Scholar] [CrossRef]
- Kalisky, T.; Oriel, S.; Bar-Lev, T.H.; Ben-Haim, N.; Trink, A.; Wineberg, Y.; Kanter, I.; Gilad, S.; Pyne, S. A brief review of single-cell transcriptomic technologies. Brief. Funct. Genom. 2018, 17, 64–76. [Google Scholar] [CrossRef]
- Hedlund, E.; Deng, Q. Single-cell RNA sequencing: Technical advancements and biological applications. Mol. Asp. Med. 2018, 59, 36–46. [Google Scholar] [CrossRef]
- Kolodziejczyk, A.A.; Kim, J.K.; Svensson, V.; Marioni, J.C.; Teichmann, S.A. The technology and biology of single-cell RNA sequencing. Mol. Cell 2015, 58, 610–620. [Google Scholar] [CrossRef]
- Chappell, L.; Russell, A.J.; Voet, T. Single-cell (multi) omics technologies. Annu. Rev. Genom. Hum. Genet. 2018, 19, 15–41. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Finck, A.; Fan, R. Single-cell omics analyses enabled by microchip technologies. Annu. Rev. Biomed. Eng. 2019, 21, 365–393. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Preissl, S.; Ren, B. Single-cell multimodal omics: The power of many. Nat. Methods 2020, 17, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Macaulay, I.C.; Ponting, C.P.; Voet, T. Single-cell multiomics: Multiple measurements from single cells. Trends Genet. 2017, 33, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Hwang, B.; Lee, J.H.; Bang, D. Single-cell RNA sequencing technologies and bioinformatics pipelines. Exp. Mol. Med. 2018, 50, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Ziegenhain, C.; Vieth, B.; Parekh, S.; Reinius, B.; Guillaumet-Adkins, A.; Smets, M.; Leonhardt, H.; Heyn, H.; Hellmann, I.; Enard, W. Comparative analysis of single-cell RNA sequencing methods. Mol. Cell 2017, 65, 631–643.e4. [Google Scholar] [CrossRef]
- Picelli, S.; Faridani, O.R.; Björklund, Å.K.; Winberg, G.; Sagasser, S.; Sandberg, R. Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc. 2014, 9, 171. [Google Scholar] [CrossRef]
- Picelli, S.; Björklund, Å.K.; Faridani, O.R.; Sagasser, S.; Winberg, G.; Sandberg, R. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat. Methods 2013, 10, 1096–1098. [Google Scholar] [CrossRef]
- Jaitin, D.A.; Kenigsberg, E.; Keren-Shaul, H.; Elefant, N.; Paul, F.; Zaretsky, I.; Mildner, A.; Cohen, N.; Jung, S.; Tanay, A. Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science 2014, 343, 776–779. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Kenigsberg, E.; Jaitin, D.A.; David, E.; Paul, F.; Tanay, A.; Amit, I. MARS-seq2.0: An experimental and analytical pipeline for indexed sorting combined with single-cell RNA sequencing. Nat. Protoc. 2019, 14, 1841. [Google Scholar] [CrossRef]
- Sasagawa, Y.; Nikaido, I.; Hayashi, T.; Danno, H.; Uno, K.D.; Imai, T.; Ueda, H.R. Quartz-Seq: A highly reproducible and sensitive single-cell RNA sequencing method, reveals non-genetic gene-expression heterogeneity. Genome Biol. 2013, 14, 3097. [Google Scholar] [CrossRef] [PubMed]
- Sasagawa, Y.; Danno, H.; Takada, H.; Ebisawa, M.; Tanaka, K.; Hayashi, T.; Kurisaki, A.; Nikaido, I. Quartz-Seq2: A high-throughput single-cell RNA-sequencing method that effectively uses limited sequence reads. Genome Biol. 2018, 19, 29. [Google Scholar] [CrossRef]
- Fan, X.; Zhang, X.; Wu, X.; Guo, H.; Hu, Y.; Tang, F.; Huang, Y. Single-cell RNA-seq transcriptome analysis of linear and circular RNAs in mouse preimplantation embryos. Genome Biol. 2015, 16, 148. [Google Scholar] [CrossRef] [PubMed]
- Sheng, K.; Cao, W.; Niu, Y.; Deng, Q.; Zong, C. Effective detection of variation in single-cell transcriptomes using MATQ-seq. Nat. Methods 2017, 14, 267. [Google Scholar] [CrossRef] [PubMed]
- Ramsköld, D.; Luo, S.; Wang, Y.-C.; Li, R.; Deng, Q.; Faridani, O.R.; Daniels, G.A.; Khrebtukova, I.; Loring, J.F.; Laurent, L.C. Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nat. Biotechnol. 2012, 30, 777. [Google Scholar] [CrossRef] [PubMed]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef]
- Kurimoto, K.; Yabuta, Y.; Ohinata, Y.; Saitou, M. Global single-cell cDNA amplification to provide a template for representative high-density oligonucleotide microarray analysis. Nat. Protoc. 2007, 2, 739. [Google Scholar] [CrossRef] [PubMed]
- Jeck, W.R.; Sharpless, N.E. Detecting and characterizing circular RNAs. Nat. Biotechnol. 2014, 32, 453. [Google Scholar] [CrossRef]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef]
- Kimmerling, R.J.; Szeto, G.L.; Li, J.W.; Genshaft, A.S.; Kazer, S.W.; Payer, K.R.; de Riba Borrajo, J.; Blainey, P.C.; Irvine, D.J.; Shalek, A.K. A microfluidic platform enabling single-cell RNA-seq of multigenerational lineages. Nat. Commun. 2016, 7, 1–7. [Google Scholar] [CrossRef]
- Sarma, M.; Lee, J.; Ma, S.; Li, S.; Lu, C. A diffusion-based microfluidic device for single-cell RNA-seq. Lab Chip 2019, 19, 1247–1256. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.-H.; Chen, Y.-C.; Lin, E.; Brien, R.; Jung, S.; Chen, Y.-T.; Lee, W.; Hao, Z.; Sahoo, S.; Kang, H.M. Hydro-Seq enables contamination-free high-throughput single-cell RNA-sequencing for circulating tumor cells. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Rotem, A.; Ram, O.; Shoresh, N.; Sperling, R.A.; Schnall-Levin, M.; Zhang, H.; Basu, A.; Bernstein, B.E.; Weitz, D.A. High-throughput single-cell labeling (Hi-SCL) for RNA-Seq using drop-based microfluidics. PLoS ONE 2015, 10, e0116328. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.X.; Terry, J.M.; Belgrader, P.; Ryvkin, P.; Bent, Z.W.; Wilson, R.; Ziraldo, S.B.; Wheeler, T.D.; McDermott, G.P.; Zhu, J. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017, 8, 1–12. [Google Scholar] [CrossRef]
- McGinnis, C.S.; Patterson, D.M.; Winkler, J.; Conrad, D.N.; Hein, M.Y.; Srivastava, V.; Hu, J.L.; Murrow, L.M.; Weissman, J.S.; Werb, Z. MULTI-seq: Sample multiplexing for single-cell RNA sequencing using lipid-tagged indices. Nat. Methods 2019, 16, 619. [Google Scholar] [CrossRef]
- Fan, H.C.; Fu, G.K.; Fodor, S.P. Combinatorial labeling of single cells for gene expression cytometry. Science 2015, 347, 1258367. [Google Scholar] [CrossRef]
- Yuan, J.; Sheng, J.; Sims, P.A. SCOPE-Seq: A scalable technology for linking live cell imaging and single-cell RNA sequencing. Genome Biol. 2018, 19, 227. [Google Scholar] [CrossRef]
- Dura, B.; Choi, J.-Y.; Zhang, K.; Damsky, W.; Thakral, D.; Bosenberg, M.; Craft, J.; Fan, R. scFTD-seq: Freeze-thaw lysis based, portable approach toward highly distributed single-cell 3′ mRNA profiling. Nucleic Acids Res. 2019, 47, e16. [Google Scholar] [CrossRef]
- Stoeckius, M.; Hafemeister, C.; Stephenson, W.; Houck-Loomis, B.; Chattopadhyay, P.K.; Swerdlow, H.; Satija, R.; Smibert, P. Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods 2017, 14, 865. [Google Scholar] [CrossRef]
- Peterson, V.M.; Zhang, K.X.; Kumar, N.; Wong, J.; Li, L.; Wilson, D.C.; Moore, R.; McClanahan, T.K.; Sadekova, S.; Klappenbach, J.A. Multiplexed quantification of proteins and transcripts in single cells. Nat. Biotechnol. 2017, 35, 936. [Google Scholar] [CrossRef]
- George, J.; Wang, J. Assay of genome-wide transcriptome and secreted proteins on the same single immune cells by microfluidics and RNA sequencing. Anal. Chem. 2016, 88, 10309–10315. [Google Scholar] [CrossRef] [PubMed]
- Timp, W.; Timp, G. Beyond mass spectrometry, the next step in proteomics. Sci. Adv. 2020, 6, eaax8978. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Wang, J.; Wu, L.; Guo, J.; Song, Y.; Tian, T.; Wang, W.; Zhu, Z.; Yang, C. Microfluidic Single-Cell Omics Analysis. Small 2019, 1903905. [Google Scholar] [CrossRef] [PubMed]
- Kong, S.L.; Li, H.; Tai, J.A.; Courtois, E.T.; Poh, H.M.; Lau, D.P.; Haw, Y.X.; Iyer, N.G.; Tan, D.S.W.; Prabhakar, S. Concurrent Single-Cell RNA and Targeted DNA Sequencing on an Automated Platform for Comeasurement of Genomic and Transcriptomic Signatures. Clin. Chem. 2019, 65, 272–281. [Google Scholar] [CrossRef]
- Dey, S.S.; Kester, L.; Spanjaard, B.; Bienko, M.; Van Oudenaarden, A. Integrated genome and transcriptome sequencing of the same cell. Nat. Biotechnol. 2015, 33, 285. [Google Scholar] [CrossRef]
- Macaulay, I.C.; Haerty, W.; Kumar, P.; Li, Y.I.; Hu, T.X.; Teng, M.J.; Goolam, M.; Saurat, N.; Coupland, P.; Shirley, L.M. G&T-seq: Parallel sequencing of single-cell genomes and transcriptomes. Nat. Methods 2015, 12, 519–522. [Google Scholar]
- Han, K.Y.; Kim, K.-T.; Joung, J.-G.; Son, D.-S.; Kim, Y.J.; Jo, A.; Jeon, H.-J.; Moon, H.-S.; Yoo, C.E.; Chung, W. SIDR: Simultaneous isolation and parallel sequencing of genomic DNA and total RNA from single cells. Genome Res. 2018, 28, 75–87. [Google Scholar] [CrossRef]
- Hou, Y.; Guo, H.; Cao, C.; Li, X.; Hu, B.; Zhu, P.; Wu, X.; Wen, L.; Tang, F.; Huang, Y. Single-cell triple omics sequencing reveals genetic, epigenetic, and transcriptomic heterogeneity in hepatocellular carcinomas. Cell Res. 2016, 26, 304–319. [Google Scholar] [CrossRef]
- Macaulay, I.C.; Teng, M.J.; Haerty, W.; Kumar, P.; Ponting, C.P.; Voet, T. Separation and parallel sequencing of the genomes and transcriptomes of single cells using G&T-seq. Nat. Protoc. 2016, 11, 2081. [Google Scholar]
- Ahadi, A.; Brennan, S.; Kennedy, P.J.; Hutvagner, G.; Tran, N. Long non-coding RNAs harboring miRNA seed regions are enriched in prostate cancer exosomes. Sci. Rep. 2016, 6, 1–14. [Google Scholar] [CrossRef]
- Smith, Z.D.; Meissner, A. DNA methylation: Roles in mammalian development. Nat. Rev. Genet. 2013, 14, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Valihrach, L.; Androvic, P.; Kubista, M. Platforms for single-cell collection and analysis. Int. J. Mol. Sci. 2018, 19, 807. [Google Scholar] [CrossRef] [PubMed]
- Shum, E.Y.; Walczak, E.M.; Chang, C.; Fan, H.C. Quantitation of mRNA Transcripts and Proteins Using the BD Rhapsody™ Single-Cell Analysis System. In Single Molecule and Single Cell Sequencing; Springer: Berlin, Germany, 2019; pp. 63–79. [Google Scholar]
- Goldstein, L.D.; Chen, Y.-J.J.; Dunne, J.; Mir, A.; Hubschle, H.; Guillory, J.; Yuan, W.; Zhang, J.; Stinson, J.; Jaiswal, B. Massively parallel nanowell-based single-cell gene expression profiling. BMC Genom. 2017, 18, 519. [Google Scholar] [CrossRef] [PubMed]
- Sanada, C.D.; Ooi, A.T. Single-Cell Dosing and mRNA Sequencing of Suspension and Adherent Cells Using the Polaris TM System. In Single Cell Methods; Springer: Berlin, Germany, 2019; pp. 185–195. [Google Scholar]
- Swennenhuis, J.F.; Tibbe, A.G.; Stevens, M.; Katika, M.R.; Van Dalum, J.; Tong, H.D.; van Rijn, C.J.; Terstappen, L.W. Self-seeding microwell chip for the isolation and characterization of single cells. Lab Chip 2015, 15, 3039–3046. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.H.; Bender, S.; Krahn, T.; Schlange, T. ctDNA and CTCs in liquid biopsy—Current status and where we need to progress. Comput. Struct. Biotechnol. J. 2018, 16, 190–195. [Google Scholar] [CrossRef]
- Rita, Z.; Rossi, E. Single-cell analysis of Circulating Tumor Cells: How far we come with omics-era? Front. Genet. 2019, 10, 958. [Google Scholar]
- Choi, J.R.; Yong, K.W.; Choi, J.Y.; Cowie, A.C. Recent advances in photo-crosslinkable hydrogels for biomedical applications. BioTechniques 2019, 66, 40–53. [Google Scholar] [CrossRef]
- Lee, I.-N.; Dobre, O.; Richards, D.; Ballestrem, C.; Curran, J.M.; Hunt, J.A.; Richardson, S.M.; Swift, J.; Wong, L.S. Photoresponsive hydrogels with photoswitchable mechanical properties allow time-resolved analysis of cellular responses to matrix stiffening. ACS Appl. Mater. Interfaces 2018, 10, 7765–7776. [Google Scholar] [CrossRef]
- Lähnemann, D.; Köster, J.; Szczurek, E.; McCarthy, D.J.; Hicks, S.C.; Robinson, M.D.; Vallejos, C.A.; Campbell, K.R.; Beerenwinkel, N.; Mahfouz, A. Eleven grand challenges in single-cell data science. Genome Biol. 2020, 21, 1–35. [Google Scholar] [CrossRef]
- Poirion, O.B.; Zhu, X.; Ching, T.; Garmire, L. Single-cell transcriptomics bioinformatics and computational challenges. Front. Genet. 2016, 7, 163. [Google Scholar] [CrossRef]
- Yau, C. pcaReduce: Hierarchical clustering of single cell transcriptional profiles. BMC Bioinform. 2016, 17, 140. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Technology | Cell Isolation Method | No. of Cells | Cell Barcode | Unique Molecular Identifiers | cDNA Coverage | Amplification Method | Advantages | Limitations | Outcomes |
|---|---|---|---|---|---|---|---|---|---|
| Smart-seq 1 & 2 [37,38] | Micropipette | 100–1000 | No | No | Full-length | Template switching-based PCR | Increased throughput and read coverage across transcripts Smart-seq 2 increases thermal stability of LNA-DNA base pairs. | Low number of cells Time-consuming cell isolation processes | Transcript enumeration Analysis of alternative splicing allelic expression Investigation of transcriptomic profile in rare cells |
| CEL-seq 1 and 2 [13,14] | Micropipette | 100–1000 | Yes | Yes | 3′ tag | In vitro transcription-based 3′ transcript amplification * The protocol is based on Smart-seq | CEL-Seq 2 adds a 5-base pair UMI upstream of the barcode to distinguish between PCR duplicates and transcript abundance in scRNA-seq, which significantly improves accuracy. | 3′ end sequencing only The use of micropipette for cell isolation makes the operational processes more difficult and time-consuming. Low number of cells are processed. | It is used to study early C. elegans embryonic development at single cell level. CEL-seq will be useful for transcriptomic analyses of complex tissues containing populations of diverse cell types. |
| SCRB-seq [15] | FACS | 1000–10,000 | Yes | Yes | 3′ tag | Template switching-based PCR * The protocol is based on Smart-seq. | High throughput | Requires skilled workers | Characterization of primary human adipose-derived stem cell differentiation system Discovery of transcriptomes across heterogeneous populations |
| MARS-seq 1 & 2 [39,40] | FACS | 1000–5000 | Yes | Yes | 3′ tag | In vitro transcription-based 3′ transcript amplification | Automated processes minimize amplification bias and labeling errors | Requires skilled workers | Analysis of in vivo transcriptional states in thousands of single cells. Identification of a unique microglia type that may restrict the development of Alzheimer’s disease |
| Quartz-seq 1 [41] | FACS | 1000–10,000 | No | No | Full length with 3′ biased | PCR after poly(A) tailing | Highly quantitative | Requires skilled workers | Detection of transcriptome heterogeneity between the cells in the same and different cell-cycle phases |
| Quartz-seq 2 [42] | FACS | 1000–10,000 | Yes | Yes | Full length with 3′ biased | PCR after poly(A) tailing | Able to detect more transcripts from limited sequence reads at a minimal cost | Requires skilled workers | Detection of transcriptome heterogeneity between embryonic stem cells and between cells in stromal vascular fraction |
| SUPeR-seq [43] | Mouth pipette | ~10 | Yes | No | Full length | PCR after poly(A) tailing | Able to detect both circular RNA (non-polyadenylated RNA) and polyadenylated RNA | Low throughput Operational processes are difficult and time-consuming | Analysis of expression dynamics of circular RNA during mammalian early embryonic development |
| MATQ-seq [44] | Mouth pipette | 10–100 | Yes | Yes | Full length | PCR after poly(A) tailing | Able to sequence both polyadenylated and non-polyadenylated RNAs with high sensitivity and accuracy | Low throughput Operational processes are difficult and time-consuming | Detection of low abundance genes and non-polyadenylated RNA extracted from a single cell |
| Technology | Cell Isolation Method | No. of Cells | Cell Barcode | Unique Molecular Identifiers | cDNA Coverage | Amplification Method | Advantages | Limitations | Outcomes |
|---|---|---|---|---|---|---|---|---|---|
| Multilayer microfluidic device and seq [19] | Valve | 10–100 | Yes | No | Full-length | PCR after poly(A) tailing | Improvement of assay sensitivity The semi-automated processes minimize technical variation and reduce risk of contamination | The requirement of off-chip amplification Complex device fabrication processes Low throughput | Identification of differentially expressed genes of single cells and measurement of biological variations in cell populations. |
| Microfluidic hydrodynamic trap array & seq [50] | Valve | 10–5000 | Yes | No | Full-length | Template switching-based PCR * The protocol is based on Smart-seq 2 | Allows multi-generational lineage tracking under controlled culture conditions | Complex device fabrication processes | Measurement of the effects of lineage and cell cycle-dependent transcriptional profiles of single cells. |
| MID-RNA-seq [51] | Valve | 1000 | Yes | No | Full length | PCR after poly(A) tailing | Allows automated processing and multiplexing | Complex device fabrication processes | Transcriptomic studies of scarce cell samples. |
| Hydro-seq [52] | Valve | 10–1000 | Yes | Yes | 3′ tag | Template switching-based PCR * The protocol is based on Drop-seq. | Improved throughput and cell capture efficiency | Complex device fabrication processes | Identification of cellular heterogeneity in critical biomarkers of tumor metastasis, understanding tumor metastasis processes, and monitoring target therapeutics in cancer patients. |
| Hi-SCL [53] | Droplet | 1000–10,000 | Yes | No | 3′ tag | PCR after poly(A) tailing | High throughput | Low cell capture efficiency | Detection and comparison of transcriptomes in mouse embryonic stem cells and mouse embryonic fibroblast populations at the single-cell level. |
| In-drop [24] | Droplet | 1000–10,000 | Yes | Yes | 3′ tag | In vitro transcription-based 3′ transcript amplification * The protocol is based on CEL-seq. | High throughput | Low cell capture efficiency Only the 3′ most terminal fragments can be used for sequencing | Sequencing of large numbers of cells from heterogeneous populations in a fast way and identification of very rare cell types. |
| Drop-seq [23] | Droplet | 1000–10,000 | Yes | Yes | 3′ tag | Template switching-based PCR | High throughput, cheaper, and faster | Only the 3′ most terminal fragments can be used for sequencing | Analysis of mRNA transcripts from thousands of individual cells concurrently while identifying the cell of origin. |
| 10x Genomics [54] | Droplet | 1000–10,000 | Yes | Yes | 3′ tag | Template switching-based PCR | The use of 10x barcodes significantly increase throughput | Only the 3′ most terminal fragments can be used for sequencing | Profile 68k peripheral blood mononuclear cells and dissect large immune populations. |
| MULTI-seq [55] | Droplet | 10,000–100,000 | Barcoded lipid-modified oligonucleotides | Yes | 3′ tag | Template switching-based PCR * The protocol is based on 10x genomics. | Readily multiplex various cell types and identify cell doublets | Only the 3′ most terminal fragments can be used for sequencing | Assessment of immune cell responses to tumor metastatic progression. |
| Cytoseq [56] | Nanowell | 100–10,000 | Yes | Yes | 3′ tag | Gene specific primers-based PCR | High throughput Simple fabrication and operation processes | Not fully automated | Characterization of cellular heterogeneity in immune response and identification of rare cells in a cell population. |
| Microwell-seq [26] | Nanowell | 100–10,000 | Yes | No | Full-length | Template switching-based PCR * The protocol is based on Smart-seq 2. | High throughput Simple fabrication and operation processes | Not fully automated | Construction of “mouse cell atlas” with more than 400k single-cell transcriptomic profiles from 51 mouse tissues, organs, and cell cultures, covering more than 800 major cell types and 1000 cell subtypes in the mouse system. |
| Seq-well [25] | Nanowell | 100–10,000 | Yes | Yes | 3′ tag | Template switching-based PCR * The protocol is based on Drop-seq. | High throughput Simple fabrication and operation processes The use of semipermeable polycarbonate membrane reduces well-to-well contamination | Not fully automated | Profile thousands of primary human macrophages exposed to Mycobacterium tuberculosis. It is compatible with on-array imaging cytometry for resolving the phenotype of cells from complex samples. |
| SCOPE-seq [57] | Nanowell | 100–10,000 | Yes | Yes | 3′ tag | Template switching-based PCR * The protocol is based on Drop-seq. | High throughput Simple fabrication and operation processes The phenotypes measured can be directly linked to expression profiles using optically decodable beads The use of perfluorinated oil prevents well-to-well contamination | Not fully automated | Combination of live cell imaging with single-cell RNA sequencing for various biomedical applications. |
| scFTD-seq [58] | Nanowell | 100–10,000 | Yes | Yes | 3′ tag | Template switching-based PCR * The protocol is based on drop-seq. | High throughput Simple fabrication and operation processes Minimizing contamination by preventing immediate cell lysis | Not fully automated | Profile circulating follicular helper T cells implicated in systemic lupus erythematosus pathogenesis |
| Technology | Cell Isolation Method | No. of Cells | Cell Barcode | Unique Molecular Identifiers | cDNA Coverage | cDNA Amplification Method | Advantages | Limitations | Outcomes |
|---|---|---|---|---|---|---|---|---|---|
| Cite-seq [59] | Droplet | 1000–10,000 | Yes | No | 3′ tag | Template switching-based PCR *The protocol is based on Drop-seq. | High throughput Allows simultaneous transcriptomic and surface protein analysis | Low cell capture efficiency Not fully automated | Simultaneous detection of about 13 surface proteins and transcripts |
| Reap-seq [60] | Droplet | 1000–10,000 | Yes | Yes | 3′ tag | Template switching-based PCR * The protocol is based on 10x genomics. | High throughput Allows simultaneous transcriptomic and surface protein analysis | Low cell capture efficiency Not fully automated | Assessment of costimulatory effects of a CD27 agonist on human CD8+ lymphocytes and characterization of an unknown cell type |
| PDMS Nanowells and seq [61] | Nanowell | 1000–10,000 | Yes | No | Full length | Template switching-based PCR * The protocol is based on Smart-seq 2. | High throughput Allows simultaneous transcriptomic and secretion analysis | Not fully automated | Study of regulation mechanisms of the immune system |
| Technology | Cell Isolation Method | No. of Cells | Cell Barcode | Unique Molecular Identifiers | cDNA Coverage | cDNA Amplification Method | Advantages | Limitations | Outcomes |
|---|---|---|---|---|---|---|---|---|---|
| DR-seq [65] | Mouth pipette | 10–50 | Yes | No | 3′ tag | In vitro transcription * The protocol is based on CEL-seq. | Allows simultaneous transcriptomic and DNA analysis | Complex work flow and low throughput Requires in silico masking of coding sequences, which complicates the data analysis processes | Study of transcriptional consequences of gDNA copy number variations in diseased and healthy tissues |
| G and T-seq [66] | FACS | 10–100 | No | No | Nearly full length | Template switching-based PCR * The protocol is based on Smart-seq 2. | Simple work flow Allows simultaneous transcriptomic and DNA analysis | Requires skilled workers Low throughput | Study of transcriptional consequences of chromosomal abnormalities in a single cell |
| SIDR-seq [67] | Micropipette | 10–100 | No | No | Nearly full-length | Template switching-based PCR * The protocol is based on Smart-seq 2. | Automated and simple work flow Allows simultaneous transcriptomic and DNA analysis | Low throughput | Assessment of cellular heterogeneity in breast and lung cancer at the singular cell level |
| CORTAD-seq [64] | Fludigm C1 | 100–1000 | No | No | Full length with weak 3′-biased | Template switching-based PCR * The protocol is based on Smart-seq. | Automated and high throughput Allows simultaneous transcriptomic and DNA analysis | Not suitable for genome-wide DNA analysis for discovery purpose | Study of transcriptional consequences of known targeted gene mutations in various types of cancer |
| scTrio-seq [68] | Mouth pipette | 10–50 | No | No | Full length with weak 3′-biased | Template switching-based PCR * The protocol is based on Smart-seq. | Allows simultaneous transcriptomic, genomic, and epigenomic analysis | Complex work flow and low throughput | Study of transcriptional consequences of genomic and epigenomic heterogeneities within a population of cells especially cancer cells |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, J.R.; Yong, K.W.; Choi, J.Y.; Cowie, A.C. Single-Cell RNA Sequencing and Its Combination with Protein and DNA Analyses. Cells 2020, 9, 1130. https://doi.org/10.3390/cells9051130
Choi JR, Yong KW, Choi JY, Cowie AC. Single-Cell RNA Sequencing and Its Combination with Protein and DNA Analyses. Cells. 2020; 9(5):1130. https://doi.org/10.3390/cells9051130
Chicago/Turabian StyleChoi, Jane Ru, Kar Wey Yong, Jean Yu Choi, and Alistair C. Cowie. 2020. "Single-Cell RNA Sequencing and Its Combination with Protein and DNA Analyses" Cells 9, no. 5: 1130. https://doi.org/10.3390/cells9051130
APA StyleChoi, J. R., Yong, K. W., Choi, J. Y., & Cowie, A. C. (2020). Single-Cell RNA Sequencing and Its Combination with Protein and DNA Analyses. Cells, 9(5), 1130. https://doi.org/10.3390/cells9051130