Molecular Dynamics Simulations Reveal Canonical Conformations in Different pMHC/TCR Interactions

 ,
,
Abstract
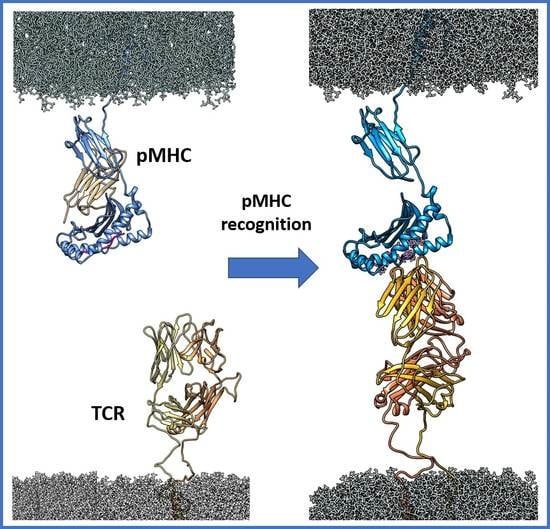
1. Introduction
2. Materials and Methods
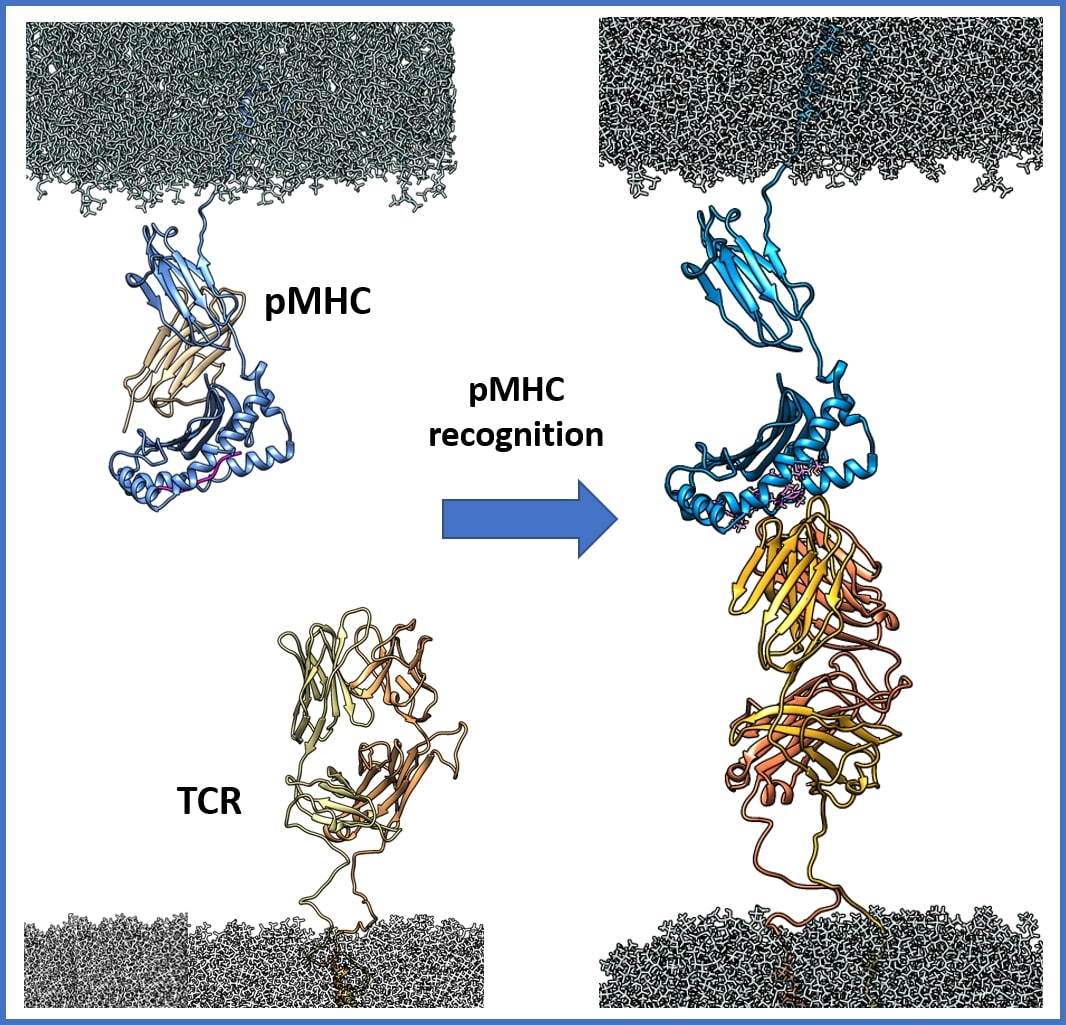
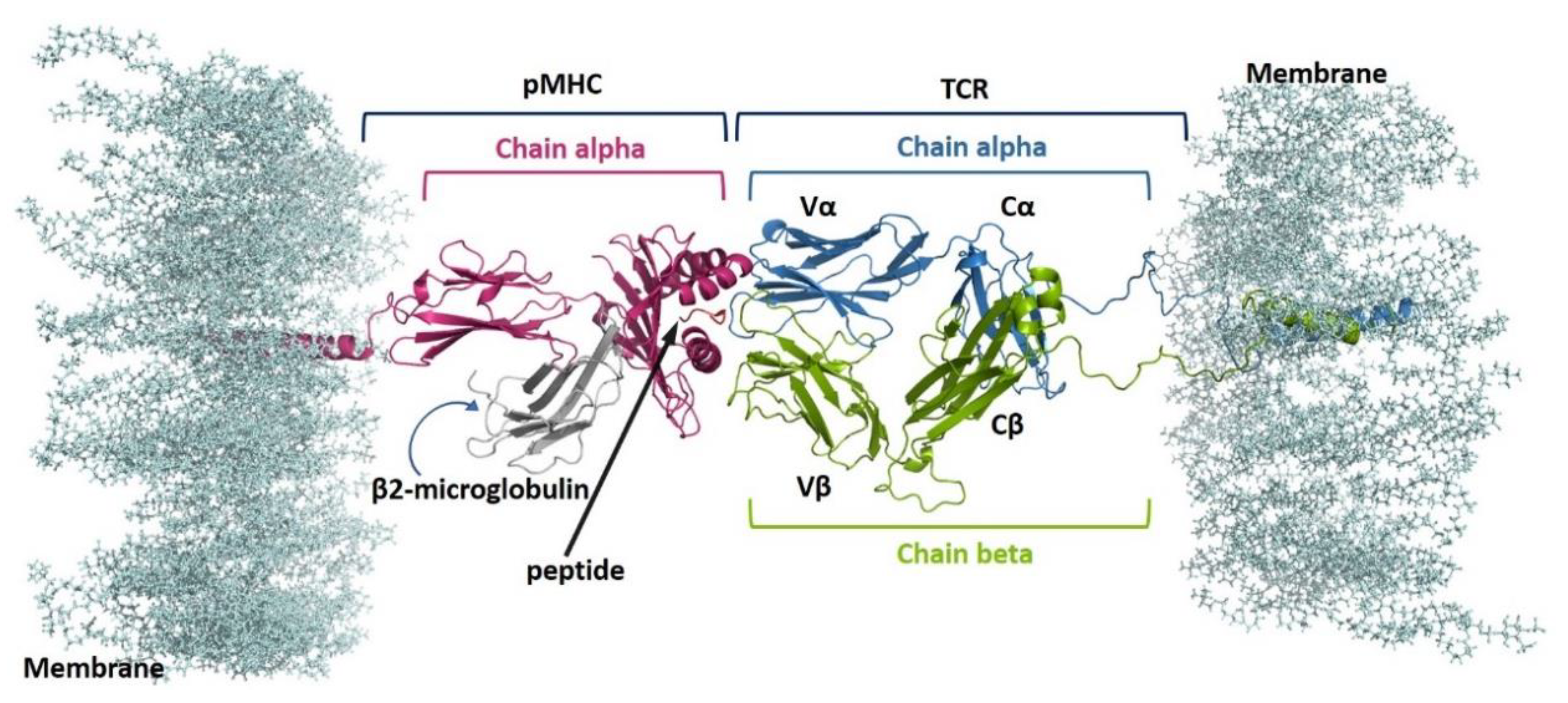
2.1. Complexes Modelling
2.2. MD Simulations
2.3. Structural Analysis
2.4. Essential Dynamics
2.5. Cross-Correlation Matrix
2.6. Construction of the Zernike Descriptor
2.7. Network Analysis
2.8. Membrane Analysis
3. Results
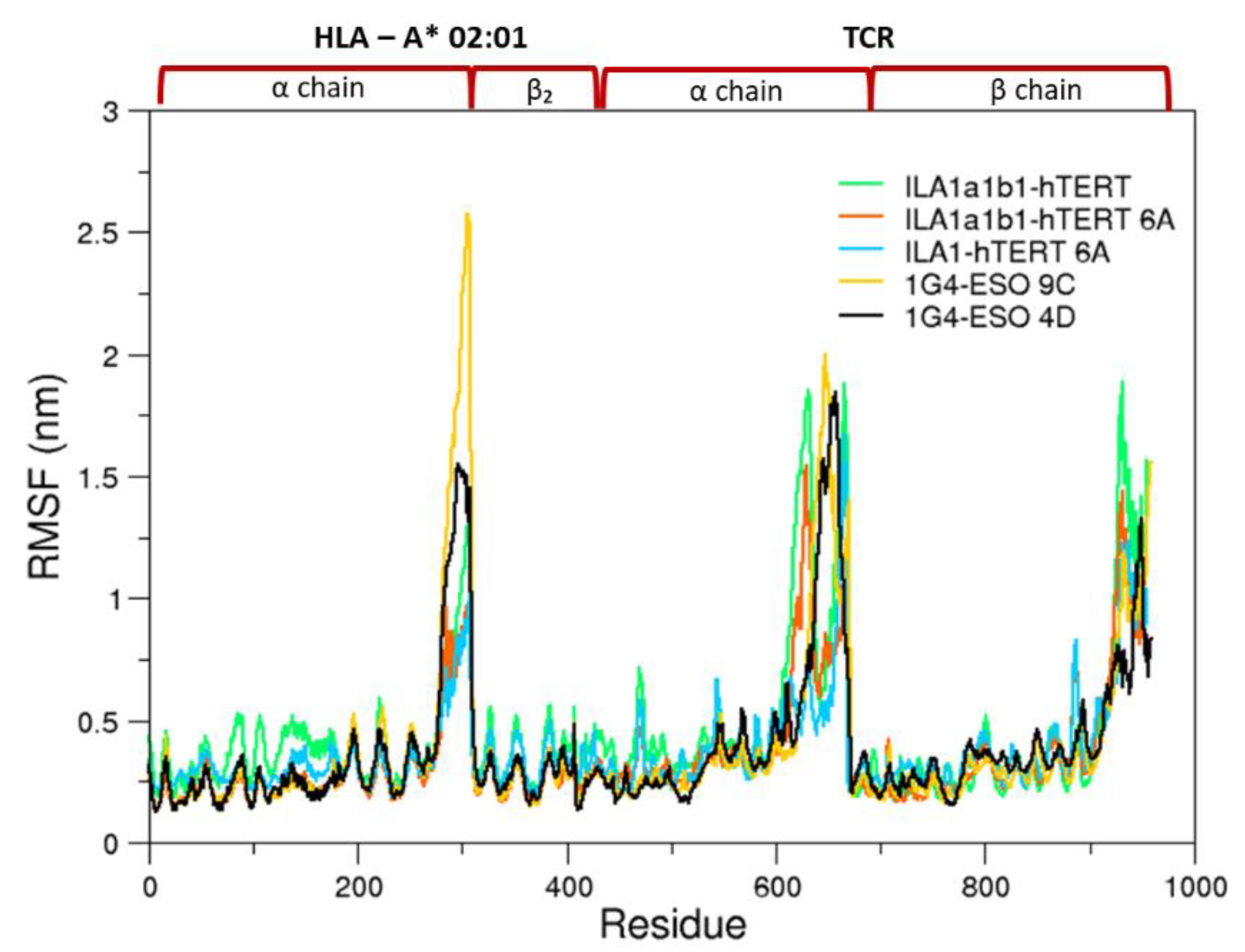
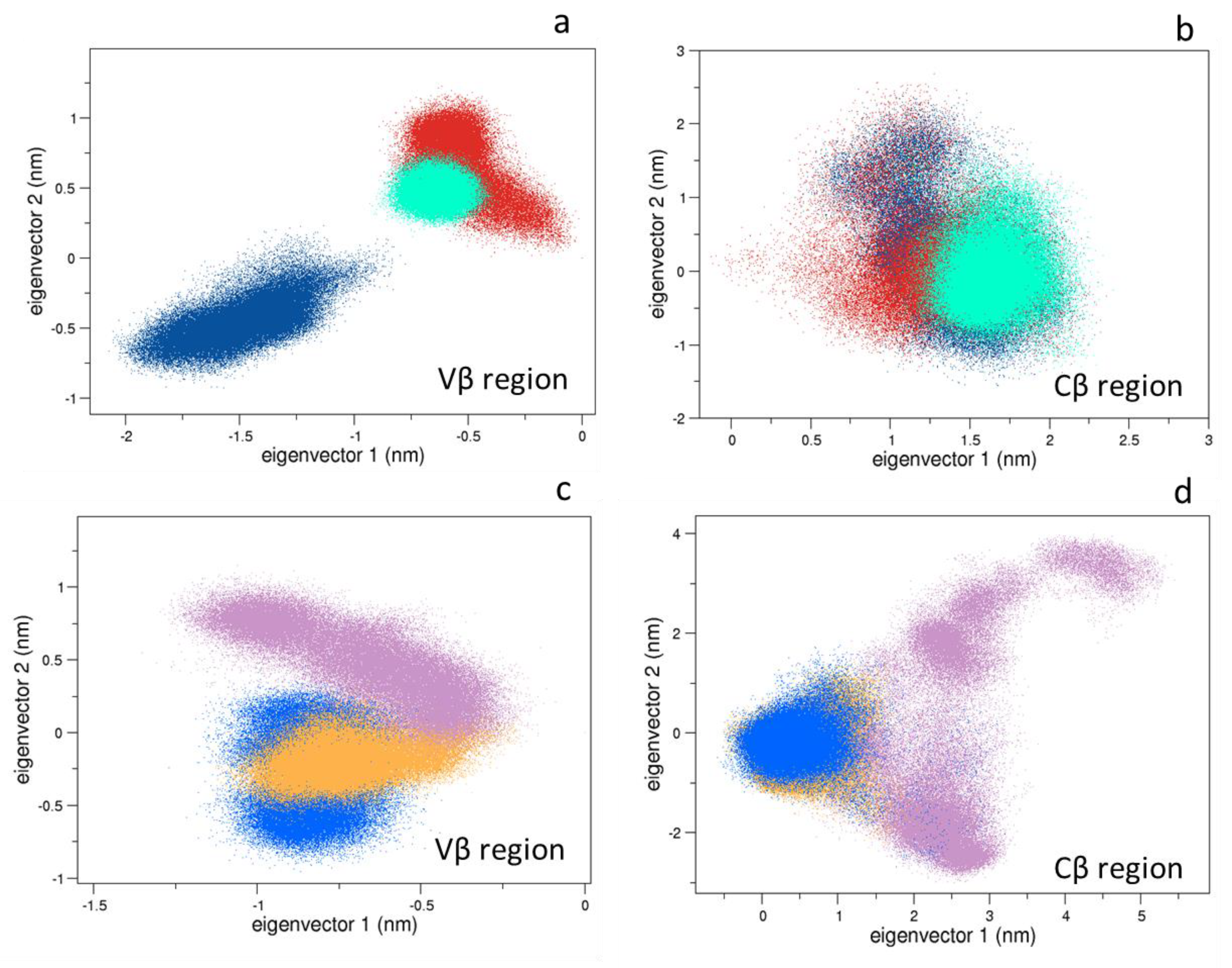
3.1. Conformational Analysis
3.1.1. Collective Motions
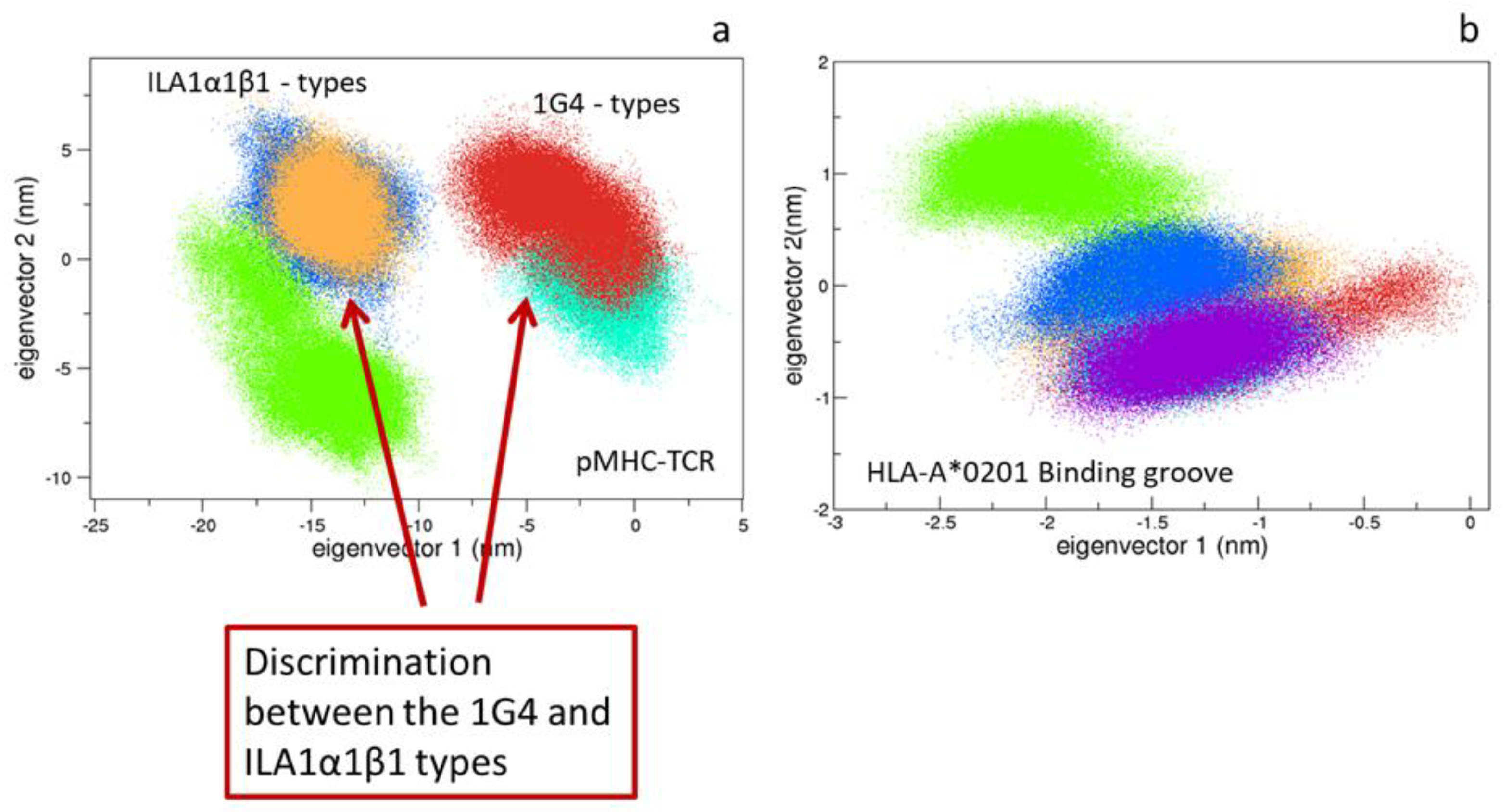
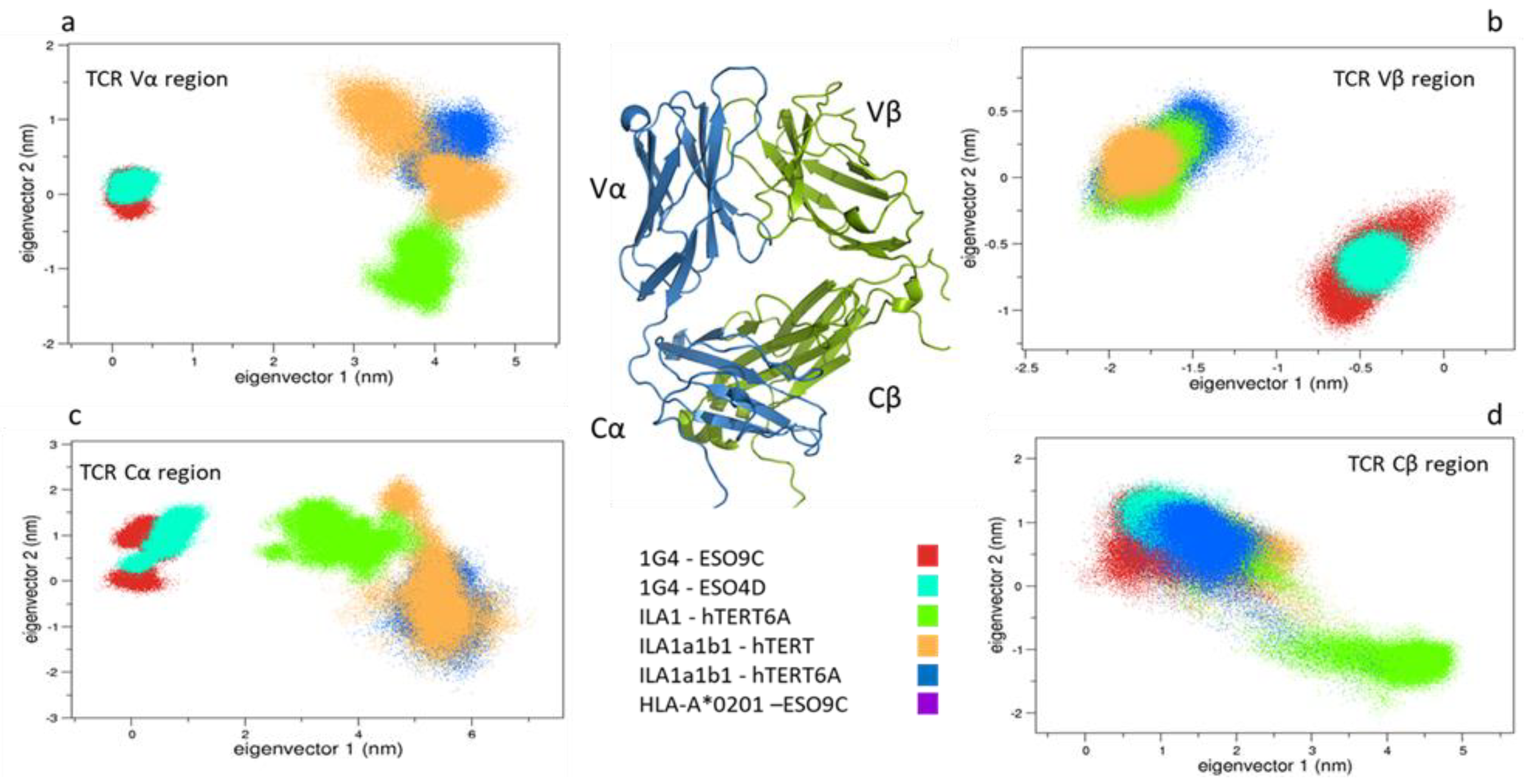
3.1.2. Comparison between the Bound and Unbound States
3.1.3. Comparison between the Bound States
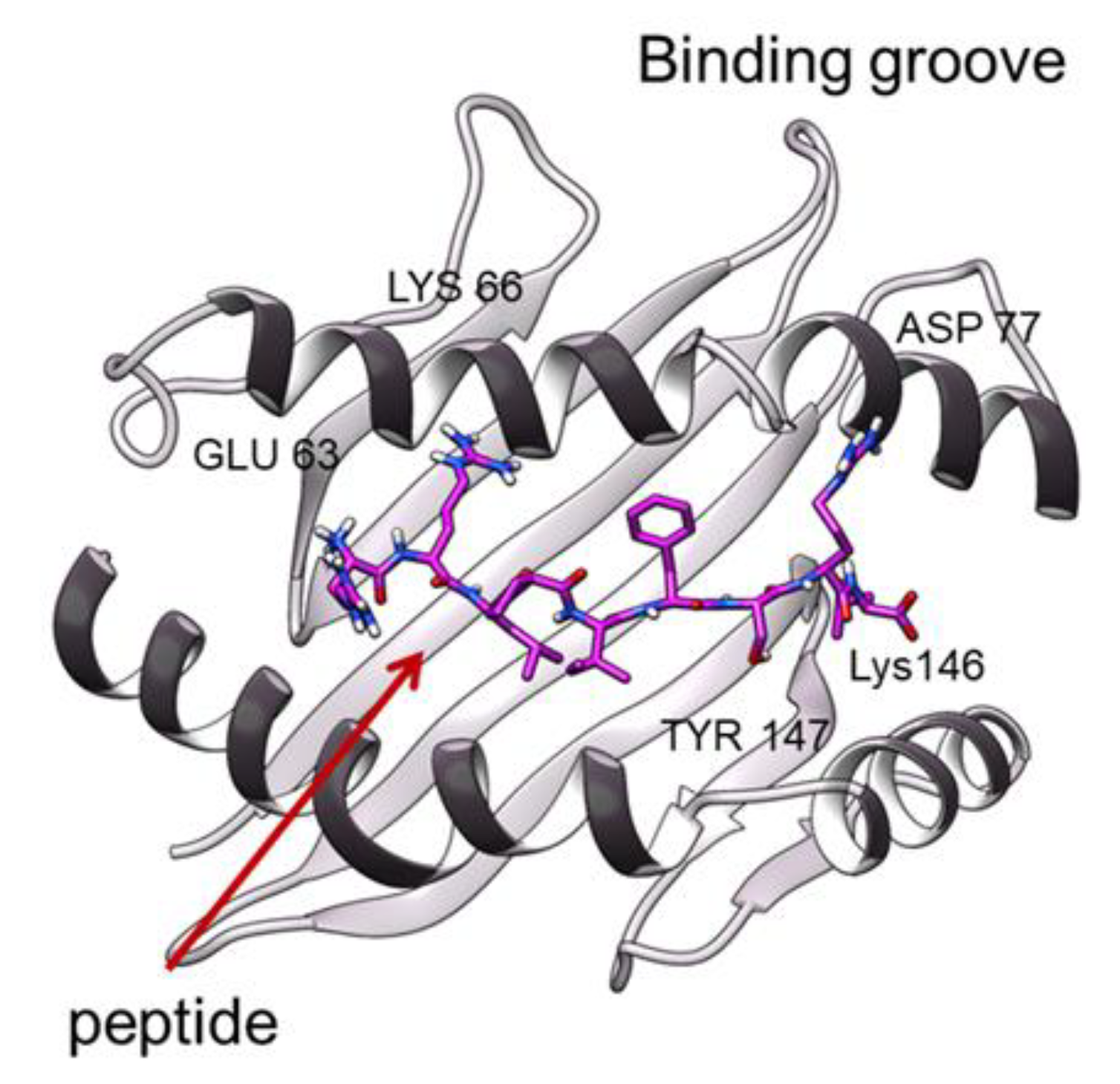
3.2. Peptide Interactions
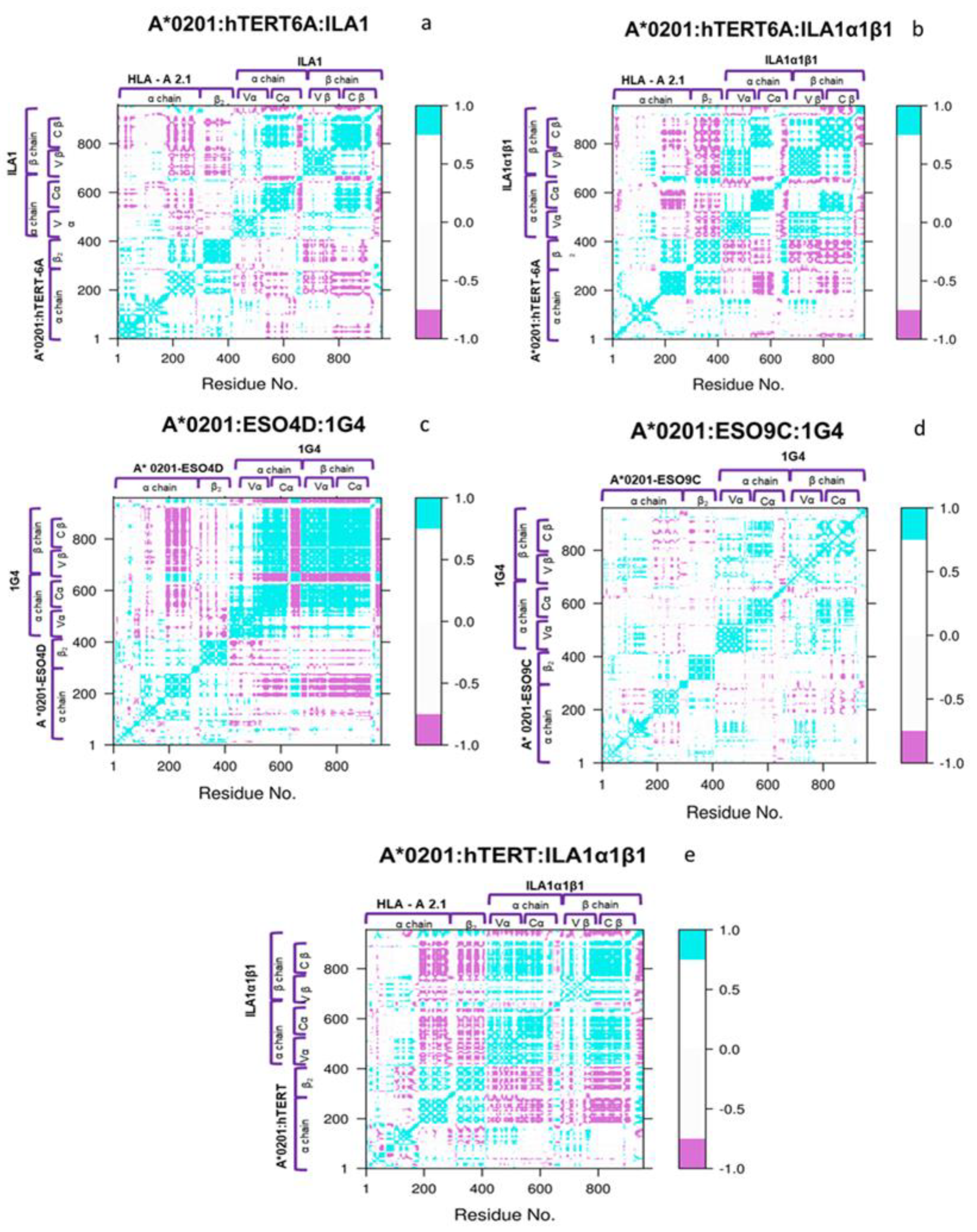
3.3. Network and Cross-Correlation Matrix Analysis
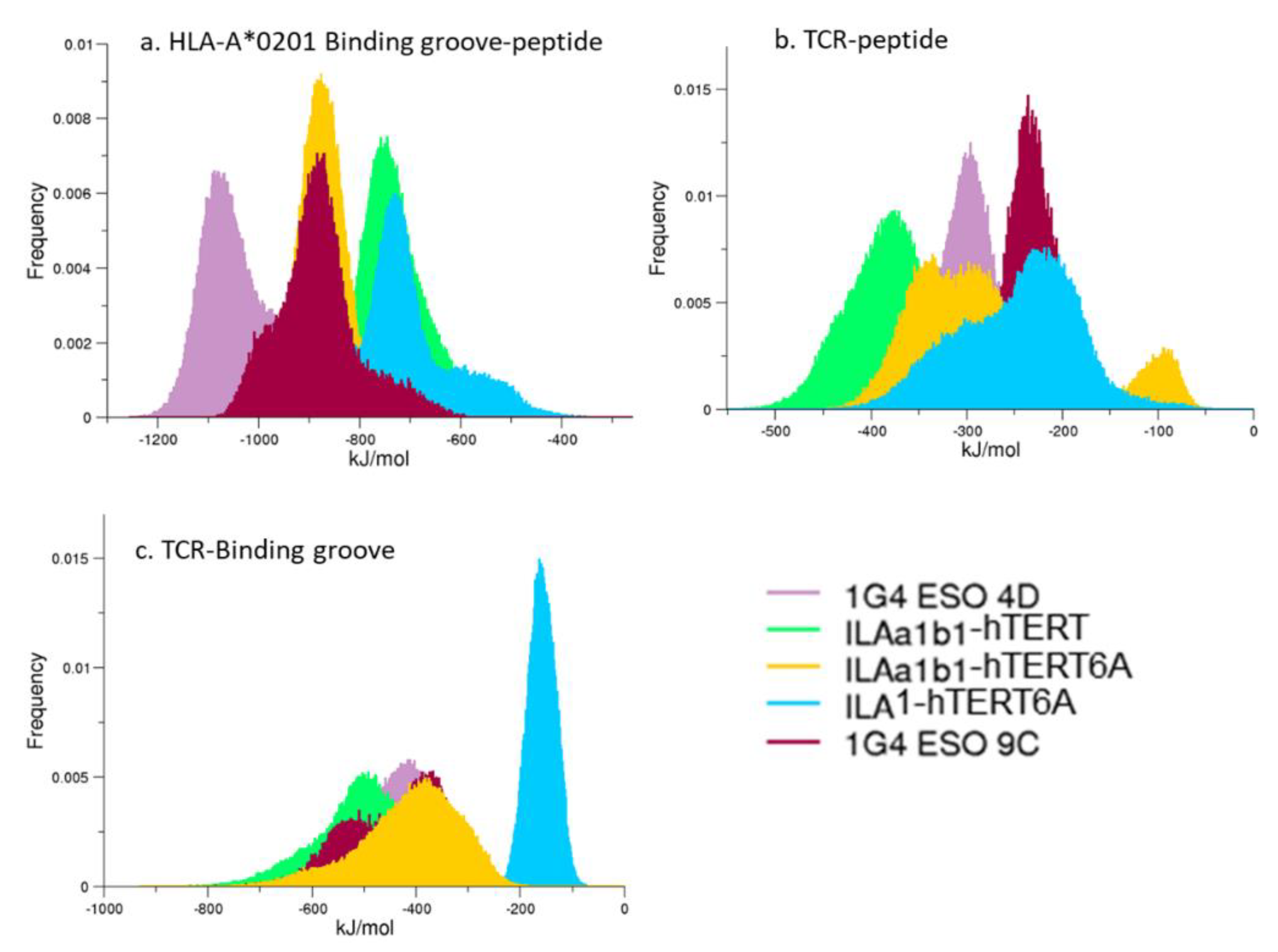
3.4. Interaction Energies
3.5. Membrane Behavior
4. Discussion
4.1. pMHC Influence on the TCRs Conformations
4.2. TCR Effects on the Binding Groove Behaviour
4.3. Dynamical Coupling of the pMHC-TCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Janeway, C.A. The immune system evolved to discriminate infectious non self from noninfectious self. Immunol. Today 1992, 13, 11–16. [Google Scholar] [CrossRef]
- Andersen, M.H.; Schrama, D.; Thor Straten, P.; Becker, J.C. Cytotoxic T Cells. J. Invest. Dermatol. 2006, 126, 32–41. [Google Scholar] [CrossRef]
- Smith, D.A.; Germolec, D.R. Introduction to immunology and autoimmunity. Environ. Health Perspect. 1999, 107, 661–665. [Google Scholar]
- Garcia, K.C.; Degano, M.; Stanfield, R.L.; Brunmark, A.; Jackson, M.R.; Peterson, P.A.; Teyton, L.; Wilson, I.A. An alphabeta T cell receptor structure at 2.5 A and its orientation in the TCR-MHC complex. Science 1996, 274, 209–219. [Google Scholar] [CrossRef]
- Garboczi, D.N.; Ghosh, P.; Utz, U.; Fan, Q.R.; Biddison, W.E.; Wiley, D.C. Structure of the complex between human T-cell receptor, viral peptide and HLA-A2. Nature 1996, 384, 134–141. [Google Scholar] [CrossRef]
- Call, M.E.; Pyrdol, J.; Wiedmann, M.; Wucherpfennig, K.W. The organizing principle in the formation of the T cell receptor-CD3 complex. Cell 2002, 111, 967–979. [Google Scholar] [CrossRef]
- Alarcón, B.; Gil, D.; Delgado, P.; Schamel, W.W.A. Initiation of TCR signaling: Regulation within CD3 dimers. Immunol. Rev. 2003, 191, 38–46. [Google Scholar] [CrossRef]
- Natarajan, K.; McShan, A.C.; Jiang, J.; Kumirov, V.K.; Wang, R.; Zhao, H.; Schuck, P.; Tilahun, M.E.; Boyd, L.F.; Ying, J.; et al. An allosteric site in the T-cell receptor Cβ domain plays a critical signalling role. Nat. Commun. 2017, 8, 1–14. [Google Scholar] [CrossRef]
- Rangarajan, S.; He, Y.; Chen, Y.; Kerzic, M.C.; Ma, B.; Gowthaman, R.; Pierce, B.G.; Nussinov, R.; Mariuzza, R.A.; Orban, J. Peptide–MHC (pMHC) binding to a human antiviral T cell receptor induces long-range allosteric communication between pMHC- and CD3-binding sites. J. Biol. Chem. 2018, 293, 15991–16005. [Google Scholar] [CrossRef]
- Tedeschi, V.; Alba, J.; Paladini, F.; Paroli, M.; Cauli, A.; Mathieu, A.; Sorrentino, R.; D’Abramo, M.; Fiorillo, M.T. Unusual Placement of an EBV Epitope into the Groove of the Ankylosing Spondylitis-Associated HLA-B27 Allele Allows CD8+ T Cell Activation. Cells 2019, 8, 572. [Google Scholar] [CrossRef]
- Šali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Bairoch, A.; Apweiler, R. The SWISS-PROT protein sequence database: Its relevance to human molecular medical research. J. Mol. Med. 1997, 75, 312–316. [Google Scholar] [PubMed]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Chavent, M.; Karia, D.; Kalli, A.C.; Domański, J.; Duncan, A.L.; Hedger, G.; Stansfeld, P.J.; Seiradake, E.; Jones, E.Y.; Sansom, M.S.P. Interactions of the EphA2 Kinase Domain with PIPs in Membranes: Implications for Receptor Function. Structure 2018, 26, 1025–1034. [Google Scholar] [CrossRef]
- Zech, T.; Ejsing, C.S.; Gaus, K.; de Wet, B.; Shevchenko, A.; Simons, K.; Harder, T. Accumulation of raft lipids in T-cell plasma membrane domains engaged in TCR signalling. EMBO J. 2009, 28, 466–476. [Google Scholar] [CrossRef]
- van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. [Google Scholar] [CrossRef]
- Wang, F.; Beck-García, K.; Zorzin, C.; Schamel, W.W.A.; Davis, M.M. Inhibition of T cell receptor signaling by cholesterol sulfate, a naturally occurring derivative of membrane cholesterol. Nat. Immunol. 2016, 17, 844–850. [Google Scholar] [CrossRef]
- Swamy, M.; Beck-Garcia, K.; Beck-Garcia, E.; Hartl, F.A.; Morath, A.; Yousefi, O.S.; Dopfer, E.P.; Molnár, E.; Schulze, A.K.; Blanco, R.; et al. A Cholesterol-Based Allostery Model of T Cell Receptor Phosphorylation. Immunity 2016, 44, 1091–1101. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD-Visual Molecular Dynamics. J. Molec. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Mark, P.; Nilsson, L. Structure and Dynamics of the TIP3P, SPC, and SPC/E Water Models at 298 K. J. Phys. Chem. 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126. [Google Scholar] [CrossRef] [PubMed]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Klauda, J.B.; Venable, R.M.; Freites, J.A.; O’Connor, J.W.; Tobias, D.J.; Mondragon-Ramirez, C.; Vorobyov, I.; MacKerell, A.D.; Pastor, R.W. Update of the CHARMM All-Atom Additive Force Field for Lipids: Validation on Six Lipid Types. J. Phys. Chem. 2010, 114, 7830–7843. [Google Scholar] [CrossRef]
- Lee, S.; Tran, A.; Allsopp, M.; Lim, J.B.; Hénin, J.; Klauda, J.B. CHARMM36 United Atom Chain Model for Lipids and Surfactants. J. Phys. Chem. 2014, 118, 547–556. [Google Scholar] [CrossRef]
- Abraham, M.J.; van der Spoel, D.; Lindahl, E.; Hess, B.; Team, D. GROMACS user manual version 2016.4 2017, Gromax Web Site. Available online: ftp://ftp.gromacs.org/pub/manual/manual-2016.4.pdf (accessed on 25 January 2020).
- Amadei, A.; Linssen, A.B.M.; Berendsen, H.J.C. Essential dynamics of proteins. Proteins Struct. Funct. Genet. 1993, 17, 412–425. [Google Scholar] [CrossRef]
- Grant, B.J.; Rodrigues, A.P.; ElSawy, K.M.; McCammon, J.A.; Caves, L.S. Bio3D: An R package for the comparative analysis of protein structures. Bioinformatics 2006, 22, 2695–2696. [Google Scholar] [CrossRef]
- RStudio Team. RStudio: Integrated Development for R. RStudio, Inc. 2015. Available online: http://www.rstudio.com/ (accessed on 25 January 2020).
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An automated pipeline for the setup of Poisson--Boltzmann electrostatics calculations. Nucleic Acids Res. 2004, 32, 665–667. [Google Scholar] [CrossRef]
- Fogolari, F.; Corazza, A.; Yarra, V.; Jalaru, A.; Viglino, P.; Esposito, G. Bluues: A program for the analysis of the electrostatic properties of proteins based on generalized Born radii. BMC Bioinform. 2012, 13. [Google Scholar] [CrossRef]
- Venkatraman, V.; Sael, L.; Kihara, D. Potential for protein surface shape analysis using spherical harmonics and 3d zernike descriptors. Cell Biochem. Biophys. 2009, 54, 23–32. [Google Scholar] [CrossRef]
- Novotni, M.; Klein, R. Shape retrieval using 3D Zernike descriptors. Comput. Des. 2004, 36, 1047–1062. [Google Scholar] [CrossRef]
- Di Rienzo, L.; Milanetti, E.; Lepore, R.; Olimpieri, P.P.; Tramontano, A. Superposition-free comparison and clustering of antibody binding sites: Implications for the prediction of the nature of their antigen. Sci. Rep. 2017, 7, 45053. [Google Scholar] [CrossRef]
- Di Rienzo, L.; Milanetti, E.; Alba, J.; D’Abramo, M. Quantitative characterization of binding pockets and binding complementarity by means of Zernike descriptors. J. Chem. Inf. Model. 2020, 60, 1390–1398. [Google Scholar] [CrossRef] [PubMed]
- Csardi, G.; Nepusz, T. The igraph software package for complex network research. InterJournal 2006, 1695, 1–9. [Google Scholar]
- Allen, W.J.; Lemkul, J.A.; Bevan, D.R. GridMAT-MD: A grid-based membrane analysis tool for use with molecular dynamics. J. Comput. Chem. 2009, 30, 1952–1958. [Google Scholar] [CrossRef] [PubMed]
- Laugel, B.; van den Berg, H.A.; Gostick, E.; Cole, D.K.; Wooldridge, L.; Boulter, J.; Milicic, A.; Price, D.A.; Sewell, A.K. Different T cell receptor affinity thresholds and CD8 coreceptor dependence govern cytotoxic T lymphocyte activation and tetramer binding properties. J. Biol. Chem. 2007, 282, 23799–23810. [Google Scholar] [CrossRef]
- Chen, J.-L.; Stewart-Jones, G.; Bossi, G.; Lissin, N.M.; Wooldridge, L.; Choi, E.M.L.; Held, G.; Dunbar, P.R.; Esnouf, R.M.; Sami, M.; et al. Structural and kinetic basis for heightened immunogenicity of T cell vaccines. J. Exp. Med. 2005, 201, 1243–1255. [Google Scholar] [CrossRef]
- Chen, J.-L.; Dunbar, P.R.; Gileadi, U.; Jäger, E.; Gnjatic, S.; Nagata, Y.; Stockert, E.; Panicali, D.L.; Chen, Y.-T.; Knuth, A.; et al. The Journal of Immunology. J. Immunol. 2000, 160, 1750–1758. [Google Scholar]
- Cole, D.K.; Miles, K.M.; Madura, F.; Holland, C.J.; Schauenburg, A.J.A.; Godkin, A.J.; Bulek, A.M.; Fuller, A.; Akpovwa, H.J.E.; Pymm, P.G.; et al. T-cell Receptor (TCR)-peptide specificity overrides affinity-enhancing TCR-major histocompatibility complex interactions. J. Biol. Chem. 2014, 289, 628–638. [Google Scholar] [CrossRef]
- Aleksic, M.; Dushek, O.; Zhang, H.; Shenderov, E.; Chen, J.-L.; Cerundolo, V.; Coombs, D.; Anton Van Der Merwe, P. Dependence of T Cell Antigen Recognition on T Cell Receptor-Peptide MHC Confinement Time. Immunity 2010, 32, 163–174. [Google Scholar] [CrossRef]
- Parkhurst, M.R.; Riley, J.P.; Igarashi, T.; Li, Y.; Robbins, P.F.; Rosenberg, S.A. Immunization of patients with the hTERT:540-548 peptide induces peptide-reactive T lymphocytes that do not recognize tumors endogenously expressing telomerase. Clin. Cancer Res. 2004, 10, 4688–4698. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Shi, X.; Xu, C. Regulation of T cell signalling by membrane lipids. Nat. Rev. Immunol. 2016, 16, 690–701. [Google Scholar] [CrossRef]
- Housset, D.; Mazza, G. The three-dimensional structure of a T-cell antigen receptor VαVβ heterodimer reveals a novel arrangement of the Vβ domain that peptide loops homologous to Ig complementarity. EMBO J. 1997, 16, 4205–4216. [Google Scholar] [CrossRef] [PubMed]
- Beddoe, T.; Chen, Z.; Clements, C.S.; Ely, L.K.; Bushell, S.R.; Vivian, J.P.; Kjer-Nielsen, L.; Pang, S.S.; Dunstone, M.A.; Liu, Y.C.; et al. Antigen Ligation Triggers a Conformational Change within the Constant Domain of the αβ T Cell Receptor. Immunity 2009, 30, 777–788. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex with HLA-A*0201 | Peptide Sequence | KD (uM) | Koff (s−1) | t ½ (s) | Kon (M−1 s−1) |
|---|---|---|---|---|---|
| 1G4-ESO9C | SLLMWITQC | 14 | 0.82 | 0.84 | 57 × 103 |
| 1G4-ESO4D | SLLDWITQV | 252 | 2.59 | 0.27 | 10 × 103 |
| ILA1α1β1-hTERT | ILAKFLHWL | 0.002 | 1.6 × 10−4 | 4260 | 8 × 104 |
| ILA1α1β1-hTERT-6A | ILAKFAHWL | 2 | 1.4 × 10−2 | 49 | 6 × 103 |
| ILA1-hTERT-6A | ILAKFAHWL | No binding | |||
| TCR | Mutation | CDR2β49-54 | CDR3α104-119 | ||
| ILA1 | Wild type | SVGAGI | CAVDSATSGTYKYIFG | ||
| ILA1α1β1 | CDR2&3 | SIHPEY | CAVDSATALPYGYIFG | ||
| GROOVE | ESO9C: HLA A*0201 | ESO4D: HLA A*0201 | hTERT: HLA A*0201 | hTERT-6A: HLA A*0201:ILA1α1β1 | hTERT-6A: HLA A*0201: ILA1 | ESO9C: HLA A*0201-Unbound |
|---|---|---|---|---|---|---|
| Tyr 7 | P1 | P1 | P1 | |||
| Glu 63 | P1 | P1 | P1 | P1 | P1 | P1 |
| Tyr 59 | P1 | |||||
| Lys 66 | P2 | P2 | P2 | P4 | P2/P4 | |
| His 70 | P3 | P3 | ||||
| Thr 73 | P8 | |||||
| Asp 77 | P8 | P8 | P8 | P8 | P8 | |
| Tyr 83 | P3 | |||||
| Tyr 84 | P9 | P9 | P9 | |||
| Tyr 99 | P2 | P2 | P2 | P2 | P2 | P2 |
| Thr 143 | P9 | P9 | P9 | P9 | P9 | P9 |
| Lys 146 | P9 | P9 | P9 | P9 | P9 | P9 |
| Trp 147 | P8 | P8 | P8 | P8 | P8 | P8 |
| Tyr 159 | P1 | P1 | P1 | P1 | P1 | |
| Tyr 171 | P1 | P1 | P1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alba, J.; Di Rienzo, L.; Milanetti, E.; Acuto, O.; D’Abramo, M. Molecular Dynamics Simulations Reveal Canonical Conformations in Different pMHC/TCR Interactions. Cells 2020, 9, 942. https://doi.org/10.3390/cells9040942
Alba J, Di Rienzo L, Milanetti E, Acuto O, D’Abramo M. Molecular Dynamics Simulations Reveal Canonical Conformations in Different pMHC/TCR Interactions. Cells. 2020; 9(4):942. https://doi.org/10.3390/cells9040942
Chicago/Turabian StyleAlba, Josephine, Lorenzo Di Rienzo, Edoardo Milanetti, Oreste Acuto, and Marco D’Abramo. 2020. "Molecular Dynamics Simulations Reveal Canonical Conformations in Different pMHC/TCR Interactions" Cells 9, no. 4: 942. https://doi.org/10.3390/cells9040942
APA StyleAlba, J., Di Rienzo, L., Milanetti, E., Acuto, O., & D’Abramo, M. (2020). Molecular Dynamics Simulations Reveal Canonical Conformations in Different pMHC/TCR Interactions. Cells, 9(4), 942. https://doi.org/10.3390/cells9040942