Protein and Mitochondria Quality Control Mechanisms and Cardiac Aging



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
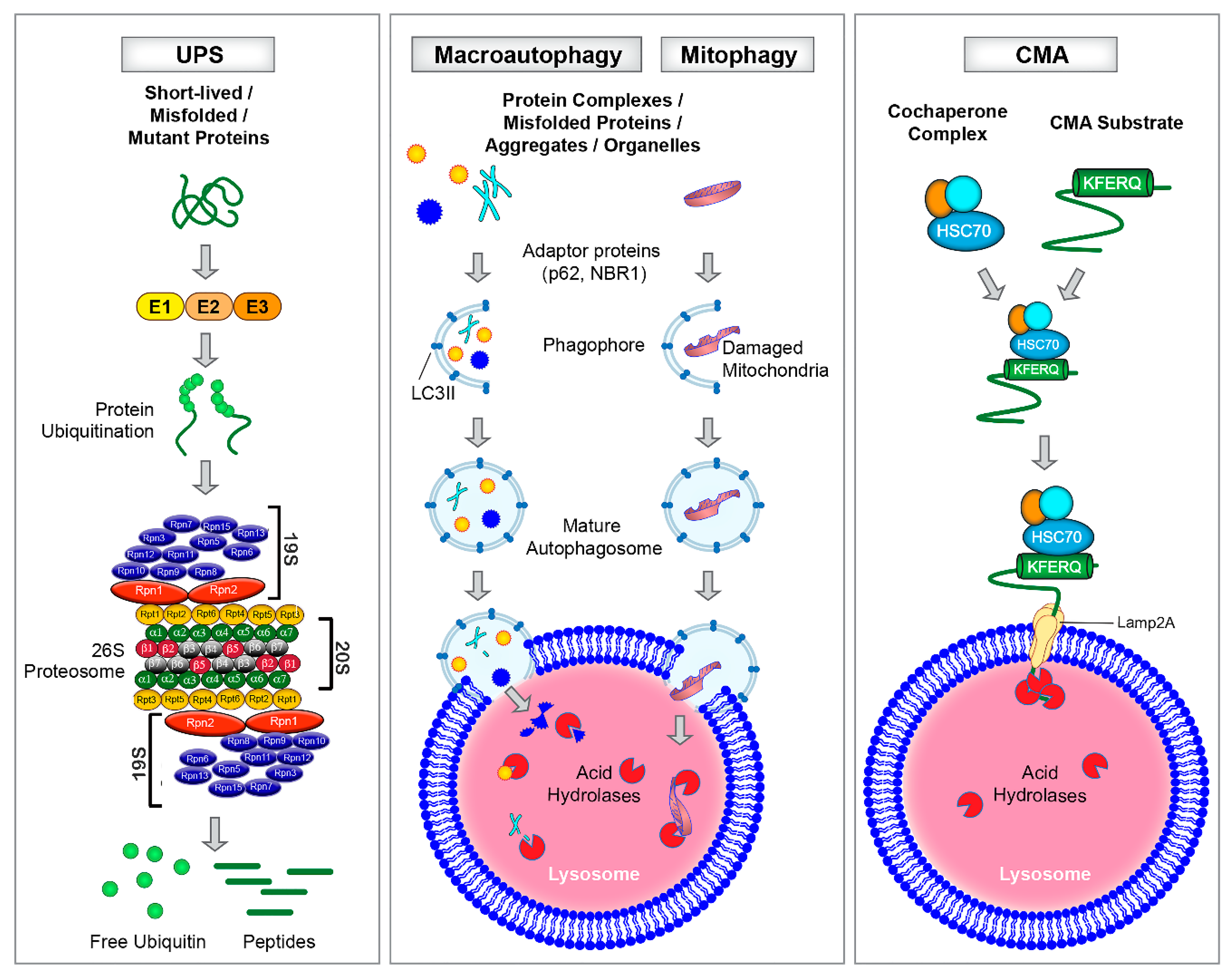
2. Protein and Mitochondrial Degradation Pathways in the Heart
2.1. Autophagy-Lysosomal Pathway
2.2. Chaperone-Mediated Autophagy (CMA)
2.3. Ubiquitin Proteasome System
2.4. The Interplay Among Protein and Mitochondria Degradation Pathways
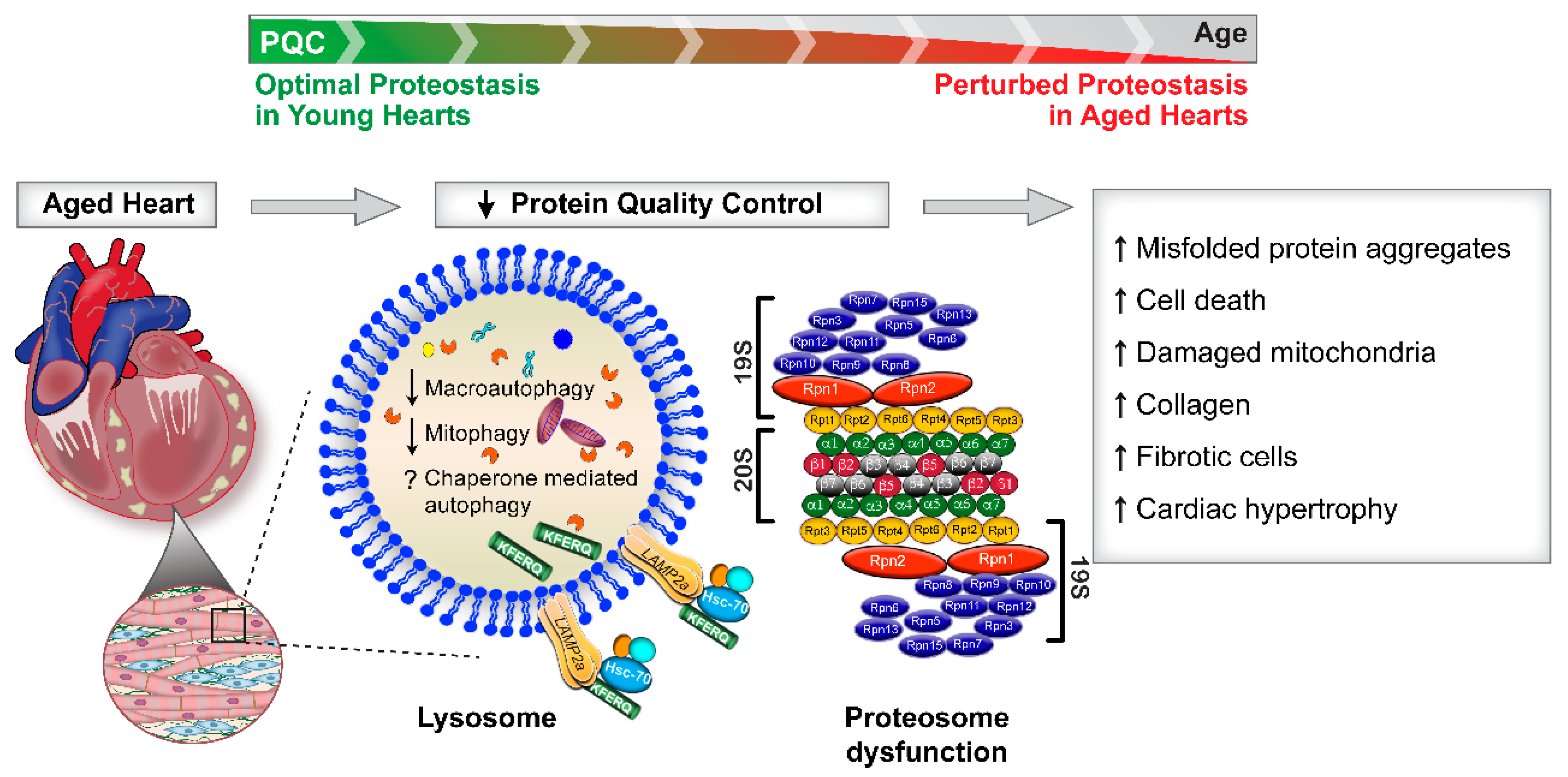
3. Mechanisms Whereby Suppression of Protein Quality Control Pathways Occurs during Cardiac Aging
3.1. Autophagy Suppression in Cardiac Aging
3.2. Mitophagy and Cardiac Aging
3.3. Chaperone-Mediated Autophagy (CMA) and Cardiac Aging
3.4. Ubiquitin Proteasome System (UPS) and Cardiac Aging
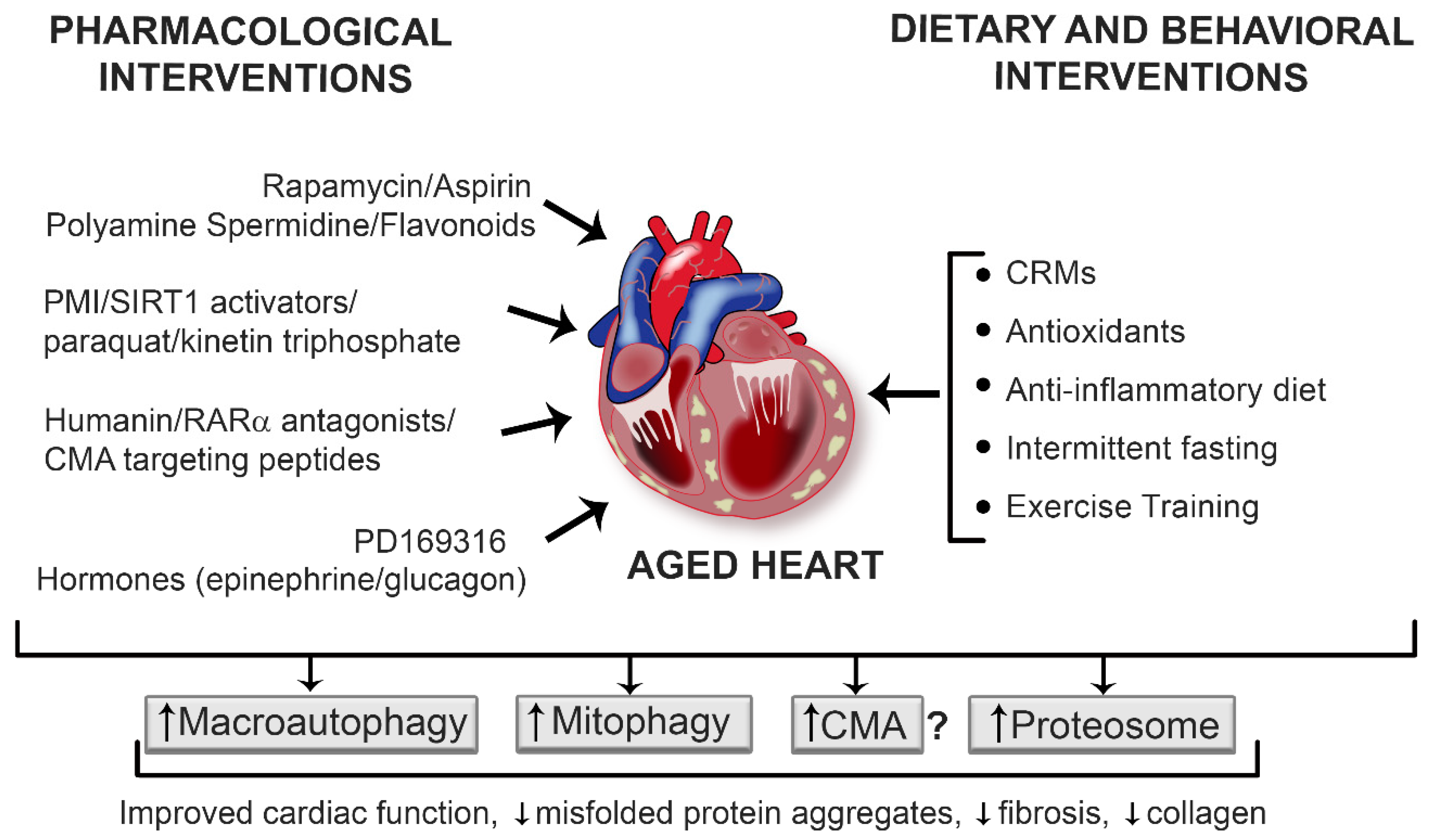
4. Strategies to Enhance Protein Quality Control Pathways
4.1. Pharmacological and Nutraceutical Interventions
4.2. Dietary and Lifestyle Interventions
4.3. Exercise
5. Conclusions and Future Directions
Funding
Acknowledgments
Conflicts of Interest
References
- Statistics FIFoA-R. Older Americans 2016: Key Indicators of Well-Being. In Federal Interagency Forum on Aging-Related Statistics; U.S. Government Printing Office: Washington, DC, USA, 2016. [Google Scholar]
- Benjamin, E.J.; Muntner, P.; Alonso, A.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Das, S.R.; et al. Heart disease and stroke statistics-2019 update: A report from the american heart association. Circulation 2019, 139, e56–e528. [Google Scholar] [PubMed]
- Strait, J.B.; Lakatta, E.G. Aging-associated cardiovascular changes and their relationship to heart failure. Heart Fail. Clin. 2012, 8, 143–164. [Google Scholar] [CrossRef] [PubMed]
- Fleg, J.L.; Strait, J. Age-associated changes in cardiovascular structure and function: A fertile milieu for future disease. Heart Fail. Rev. 2012, 17, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Paneni, F.; Diaz Canestro, C.; Libby, P.; Luscher, T.F.; Camici, G.G. The aging cardiovascular system: Understanding it at the cellular and clinical levels. J. Am. Coll. Cardiol. 2017, 69, 1952–1967. [Google Scholar] [CrossRef] [PubMed]
- Kovacic, J.C.; Moreno, P.; Nabel, E.G.; Hachinski, V.; Fuster, V. Cellular senescence, vascular disease, and aging: Part 2 of a 2-part review: Clinical vascular disease in the elderly. Circulation 2011, 123, 1900–1910. [Google Scholar] [CrossRef]
- Chiao, Y.A.; Rabinovitch, P.S. The aging heart. Cold Spring Harb. Perspect. Med. 2015, 5, a025148. [Google Scholar] [CrossRef]
- Biernacka, A.; Frangogiannis, N.G. Aging and cardiac fibrosis. Aging Dis. 2011, 2, 158–173. [Google Scholar]
- Lindsey, M.L.; Goshorn, D.K.; Squires, C.E.; Escobar, G.P.; Hendrick, J.W.; Mingoia, J.T.; Sweterlitsch, S.E.; Spinale, F.G. Age-dependent changes in myocardial matrix metalloproteinase/tissue inhibitor of metalloproteinase profiles and fibroblast function. Cardiovasc. Res. 2005, 66, 410–419. [Google Scholar] [CrossRef]
- Salminen, A.; Ojala, J.; Kaarniranta, K.; Kauppinen, A. Mitochondrial dysfunction and oxidative stress activate inflammasomes: Impact on the aging process and age-related diseases. Cell. Mol. Life Sci. Cmls 2012, 69, 2999–3013. [Google Scholar] [CrossRef]
- Chang, C.C.; Chang, Y.C.; Hu, W.L.; Hung, Y.C. Oxidative stress and salvia miltiorrhiza in aging-associated cardiovascular diseases. Oxidative Med. Cell. Longev. 2016, 2016, 4797102. [Google Scholar] [CrossRef]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef]
- Kajstura, J.; Cheng, W.; Sarangarajan, R.; Li, P.; Li, B.; Nitahara, J.A.; Chapnick, S.; Reiss, K.; Olivetti, G.; Anversa, P. Necrotic and apoptotic myocyte cell death in the aging heart of fischer 344 rats. Am. J. Physiol. 1996, 271, H1215–H1228. [Google Scholar] [CrossRef]
- Robert, V.; Besse, S.; Sabri, A.; Silvestre, J.S.; Assayag, P.; Nguyen, V.T.; Swynghedauw, B.; Delcayre, C. Differential regulation of matrix metalloproteinases associated with aging and hypertension in the rat heart. Lab. Investig. J. Tech. Methods Pathol. 1997, 76, 729–738. [Google Scholar]
- Bharath, L.P.; Mueller, R.; Li, Y.; Ruan, T.; Kunz, D.; Goodrich, R.; Mills, T.; Deeter, L.; Sargsyan, A.; Anandh Babu, P.V.; et al. Impairment of autophagy in endothelial cells prevents shear-stress-induced increases in nitric oxide bioavailability. Can. J. Physiol. Pharmacol. 2014, 92, 605–612. [Google Scholar] [CrossRef]
- Brady, N.R.; Hamacher-Brady, A.; Yuan, H.; Gottlieb, R.A. The autophagic response to nutrient deprivation in the hl-1 cardiac myocyte is modulated by bcl-2 and sarco/endoplasmic reticulum calcium stores. FEBS J. 2007, 274, 3184–3197. [Google Scholar] [CrossRef]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef]
- Ghosh, R.; Pattison, J.S. Macroautophagy and chaperone-mediated autophagy in heart failure: The known and the unknown. Oxidative Med. Cell. Longev. 2018, 2018, 8602041. [Google Scholar] [CrossRef]
- Bjorkoy, G.; Lamark, T.; Pankiv, S.; Overvatn, A.; Brech, A.; Johansen, T. Monitoring autophagic degradation of p62/sqstm1. Methods Enzymol. 2009, 452, 181–197. [Google Scholar]
- Kirkin, V.; McEwan, D.G.; Novak, I.; Dikic, I. A role for ubiquitin in selective autophagy. Mol. Cell 2009, 34, 259–269. [Google Scholar] [CrossRef]
- Oku, M.; Sakai, Y. Three distinct types of microautophagy based on membrane dynamics and molecular machineries. Bioessays News Rev. Mol. Cell. Dev. Biol. 2018, 40, e1800008. [Google Scholar] [CrossRef]
- Kubli, D.A.; Gustafsson, A.B. Mitochondria and mitophagy: The yin and yang of cell death control. Circ. Res. 2012, 111, 1208–1221. [Google Scholar] [CrossRef]
- Rodriguez-Enriquez, S.; Kim, I.; Currin, R.T.; Lemasters, J.J. Tracker dyes to probe mitochondrial autophagy (mitophagy) in rat hepatocytes. Autophagy 2006, 2, 39–46. [Google Scholar] [CrossRef]
- Kissova, I.; Deffieu, M.; Manon, S.; Camougrand, N. Uth1p is involved in the autophagic degradation of mitochondria. J. Biol. Chem. 2004, 279, 39068–39074. [Google Scholar] [CrossRef]
- Ding, W.X.; Yin, X.M. Mitophagy: Mechanisms, pathophysiological roles, and analysis. Biol. Chem. 2012, 393, 547–564. [Google Scholar] [CrossRef]
- Jin, S.M.; Youle, R.J. Pink1- and parkin-mediated mitophagy at a glance. J. Cell Sci. 2012, 125, 795–799. [Google Scholar] [CrossRef]
- Deas, E.; Plun-Favreau, H.; Wood, N.W. Pink1 function in health and disease. Embo Mol. Med. 2009, 1, 152–165. [Google Scholar] [CrossRef]
- Durcan, T.M.; Fon, E.A. The three ‘p’s of mitophagy: Parkin, pink1, and post-translational modifications. Genes Dev. 2015, 29, 989–999. [Google Scholar] [CrossRef]
- Solaini, G.; Baracca, A.; Lenaz, G.; Sgarbi, G. Hypoxia and mitochondrial oxidative metabolism. Biochim. Biophys. Acta 2010, 1797, 1171–1177. [Google Scholar] [CrossRef]
- Giordano, F.J. Oxygen, oxidative stress, hypoxia, and heart failure. J. Clin. Investig. 2005, 115, 500–508. [Google Scholar] [CrossRef]
- Lakatta, E.G. Arterial and cardiac aging: Major shareholders in cardiovascular disease enterprises: Part iii: Cellular and molecular clues to heart and arterial aging. Circulation 2003, 107, 490–497. [Google Scholar] [CrossRef]
- Cadenas, E.; Davies, K.J. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. Chaperone-mediated autophagy: A unique way to enter the lysosome world. Trends Cell Biol. 2012, 22, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M. Autophagy and aging--when “all you can eat” is yourself. Sci. Aging Knowl. Environ. Sage Ke 2003, 2003, pe25. [Google Scholar] [CrossRef]
- Li, W.; Yang, Q.; Mao, Z. Chaperone-mediated autophagy: Machinery, regulation and biological consequences. Cell. Mol. Life Sci. Cmls 2011, 68, 749–763. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Dice, J.F. Regulation of lamp2a levels in the lysosomal membrane. Traffic 2000, 1, 570–583. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Wong, E. Chaperone-mediated autophagy: Roles in disease and aging. Cell Res. 2014, 24, 92–104. [Google Scholar] [CrossRef]
- Zhou, J.; Chong, S.Y.; Lim, A.; Singh, B.K.; Sinha, R.A.; Salmon, A.B.; Yen, P.M. Changes in macroautophagy, chaperone-mediated autophagy, and mitochondrial metabolism in murine skeletal and cardiac muscle during aging. Aging 2017, 9, 583–599. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Yu, H.; Mim, C.; Matouschek, A. Regulated protein turnover: Snapshots of the proteasome in action. Nat. Rev. Mol. Cell Biol. 2014, 15, 122–133. [Google Scholar] [CrossRef]
- Schwartz, A.L.; Ciechanover, A. Targeting proteins for destruction by the ubiquitin system: Implications for human pathobiology. Annu. Rev. Pharmacol. Toxicol. 2009, 49, 73–96. [Google Scholar] [CrossRef]
- Voges, D.; Zwickl, P.; Baumeister, W. The 26s proteasome: A molecular machine designed for controlled proteolysis. Annu. Rev. Biochem. 1999, 68, 1015–1068. [Google Scholar] [CrossRef]
- Kumar Deshmukh, F.; Yaffe, D.; Olshina, M.A.; Ben-Nissan, G.; Sharon, M. The contribution of the 20s proteasome to proteostasis. Biomolecules 2019, 9, 190. [Google Scholar] [CrossRef]
- Zhang, T.; Ghaemmaghami, S. Global analysis of cellular protein flux quantifies the selectivity of basal autophagy. Autophagy 2016, 12, 1411–1412. [Google Scholar] [CrossRef]
- Ding, W.X.; Yin, X.M. Sorting, recognition and activation of the misfolded protein degradation pathways through macroautophagy and the proteasome. Autophagy 2008, 4, 141–150. [Google Scholar] [CrossRef]
- Webb, J.L.; Ravikumar, B.; Atkins, J.; Skepper, J.N.; Rubinsztein, D.C. Alpha-synuclein is degraded by both autophagy and the proteasome. J. Biol. Chem. 2003, 278, 25009–25013. [Google Scholar] [CrossRef]
- Pattison, J.S.; Robbins, J. Autophagy and proteotoxicity in cardiomyocytes. Autophagy 2011, 7, 1259–1260. [Google Scholar] [CrossRef]
- Zhang, H.; Pan, B.; Wu, P.; Parajuli, N.; Rekhter, M.D.; Goldberg, A.L.; Wang, X. Pde1 inhibition facilitates proteasomal degradation of misfolded proteins and protects against cardiac proteinopathy. Sci. Adv. 2019, 5, eaaw5870. [Google Scholar] [CrossRef]
- Cohen-Kaplan, V.; Livneh, I.; Avni, N.; Fabre, B.; Ziv, T.; Kwon, Y.T.; Ciechanover, A. P62- and ubiquitin-dependent stress-induced autophagy of the mammalian 26s proteasome. Proc. Natl. Acad. Sci. USA 2016, 113, E7490–E7499. [Google Scholar] [CrossRef]
- Blasiak, J.; Pawlowska, E.; Szczepanska, J.; Kaarniranta, K. Interplay between autophagy and the ubiquitin-proteasome system and its role in the pathogenesis of age-related macular degeneration. Int. J. Mol. Sci. 2019, 20, 210. [Google Scholar] [CrossRef]
- Lu, K.; den Brave, F.; Jentsch, S. Pathway choice between proteasomal and autophagic degradation. Autophagy 2017, 13, 1799–1800. [Google Scholar] [CrossRef]
- Wu, H.; Chen, S.; Ammar, A.B.; Xu, J.; Wu, Q.; Pan, K.; Zhang, J.; Hong, Y. Crosstalk between macroautophagy and chaperone-mediated autophagy: Implications for the treatment of neurological diseases. Mol. Neurobiol. 2015, 52, 1284–1296. [Google Scholar] [CrossRef]
- Zheng, Q.; Wang, X. Autophagy and the ubiquitin-proteasome system in cardiac dysfunction. Panminerva Med. 2010, 52, 9–25. [Google Scholar]
- Guo, F.; He, X.B.; Li, S.; Le, W. A central role for phosphorylated p38alpha in linking proteasome inhibition-induced apoptosis and autophagy. Mol. Neurobiol. 2017, 54, 7597–7609. [Google Scholar] [CrossRef]
- Zhang, Y.; Nicholatos, J.; Dreier, J.R.; Ricoult, S.J.; Widenmaier, S.B.; Hotamisligil, G.S.; Kwiatkowski, D.J.; Manning, B.D. Coordinated regulation of protein synthesis and degradation by mtorc1. Nature 2014, 513, 440–443. [Google Scholar] [CrossRef]
- Korolchuk, V.I.; Mansilla, A.; Menzies, F.M.; Rubinsztein, D.C. Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol. Cell 2009, 33, 517–527. [Google Scholar] [CrossRef]
- Yoshii, S.R.; Kishi, C.; Ishihara, N.; Mizushima, N. Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane. J. Biol. Chem. 2011, 286, 19630–19640. [Google Scholar] [CrossRef]
- Nakagawa, T.; Shirane, M.; Iemura, S.; Natsume, T.; Nakayama, K.I. Anchoring of the 26s proteasome to the organellar membrane by fkbp38. Genes Cells Devoted Mol. Cell. Mech. 2007, 12, 709–719. [Google Scholar] [CrossRef]
- Agarraberes, F.A.; Terlecky, S.R.; Dice, J.F. An intralysosomal hsp70 is required for a selective pathway of lysosomal protein degradation. J. Cell Biol. 1997, 137, 825–834. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Knecht, E.; Terlecky, S.R.; Dice, J.F. Activation of a selective pathway of lysosomal proteolysis in rat liver by prolonged starvation. Am. J. Physiol. 1995, 269, C1200–C1208. [Google Scholar] [CrossRef]
- Massey, A.C.; Kaushik, S.; Sovak, G.; Kiffin, R.; Cuervo, A.M. Consequences of the selective blockage of chaperone-mediated autophagy. Proc. Natl. Acad. Sci. USA 2006, 103, 5805–5810. [Google Scholar] [CrossRef]
- Kaushik, S.; Massey, A.C.; Mizushima, N.; Cuervo, A.M. Constitutive activation of chaperone-mediated autophagy in cells with impaired macroautophagy. Mol. Biol. Cell 2008, 19, 2179–2192. [Google Scholar] [CrossRef]
- Wang, C.; Wang, H.; Zhang, D.; Luo, W.; Liu, R.; Xu, D.; Diao, L.; Liao, L.; Liu, Z. Phosphorylation of ulk1 affects autophagosome fusion and links chaperone-mediated autophagy to macroautophagy. Nat. Commun. 2018, 9, 3492. [Google Scholar] [CrossRef]
- Taneike, M.; Yamaguchi, O.; Nakai, A.; Hikoso, S.; Takeda, T.; Mizote, I.; Oka, T.; Tamai, T.; Oyabu, J.; Murakawa, T.; et al. Inhibition of autophagy in the heart induces age-related cardiomyopathy. Autophagy 2010, 6, 600–606. [Google Scholar] [CrossRef]
- Hua, Y.; Zhang, Y.; Ceylan-Isik, A.F.; Wold, L.E.; Nunn, J.M.; Ren, J. Chronic akt activation accentuates aging-induced cardiac hypertrophy and myocardial contractile dysfunction: Role of autophagy. Basic Res. Cardiol. 2011, 106, 1173–1191. [Google Scholar] [CrossRef]
- Ren, J.; Yang, L.; Zhu, L.; Xu, X.; Ceylan, A.F.; Guo, W.; Yang, J.; Zhang, Y. Akt2 ablation prolongs life span and improves myocardial contractile function with adaptive cardiac remodeling: Role of sirt1-mediated autophagy regulation. Aging Cell 2017, 16, 976–987. [Google Scholar] [CrossRef]
- Linton, P.J.; Gurney, M.; Sengstock, D.; Mentzer, R.M.; Gottlieb, R.A. This old heart: Cardiac aging and autophagy. J. Mol. Cell Cardiol. 2015, 83, 44–54. [Google Scholar] [CrossRef]
- Hoshino, A.; Mita, Y.; Okawa, Y.; Ariyoshi, M.; Iwai-Kanai, E.; Ueyama, T.; Ikeda, K.; Ogata, T.; Matoba, S. Cytosolic p53 inhibits parkin-mediated mitophagy and promotes mitochondrial dysfunction in the mouse heart. Nat. Commun. 2013, 4, 2308. [Google Scholar] [CrossRef]
- Boyle, A.J.; Shih, H.; Hwang, J.; Ye, J.; Lee, B.; Zhang, Y.; Kwon, D.; Jun, K.; Zheng, D.; Sievers, R.; et al. Cardiomyopathy of aging in the mammalian heart is characterized by myocardial hypertrophy, fibrosis and a predisposition towards cardiomyocyte apoptosis and autophagy. Exp. Gerontol. 2011, 46, 549–559. [Google Scholar] [CrossRef]
- Inuzuka, Y.; Okuda, J.; Kawashima, T.; Kato, T.; Niizuma, S.; Tamaki, Y.; Iwanaga, Y.; Yoshida, Y.; Kosugi, R.; Watanabe-Maeda, K.; et al. Suppression of phosphoinositide 3-kinase prevents cardiac aging in mice. Circulation 2009, 120, 1695–1703. [Google Scholar] [CrossRef]
- Zhou, J.; Freeman, T.A.; Ahmad, F.; Shang, X.; Mangano, E.; Gao, E.; Farber, J.; Wang, Y.; Ma, X.L.; Woodgett, J.; et al. Gsk-3alpha is a central regulator of age-related pathologies in mice. J. Clin. Investig. 2013, 123, 1821–1832. [Google Scholar] [CrossRef]
- Li, Z.; Song, Y.; Liu, L.; Hou, N.; An, X.; Zhan, D.; Li, Y.; Zhou, L.; Li, P.; Yu, L.; et al. Mir-199a impairs autophagy and induces cardiac hypertrophy through mtor activation. Cell Death Differ. 2017, 24, 1205–1213. [Google Scholar] [CrossRef]
- Taneike, M.; Nishida, K.; Omiya, S.; Zarrinpashneh, E.; Misaka, T.; Kitazume-Taneike, R.; Austin, R.; Takaoka, M.; Yamaguchi, O.; Gambello, M.J.; et al. Mtor hyperactivation by ablation of tuberous sclerosis complex 2 in the mouse heart induces cardiac dysfunction with the increased number of small mitochondria mediated through the down-regulation of autophagy. PLoS ONE 2016, 11, e0152628. [Google Scholar] [CrossRef]
- Upadhyay, A.; Moss-Taylor, L.; Kim, M.J.; Ghosh, A.C.; O’Connor, M.B. Tgf-beta family signaling in drosophila. Cold Spring Harbor Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef]
- Marino, G.; Lopez-Otin, C. Autophagy: Molecular mechanisms, physiological functions and relevance in human pathology. Cell. Mol. Life Sci. CMLS 2004, 61, 1439–1454. [Google Scholar] [CrossRef]
- Chang, K.; Kang, P.; Liu, Y.; Huang, K.; Miao, T.; Sagona, A.P.; Nezis, I.P.; Bodmer, R.; Ocorr, K.; Bai, H. Tgfb-inhb/activin signaling regulates age-dependent autophagy and cardiac health through inhibition of mtorc2. Autophagy 2019, 1–16. [Google Scholar] [CrossRef]
- Mauro, A.G.; Bonaventura, A.; Mezzaroma, E.; Quader, M.; Toldo, S. Nlrp3 inflammasome in acute myocardial infarction. J. Cardiovasc. Pharmacol. 2019, 74, 175–187. [Google Scholar] [CrossRef]
- Buckley, L.F.; Libby, P. Inhibiting nlrp3 inflammasome activity in acute myocardial infarction: A review of pharmacologic agents and clinical outcomes. J. Cardiovasc. Pharmacol. 2019, 74, 297–305. [Google Scholar] [CrossRef]
- Bullon, P.; Cano-Garcia, F.J.; Alcocer-Gomez, E.; Varela-Lopez, A.; Roman-Malo, L.; Ruiz-Salmeron, R.J.; Quiles, J.L.; Navarro-Pando, J.M.; Battino, M.; Ruiz-Cabello, J.; et al. Could nlrp3-inflammasome be a cardiovascular risk biomarker in acute myocardial infarction patients? Antioxid. Redox Signal. 2017, 27, 269–275. [Google Scholar] [CrossRef]
- Marin-Aguilar, F.; Lechuga-Vieco, A.V.; Alcocer-Gomez, E.; Castejon-Vega, B.; Lucas, J.; Garrido, C.; Peralta-Garcia, A.; Perez-Pulido, A.J.; Varela-Lopez, A.; Quiles, J.L.; et al. Nlrp3 inflammasome suppression improves longevity and prevents cardiac aging in male mice. Aging Cell 2020, 19, e13050. [Google Scholar] [CrossRef]
- Chang, J.; Xie, M.; Shah, V.R.; Schneider, M.D.; Entman, M.L.; Wei, L.; Schwartz, R.J. Activation of rho-associated coiled-coil protein kinase 1 (rock-1) by caspase-3 cleavage plays an essential role in cardiac myocyte apoptosis. Proc. Natl. Acad. Sci. USA 2006, 103, 14495–14500. [Google Scholar] [CrossRef]
- Shi, J.; Zhang, L.; Wei, L. Rho-kinase in development and heart failure: Insights from genetic models. Pediatric Cardiol. 2011, 32, 297–304. [Google Scholar] [CrossRef]
- Shi, J.J.; Surma, M.; Yang, Y.; Wei, L. Disruption of both rock1 and rock2 genes in cardiomyocytes promotes autophagy and reduces cardiac fibrosis during aging. FASEB J. 2019, 33, 7348–7362. [Google Scholar] [CrossRef]
- Filomeni, G.; De Zio, D.; Cecconi, F. Oxidative stress and autophagy: The clash between damage and metabolic needs. Cell Death Differ. 2015, 22, 377–388. [Google Scholar] [CrossRef]
- Shirakabe, A.; Ikeda, Y.; Sciarretta, S.; Zablocki, D.K.; Sadoshima, J. Aging and autophagy in the heart. Circ. Res. 2016, 118, 1563–1576. [Google Scholar] [CrossRef]
- Cervantes Gracia, K.; Llanas-Cornejo, D.; Husi, H. Cvd and oxidative stress. J. Clin. Med. 2017, 6, 22. [Google Scholar] [CrossRef]
- Frudd, K.; Burgoyne, T.; Burgoyne, J.R. Oxidation of atg3 and atg7 mediates inhibition of autophagy. Nat. Commun. 2018, 9, 95. [Google Scholar] [CrossRef]
- Gouveia, M.; Xia, K.; Colon, W.; Vieira, S.I.; Ribeiro, F. Protein aggregation, cardiovascular diseases, and exercise training: Where do we stand? Ageing Res. Rev. 2017, 40, 1–10. [Google Scholar] [CrossRef]
- Ayyadevara, S.; Mercanti, F.; Wang, X.; Mackintosh, S.G.; Tackett, A.J.; Prayaga, S.V.; Romeo, F.; Shmookler Reis, R.J.; Mehta, J.L. Age- and hypertension-associated protein aggregates in mouse heart have similar proteomic profiles. Hypertension 2016, 67, 1006–1013. [Google Scholar] [CrossRef]
- Scaffidi, P.; Misteli, T. Lamin a-dependent nuclear defects in human aging. Science. 2006, 312, 1059–1063. [Google Scholar] [CrossRef]
- Henkel, V.; Schurmanns, L.; Brunner, M.; Hamann, A.; Osiewacz, H.D. Role of sorting nexin paatg24 in autophagy, aging and development of podospora anserina. Mech. Ageing Dev. 2020, 186, 111211. [Google Scholar] [CrossRef]
- Heller, F.; Dabaj, I.; Mah, J.K.; Bergounioux, J.; Essid, A.; Bonnemann, C.G.; Rutkowski, A.; Bonne, G.; Quijano-Roy, S.; Wahbi, K. Cardiac manifestations of congenital lmna-related muscular dystrophy in children: Three case reports and recommendations for care. Cardiol. Young 2017, 27, 1076–1082. [Google Scholar] [CrossRef]
- Bhide, S.; Trujillo, A.S.; O’Connor, M.T.; Young, G.H.; Cryderman, D.E.; Chandran, S.; Nikravesh, M.; Wallrath, L.L.; Melkani, G.C. Increasing autophagy and blocking nrf2 suppress laminopathy-induced age-dependent cardiac dysfunction and shortened lifespan. Aging Cell 2018, 17, e12747. [Google Scholar] [CrossRef]
- Bravo-San Pedro, J.M.; Kroemer, G.; Galluzzi, L. Autophagy and mitophagy in cardiovascular disease. Circ. Res. 2017, 120, 1812–1824. [Google Scholar] [CrossRef]
- Moyzis, A.G.; Sadoshima, J.; Gustafsson, A.B. Mending a broken heart: The role of mitophagy in cardioprotection. Am. J. Physiol.-Heart C 2015, 308, H183–H192. [Google Scholar] [CrossRef]
- Shires, S.E.; Gustafsson, A.B. Mitophagy and heart failure. J. Mol. Med. 2015, 93, 253–262. [Google Scholar] [CrossRef]
- Wang, B.; Nie, J.; Wu, L.; Hu, Y.; Wen, Z.; Dong, L.; Zou, M.H.; Chen, C.; Wang, D.W. Ampkalpha2 protects against the development of heart failure by enhancing mitophagy via pink1 phosphorylation. Circ. Res. 2018, 122, 712–729. [Google Scholar] [CrossRef]
- Nakai, A.; Yamaguchi, O.; Takeda, T.; Higuchi, Y.; Hikoso, S.; Taniike, M.; Omiya, S.; Mizote, I.; Matsumura, Y.; Asahi, M.; et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat. Med. 2007, 13, 619–624. [Google Scholar] [CrossRef]
- Chen, Y.; Dorn, G.W. Pink1-phosphorylated mitofusin 2 is a parkin receptor for culling damaged mitochondria. Science 2013, 340, 471–475. [Google Scholar] [CrossRef]
- Bhandari, P.; Song, M.; Chen, Y.; Burelle, Y.; Dorn, G.W. Mitochondrial contagion induced by parkin deficiency in drosophila hearts and its containment by suppressing mitofusin. Circ. Res. 2014, 114, 257–265. [Google Scholar] [CrossRef]
- Lin, S.; Wang, Y.; Zhang, X.; Kong, Q.; Li, C.; Li, Y.; Ding, Z.; Liu, L. Hsp27 alleviates cardiac aging in mice via a mechanism involving antioxidation and mitophagy activation. Oxidative Med. Cell. Longev. 2016, 2016, 2586706. [Google Scholar] [CrossRef]
- Yang, S.; Long, L.H.; Li, D.; Zhang, J.K.; Jin, S.; Wang, F.; Chen, J.G. Beta-guanidinopropionic acid extends the lifespan of drosophila melanogaster via an amp-activated protein kinase-dependent increase in autophagy. Aging Cell 2015, 14, 1024–1033. [Google Scholar] [CrossRef]
- Wang, S.; Kandadi, M.R.; Ren, J. Double knockout of akt2 and ampk predisposes cardiac aging without affecting lifespan: Role of autophagy and mitophagy. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1865–1875. [Google Scholar] [CrossRef]
- Xu, P.; Damschroder, D.; Zhang, M.; Ryall, K.A.; Adler, P.N.; Saucerman, J.J.; Wessells, R.J.; Yan, Z. Atg2, atg9 and atg18 in mitochondrial integrity, cardiac function and healthspan in drosophila. J. Mol. Cell. Cardiol. 2019, 127, 116–124. [Google Scholar] [CrossRef]
- Laker, R.C.; Xu, P.; Ryall, K.A.; Sujkowski, A.; Kenwood, B.M.; Chain, K.H.; Zhang, M.; Royal, M.A.; Hoehn, K.L.; Driscoll, M.; et al. A novel mitotimer reporter gene for mitochondrial content, structure, stress, and damage in vivo. J. Biol. Chem. 2014, 289, 12005–12015. [Google Scholar] [CrossRef]
- Terskikh, A.; Fradkov, A.; Ermakova, G.; Zaraisky, A.; Tan, P.; Kajava, A.V.; Zhao, X.; Lukyanov, S.; Matz, M.; Kim, S.; et al. “Fluorescent timer”: Protein that changes color with time. Science 2000, 290, 1585–1588. [Google Scholar] [CrossRef]
- Diot, A.; Morten, K.; Poulton, J. Mitophagy plays a central role in mitochondrial ageing. Mamm. Genome Off. J. Int. Mamm. Genome Soc. 2016, 27, 381–395. [Google Scholar] [CrossRef]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Manzella, N.; Santin, Y.; Maggiorani, D.; Martini, H.; Douin-Echinard, V.; Passos, J.F.; Lezoualc’h, F.; Binda, C.; Parini, A.; Mialet-Perez, J. Monoamine oxidase-a is a novel driver of stress-induced premature senescence through inhibition of parkin-mediated mitophagy. Aging Cell 2018, 17, e12811. [Google Scholar] [CrossRef]
- Martinez-Lopez, N.; Athonvarangkul, D.; Singh, R. Autophagy and aging. Adv. Exp. Med. Biol. 2015, 847, 73–87. [Google Scholar]
- Cuervo, A.M.; Dice, J.F. Age-related decline in chaperone-mediated autophagy. J. Biol. Chem. 2000, 275, 31505–31513. [Google Scholar] [CrossRef]
- Tanaka, Y.; Guhde, G.; Suter, A.; Eskelinen, E.L.; Hartmann, D.; Lullmann-Rauch, R.; Janssen, P.M.; Blanz, J.; von Figura, K.; Saftig, P. Accumulation of autophagic vacuoles and cardiomyopathy in lamp-2-deficient mice. Nature 2000, 406, 902–906. [Google Scholar] [CrossRef]
- Raynes, R.; Pomatto, L.C.; Davies, K.J. Degradation of oxidized proteins by the proteasome: Distinguishing between the 20s, 26s, and immunoproteasome proteolytic pathways. Mol. Asp. Med. 2016, 50, 41–55. [Google Scholar] [CrossRef] [PubMed]
- Kisselev, A.F.; Goldberg, A.L. Monitoring activity and inhibition of 26s proteasomes with fluorogenic peptide substrates. Methods Enzymol. 2005, 398, 364–378. [Google Scholar]
- Kumarapeli, A.R.; Horak, K.M.; Glasford, J.W.; Li, J.; Chen, Q.; Liu, J.; Zheng, H.; Wang, X. A novel transgenic mouse model reveals deregulation of the ubiquitin-proteasome system in the heart by doxorubicin. FASEB J. 2005, 19, 2051–2053. [Google Scholar] [CrossRef] [PubMed]
- Bulteau, A.L.; Szweda, L.I.; Friguet, B. Age-dependent declines in proteasome activity in the heart. Arch. Biochem. Biophys. 2002, 397, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Blice-Baum, A.C.; Zambon, A.C.; Kaushik, G.; Viswanathan, M.C.; Engler, A.J.; Bodmer, R.; Cammarato, A. Modest overexpression of foxo maintains cardiac proteostasis and ameliorates age-associated functional decline. Aging Cell 2017, 16, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Webb, A.E.; Brunet, A. Foxo transcription factors: Key regulators of cellular quality control. Trends Biochem. Sci. 2014, 39, 159–169. [Google Scholar] [CrossRef]
- Nakayama, H.; Nishida, K.; Otsu, K. Macromolecular degradation systems and cardiovascular aging. Circ. Res. 2016, 118, 1577–1592. [Google Scholar] [CrossRef]
- Riehle, C.; Wende, A.R.; Sena, S.; Pires, K.M.; Pereira, R.O.; Zhu, Y.; Bugger, H.; Frank, D.; Bevins, J.; Chen, D.; et al. Insulin receptor substrate signaling suppresses neonatal autophagy in the heart. J. Clin. Investig. 2013, 123, 5319–5333. [Google Scholar] [CrossRef]
- Flynn, J.M.; O’Leary, M.N.; Zambataro, C.A.; Academia, E.C.; Presley, M.P.; Garrett, B.J.; Zykovich, A.; Mooney, S.D.; Strong, R.; Rosen, C.J.; et al. Late-life rapamycin treatment reverses age-related heart dysfunction. Aging Cell 2013, 12, 851–862. [Google Scholar] [CrossRef]
- Gu, J.; Hu, W.; Song, Z.P.; Chen, Y.G.; Zhang, D.D.; Wang, C.Q. Rapamycin inhibits cardiac hypertrophy by promoting autophagy via the mek/erk/beclin-1 pathway. Front. Physiol. 2016, 7, 104. [Google Scholar] [CrossRef]
- Dai, D.F.; Karunadharma, P.P.; Chiao, Y.A.; Basisty, N.; Crispin, D.; Hsieh, E.J.; Chen, T.; Gu, H.; Djukovic, D.; Raftery, D.; et al. Altered proteome turnover and remodeling by short-term caloric restriction or rapamycin rejuvenate the aging heart. Aging Cell 2014, 13, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Quarles, E.; Basisty, N.; Chiao, Y.A.; Merrihew, G.; Gu, H.; Sweetwyne, M.T.; Fredrickson, J.; Nguyen, N.H.; Razumova, M.; Kooiker, K.; et al. Rapamycin persistently improves cardiac function in aged, male and female mice, even following cessation of treatment. Aging Cell 2020, 19, e13086. [Google Scholar] [CrossRef] [PubMed]
- Chiao, Y.A.; Kolwicz, S.C.; Basisty, N.; Gagnidze, A.; Zhang, J.; Gu, H.; Djukovic, D.; Beyer, R.P.; Raftery, D.; MacCoss, M.; et al. Rapamycin transiently induces mitochondrial remodeling to reprogram energy metabolism in old hearts. Aging 2016, 8, 314–327. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Ali, S.M.; Sengupta, S.; Sheen, J.H.; Hsu, P.P.; Bagley, A.F.; Markhard, A.L.; Sabatini, D.M. Prolonged rapamycin treatment inhibits mtorc2 assembly and akt/pkb. Mol. Cell 2006, 22, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Sciarretta, S.; Zhai, P.; Maejima, Y.; Del Re, D.P.; Nagarajan, N.; Yee, D.; Liu, T.; Magnuson, M.A.; Volpe, M.; Frati, G.; et al. Mtorc2 regulates cardiac response to stress by inhibiting mst1. Cell Rep. 2015, 11, 125–136. [Google Scholar] [CrossRef]
- Gyurus, E.; Kaposztas, Z.; Kahan, B.D. Sirolimus therapy predisposes to new-onset diabetes mellitus after renal transplantation: A long-term analysis of various treatment regimens. Transplant. Proc. 2011, 43, 1583–1592. [Google Scholar] [CrossRef]
- Yoon, H.Y.; Hwang, J.J.; Kim, D.S.; Song, J.W. Efficacy and safety of low-dose sirolimus in lymphangioleiomyomatosis. Orphanet J. Rare Dis. 2018, 13, 204. [Google Scholar] [CrossRef]
- Turdi, S.; Fan, X.; Li, J.; Zhao, J.; Huff, A.F.; Du, M.; Ren, J. Amp-activated protein kinase deficiency exacerbates aging-induced myocardial contractile dysfunction. Aging Cell 2010, 9, 592–606. [Google Scholar] [CrossRef]
- Pu, Y.; Zhang, H.; Wang, P.; Zhao, Y.; Li, Q.; Wei, X.; Cui, Y.; Sun, J.; Shang, Q.; Liu, D.; et al. Dietary curcumin ameliorates aging-related cerebrovascular dysfunction through the ampk/uncoupling protein 2 pathway. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2013, 32, 1167–1177. [Google Scholar] [CrossRef]
- Hawley, S.A.; Gadalla, A.E.; Olsen, G.S.; Hardie, D.G. The antidiabetic drug metformin activates the amp-activated protein kinase cascade via an adenine nucleotide-independent mechanism. Diabetes 2002, 51, 2420–2425. [Google Scholar] [CrossRef]
- Xie, Z.; Lau, K.; Eby, B.; Lozano, P.; He, C.; Pennington, B.; Li, H.; Rathi, S.; Dong, Y.; Tian, R.; et al. Improvement of cardiac functions by chronic metformin treatment is associated with enhanced cardiac autophagy in diabetic ove26 mice. Diabetes 2011, 60, 1770–1778. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Song, Y.; Li, H.; Shen, Q.; Shen, J.; An, X.; Wu, J.; Zhang, J.; Wu, Y.; Xiao, H.; et al. Exacerbated cardiac fibrosis induced by beta-adrenergic activation in old mice due to decreased ampk activity. Clin. Exp. Pharmacol. Physiol. 2016, 43, 1029–1037. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Mu, N.; Gu, C.; Liu, M.; Yang, Z.; Yin, Y.; Chen, M.; Wang, Y.; Han, Y.; Yu, L.; et al. Metformin mediates cardioprotection against aging-induced ischemic necroptosis. Aging Cell 2020, 19, e13096. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.P.; Lorenzo, C.; Habib, S.L.; Jo, B.; Espinoza, S.E. Differential effects of metformin on age related comorbidities in older men with type 2 diabetes. J. Diabetes Complicat. 2017, 31, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Din, F.V.; Valanciute, A.; Houde, V.P.; Zibrova, D.; Green, K.A.; Sakamoto, K.; Alessi, D.R.; Dunlop, M.G. Aspirin inhibits mtor signaling, activates amp-activated protein kinase, and induces autophagy in colorectal cancer cells. Gastroenterology 2012, 142, 1504–1515.e3. [Google Scholar] [CrossRef] [PubMed]
- Tung, B.T.; Rodriguez-Bies, E.; Thanh, H.N.; Le-Thi-Thu, H.; Navas, P.; Sanchez, V.M.; Lopez-Lluch, G. Organ and tissue-dependent effect of resveratrol and exercise on antioxidant defenses of old mice. Aging Clin. Exp. Res. 2015, 27, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, T.; Knauer, H.; Schauer, A.; Buttner, S.; Ruckenstuhl, C.; Carmona-Gutierrez, D.; Ring, J.; Schroeder, S.; Magnes, C.; Antonacci, L.; et al. Induction of autophagy by spermidine promotes longevity. Nat. Cell Biol. 2009, 11, 1305–1314. [Google Scholar] [CrossRef]
- Gupta, V.K.; Scheunemann, L.; Eisenberg, T.; Mertel, S.; Bhukel, A.; Koemans, T.S.; Kramer, J.M.; Liu, K.S.; Schroeder, S.; Stunnenberg, H.G.; et al. Restoring polyamines protects from age-induced memory impairment in an autophagy-dependent manner. Nat. Neurosci. 2013, 16, 1453–1460. [Google Scholar] [CrossRef]
- Wang, I.F.; Guo, B.S.; Liu, Y.C.; Wu, C.C.; Yang, C.H.; Tsai, K.J.; Shen, C.K. Autophagy activators rescue and alleviate pathogenesis of a mouse model with proteinopathies of the tar DNA-binding protein 43. Proc. Natl. Acad. Sci. USA 2012, 109, 15024–15029. [Google Scholar] [CrossRef]
- Eisenberg, T.; Abdellatif, M.; Schroeder, S.; Primessnig, U.; Stekovic, S.; Pendl, T.; Harger, A.; Schipke, J.; Zimmermann, A.; Schmidt, A.; et al. Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat. Med. 2016, 22, 1428–1438. [Google Scholar] [CrossRef]
- Kiechl, S.; Pechlaner, R.; Willeit, P.; Notdurfter, M.; Paulweber, B.; Willeit, K.; Werner, P.; Ruckenstuhl, C.; Iglseder, B.; Weger, S.; et al. Higher spermidine intake is linked to lower mortality: A prospective population-based study. Am. J. Clin. Nutr. 2018, 108, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Wirth, M.; Schwarz, C.; Benson, G.; Horn, N.; Buchert, R.; Lange, C.; Kobe, T.; Hetzer, S.; Maglione, M.; Michael, E.; et al. Effects of spermidine supplementation on cognition and biomarkers in older adults with subjective cognitive decline (smartage)-study protocol for a randomized controlled trial. Alzheimer Res. Therapy 2019, 11, 36. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Gutierrez, D.; Zimmermann, A.; Kainz, K.; Pietrocola, F.; Chen, G.; Maglioni, S.; Schiavi, A.; Nah, J.; Mertel, S.; Beuschel, C.B.; et al. The flavonoid 4,4′-dimethoxychalcone promotes autophagy-dependent longevity across species. Nat. Commun. 2019, 10, 651. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, A.; Kainz, K.; Hofer, S.J.; Bauer, M.A.; Schroeder, S.; Dengjel, J.; Pietrocola, F.; Kepp, O.; Ruckenstuhl, C.; Eisenberg, T.; et al. 4,4′dimethoxychalcone: A natural flavonoid that promotes health through autophagy-dependent and -independent effects. Autophagy 2019, 15, 1662–1664. [Google Scholar] [CrossRef]
- Georgakopoulos, N.D.; Wells, G.; Campanella, M. The pharmacological regulation of cellular mitophagy. Nat. Chem. Biol. 2017, 13, 136–146. [Google Scholar] [CrossRef]
- East, D.A.; Fagiani, F.; Crosby, J.; Georgakopoulos, N.D.; Bertrand, H.; Schaap, M.; Fowkes, A.; Wells, G.; Campanella, M. Pmi: A deltapsim independent pharmacological regulator of mitophagy. Chem. Biol. 2014, 21, 1585–1596. [Google Scholar] [CrossRef]
- Hertz, N.T.; Berthet, A.; Sos, M.L.; Thorn, K.S.; Burlingame, A.L.; Nakamura, K.; Shokat, K.M. A neo-substrate that amplifies catalytic activity of parkinson’s-disease-related kinase pink1. Cell 2013, 154, 737–747. [Google Scholar] [CrossRef]
- Niikura, T.; Chiba, T.; Aiso, S.; Matsuoka, M.; Nishimoto, I. Humanin: After the discovery. Mol. Neurobiol. 2004, 30, 327–340. [Google Scholar] [CrossRef]
- Muzumdar, R.H.; Huffman, D.M.; Calvert, J.W.; Jha, S.; Weinberg, Y.; Cui, L.; Nemkal, A.; Atzmon, G.; Klein, L.; Gundewar, S.; et al. Acute humanin therapy attenuates myocardial ischemia and reperfusion injury in mice. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1940–1948. [Google Scholar] [CrossRef]
- Gong, Z.; Tasset, I.; Diaz, A.; Anguiano, J.; Tas, E.; Cui, L.; Kuliawat, R.; Liu, H.; Kuhn, B.; Cuervo, A.M.; et al. Humanin is an endogenous activator of chaperone-mediated autophagy. J. Cell Biol. 2018, 217, 635–647. [Google Scholar] [CrossRef]
- Koga, H.; Martinez-Vicente, M.; Macian, F.; Verkhusha, V.V.; Cuervo, A.M. A photoconvertible fluorescent reporter to track chaperone-mediated autophagy. Nat. Commun. 2011, 2, 386. [Google Scholar] [CrossRef] [PubMed]
- Qin, Q.; Mehta, H.; Yen, K.; Navarrete, G.; Brandhorst, S.; Wan, J.; Delrio, S.; Zhang, X.; Lerman, L.O.; Cohen, P.; et al. Chronic treatment with the mitochondrial peptide humanin prevents age-related myocardial fibrosis in mice. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1127–H1136. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Jin, W.Y.; Lu, J.; Wang, J.; Wang, Y.T. Rapid and reversible knockdown of endogenous proteins by peptide-directed lysosomal degradation. Nat. Neurosci. 2014, 17, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Leestemaker, Y.; de Jong, A.; Witting, K.F.; Penning, R.; Schuurman, K.; Rodenko, B.; Zaal, E.A.; van de Kooij, B.; Laufer, S.; Heck, A.J.R.; et al. Proteasome activation by small molecules. Cell Chem. Biol. 2017, 24, 725–736.e7. [Google Scholar] [CrossRef] [PubMed]
- VerPlank, J.J.S.; Lokireddy, S.; Zhao, J.H.; Goldberg, A.L. 26s proteasomes are rapidly activated by diverse hormones and physiological states that raise camp and cause rpn6 phosphorylation. Proc. Natl. Acad. Sci. USA 2019, 116, 4228–4237. [Google Scholar] [CrossRef]
- Abdellatif, M.; Sedej, S.; Carmona-Gutierrez, D.; Madeo, F.; Kroemer, G. Autophagy in cardiovascular aging. Circ. Res. 2018, 123, 803–824. [Google Scholar] [CrossRef]
- Shinmura, K.; Tamaki, K.; Sano, M.; Murata, M.; Yamakawa, H.; Ishida, H.; Fukuda, K. Impact of long-term caloric restriction on cardiac senescence: Caloric restriction ameliorates cardiac diastolic dysfunction associated with aging. J. Mol. Cell. Cardiol. 2011, 50, 117–127. [Google Scholar] [CrossRef]
- Sheng, Y.; Lv, S.; Huang, M.; Lv, Y.; Yu, J.; Liu, J.; Tang, T.; Qi, H.; Di, W.; Ding, G. Opposing effects on cardiac function by calorie restriction in different-aged mice. Aging Cell 2017, 16, 1155–1167. [Google Scholar] [CrossRef]
- Yan, L.; Gao, S.; Ho, D.; Park, M.; Ge, H.; Wang, C.; Tian, Y.; Lai, L.; De Lorenzo, M.S.; Vatner, D.E.; et al. Calorie restriction can reverse, as well as prevent, aging cardiomyopathy. Age 2013, 35, 2177–2182. [Google Scholar] [CrossRef]
- Meyer, T.E.; Kovacs, S.J.; Ehsani, A.A.; Klein, S.; Holloszy, J.O.; Fontana, L. Long-term caloric restriction ameliorates the decline in diastolic function in humans. J. Am. Coll. Cardiol. 2006, 47, 398–402. [Google Scholar] [CrossRef]
- Castello, L.; Maina, M.; Testa, G.; Cavallini, G.; Biasi, F.; Donati, A.; Leonarduzzi, G.; Bergamini, E.; Poli, G.; Chiarpotto, E. Alternate-day fasting reverses the age-associated hypertrophy phenotype in rat heart by influencing the erk and pi3k signaling pathways. Mech. Ageing Dev. 2011, 132, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Castello, L.; Froio, T.; Maina, M.; Cavallini, G.; Biasi, F.; Leonarduzzi, G.; Donati, A.; Bergamini, E.; Poli, G.; Chiarpotto, E. Alternate-day fasting protects the rat heart against age-induced inflammation and fibrosis by inhibiting oxidative damage and nf-kb activation. Free Radic. Biol. Med. 2010, 48, 47–54. [Google Scholar] [CrossRef]
- Alirezaei, M.; Kemball, C.C.; Flynn, C.T.; Wood, M.R.; Whitton, J.L.; Kiosses, W.B. Short-term fasting induces profound neuronal autophagy. Autophagy 2010, 6, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Usman Aslam, S.K.; Liang, Q.; Martin, G. Time-dependent differential effects of fasting on cardiac autophagy and mitophagy. FASEB J. 2016, 30 (Suppl. 1), 1015-1. [Google Scholar]
- Gill, S.; Le, H.D.; Melkani, G.C.; Panda, S. Time-restricted feeding attenuates age-related cardiac decline in drosophila. Science 2015, 347, 1265–1269. [Google Scholar] [CrossRef] [PubMed]
- Longo, V.D.; Panda, S. Fasting, circadian rhythms, and time-restricted feeding in healthy lifespan. Cell Metab. 2016, 23, 1048–1059. [Google Scholar] [CrossRef] [PubMed]
- Manoogian, E.N.C.; Chaix, A.; Panda, S. When to eat: The importance of eating patterns in health and disease. J. Biol. Rhythm. 2019, 34, 579–581. [Google Scholar] [CrossRef]
- Giannuzzi, P.; Temporelli, P.L.; Corra, U.; Tavazzi, L.; Group, E.-C.S. Antiremodeling effect of long-term exercise training in patients with stable chronic heart failure: Results of the exercise in left ventricular dysfunction and chronic heart failure (elvd-chf) trial. Circulation 2003, 108, 554–559. [Google Scholar] [CrossRef]
- Haykowsky, M.J.; Liang, Y.; Pechter, D.; Jones, L.W.; McAlister, F.A.; Clark, A.M. A meta-analysis of the effect of exercise training on left ventricular remodeling in heart failure patients: The benefit depends on the type of training performed. J. Am. Coll. Cardiol. 2007, 49, 2329–2336. [Google Scholar] [CrossRef]
- He, C.; Bassik, M.C.; Moresi, V.; Sun, K.; Wei, Y.; Zou, Z.; An, Z.; Loh, J.; Fisher, J.; Sun, Q.; et al. Exercise-induced bcl2-regulated autophagy is required for muscle glucose homeostasis. Nature 2012, 481, 511–515. [Google Scholar] [CrossRef]
- Jamart, C.; Benoit, N.; Raymackers, J.M.; Kim, H.J.; Kim, C.K.; Francaux, M. Autophagy-related and autophagy-regulatory genes are induced in human muscle after ultraendurance exercise. Eur. J. Appl. Physiol. 2012, 112, 3173–3177. [Google Scholar] [CrossRef] [PubMed]
- Bhuiyan, M.S.; Pattison, J.S.; Osinska, H.; James, J.; Gulick, J.; McLendon, P.M.; Hill, J.A.; Sadoshima, J.; Robbins, J. Enhanced autophagy ameliorates cardiac proteinopathy. J. Clin. Investig. 2013, 123, 5284–5297. [Google Scholar] [CrossRef] [PubMed]
- Campos, J.C.; Queliconi, B.B.; Bozi, L.H.M.; Bechara, L.R.G.; Dourado, P.M.M.; Andres, A.M.; Jannig, P.R.; Gomes, K.M.S.; Zambelli, V.O.; Rocha-Resende, C.; et al. Exercise reestablishes autophagic flux and mitochondrial quality control in heart failure. Autophagy 2017, 13, 1304–1317. [Google Scholar] [CrossRef]
- Chen, C.Y.; Hsu, H.C.; Lee, B.C.; Lin, H.J.; Chen, Y.H.; Huang, H.C.; Ho, Y.L.; Chen, M.F. Exercise training improves cardiac function in infarcted rabbits: Involvement of autophagic function and fatty acid utilization. Eur. J. Heart Fail. 2010, 12, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Sun, Y.; Tan, Y.; Zhang, Z.; Hou, Z.; Gao, C.; Feng, P.; Zhang, X.; Yi, W.; Gao, F. Short-duration swimming exercise after myocardial infarction attenuates cardiac dysfunction and regulates mitochondrial quality control in aged mice. Oxidative Med. Cell. Longev. 2018, 2018, 4079041. [Google Scholar] [CrossRef] [PubMed]
- Mihm, M.J.; Amann, D.M.; Schanbacher, B.L.; Altschuld, R.A.; Bauer, J.A.; Hoyt, K.R. Cardiac dysfunction in the r6/2 mouse model of huntington’s disease. Neurobiol. Dis. 2007, 25, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Pyo, J.O.; Yoo, S.M.; Ahn, H.H.; Nah, J.; Hong, S.H.; Kam, T.I.; Jung, S.; Jung, Y.K. Overexpression of atg5 in mice activates autophagy and extends lifespan. Nat. Commun. 2013, 4, 2300. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghosh, R.; Vinod, V.; Symons, J.D.; Boudina, S. Protein and Mitochondria Quality Control Mechanisms and Cardiac Aging. Cells 2020, 9, 933. https://doi.org/10.3390/cells9040933
Ghosh R, Vinod V, Symons JD, Boudina S. Protein and Mitochondria Quality Control Mechanisms and Cardiac Aging. Cells. 2020; 9(4):933. https://doi.org/10.3390/cells9040933
Chicago/Turabian StyleGhosh, Rajeshwary, Vishaka Vinod, J. David Symons, and Sihem Boudina. 2020. "Protein and Mitochondria Quality Control Mechanisms and Cardiac Aging" Cells 9, no. 4: 933. https://doi.org/10.3390/cells9040933
APA StyleGhosh, R., Vinod, V., Symons, J. D., & Boudina, S. (2020). Protein and Mitochondria Quality Control Mechanisms and Cardiac Aging. Cells, 9(4), 933. https://doi.org/10.3390/cells9040933