Failure to Down-Regulate miR-154 Expression in Early Postnatal Mouse Lung Epithelium Suppresses Alveologenesis, with Changes in Tgf-β Signaling Similar to those Induced by Exposure to Hyperoxia


 ,
,  , and
, and
Abstract
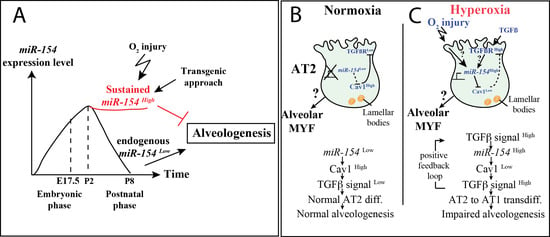
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Material and Methods
2.1. Study Approval
2.2. Mice
2.3. Hyperoxia Injury (BPD Mouse Model)
2.4. Left Lobe Perfusion, Isolation and Tissue Processing, Alveolar Morphometry (Mean Linear Intercept, Air Space, Septal-Wall Thickness)
2.5. RNA Extraction and Quantitative Real-Time RT-qPCR
2.6. Isolation of Primary Alveolar Type II Cells and Microarray Experiments
2.7. Immunofluorescence Staining
2.8. Fluorescence activated Cell Sorting
2.9. Fluorescence In-Situ-Hybridization (FISH)
2.10. Pull Down Assay with Biotinylated miR-154-3p
2.11. Statistical Analyses
3. Results
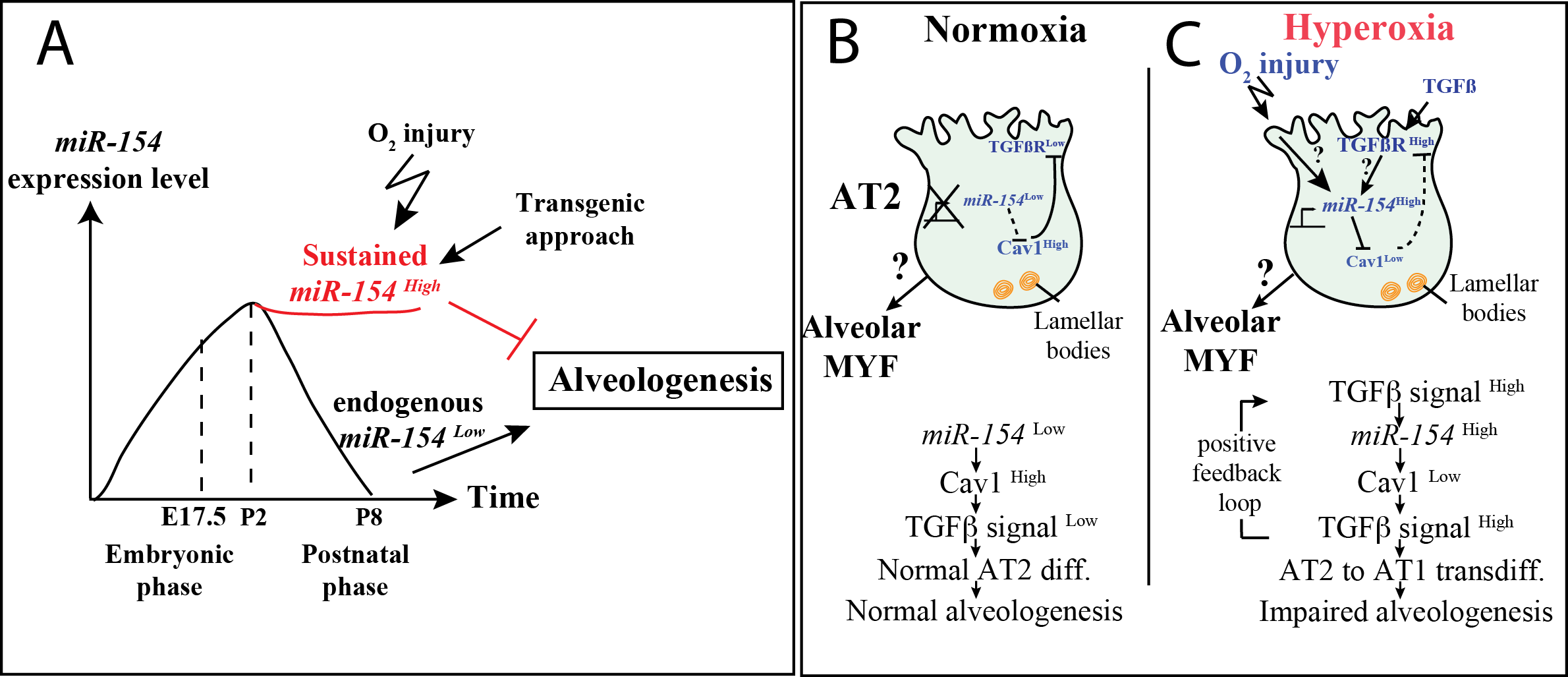
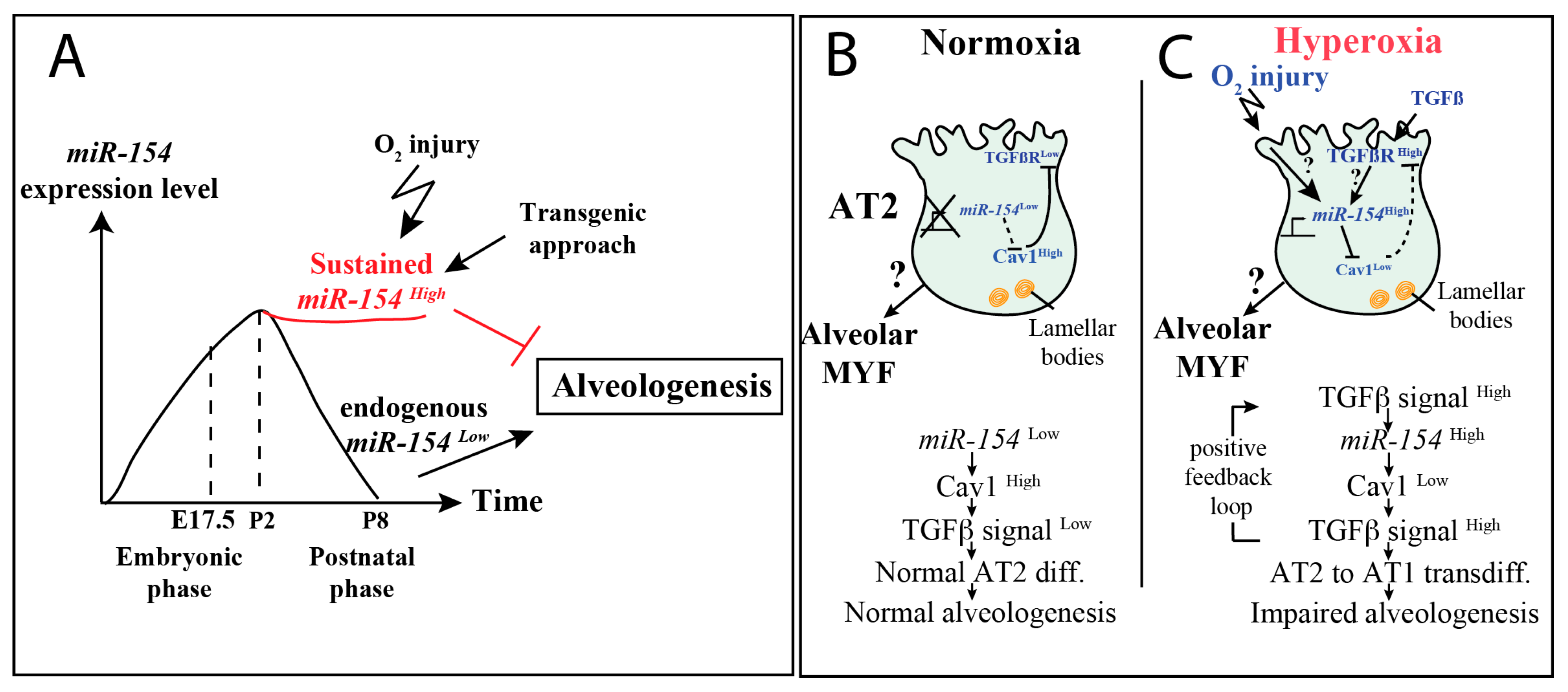
3.1. Normal Postnatal Decrease of miR-154-3p Expression Is Prevented by Hyperoxia Treatment
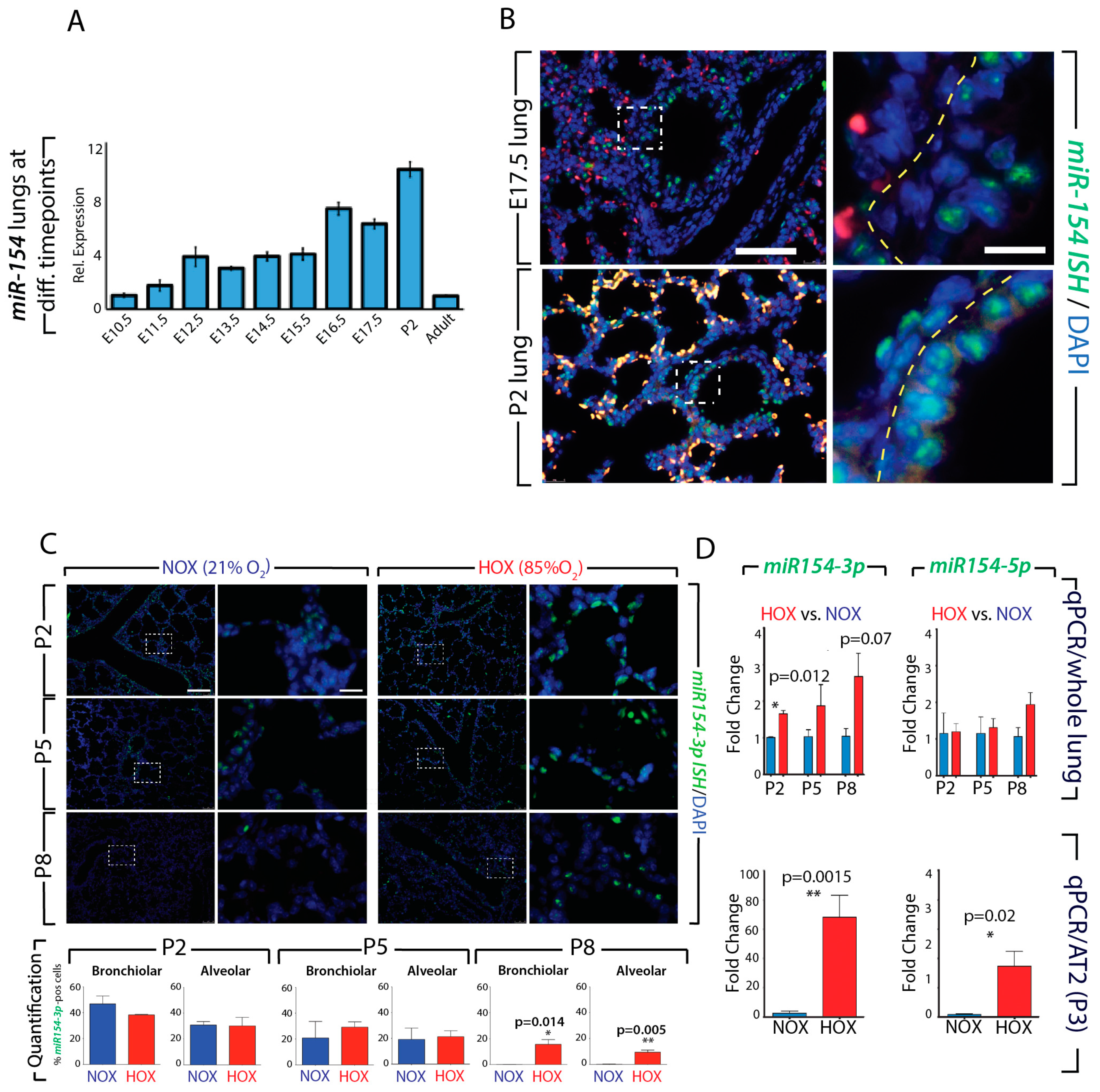
3.2. Generation and Validation of a Mouse Transgenic Model Allowing Postnatal miR-154 Overexpression
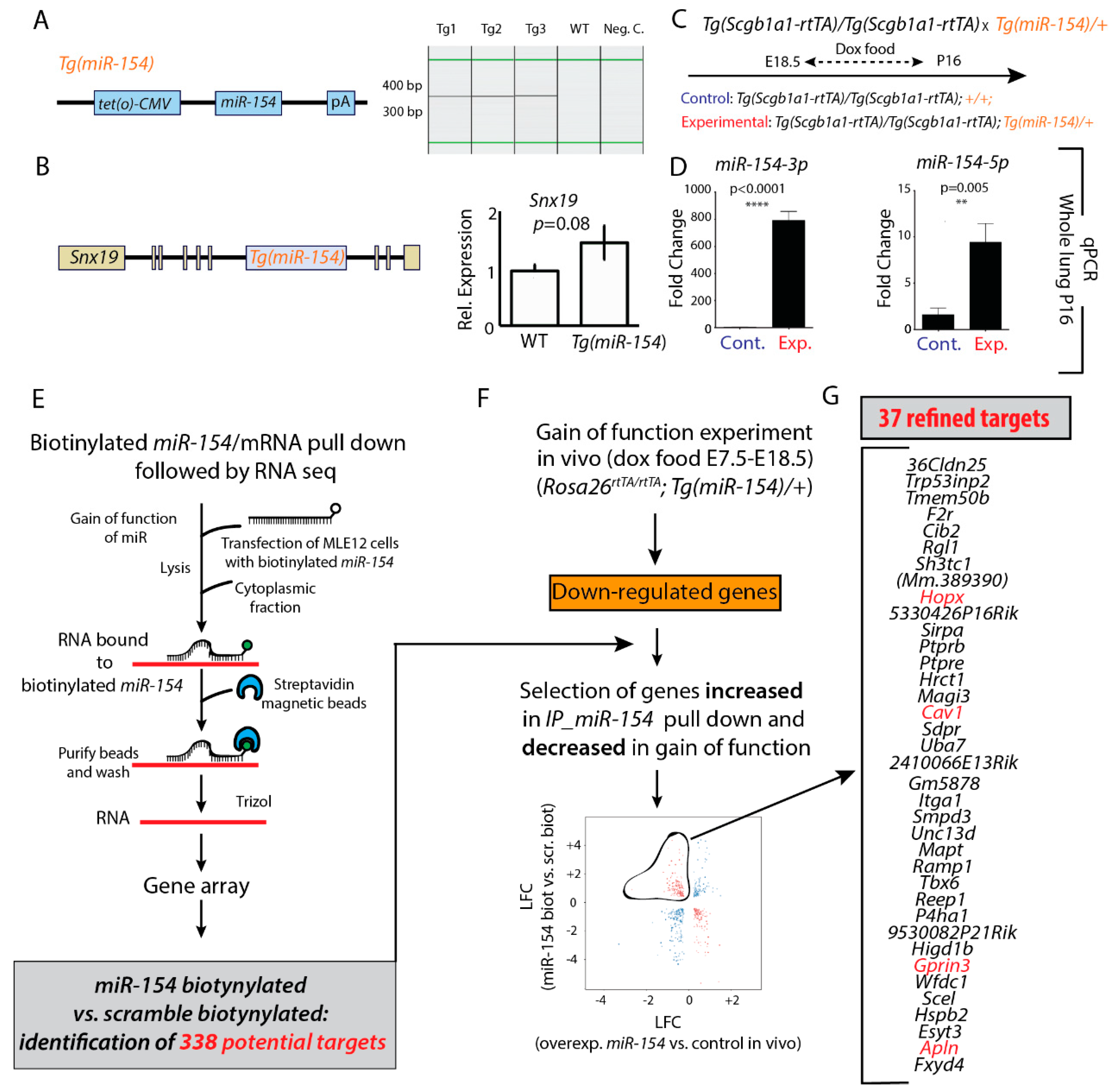
3.3. Identification of Potential miR-154 Targets Using a Biotinylated Pull-Down Assay Followed by Gene Arrays
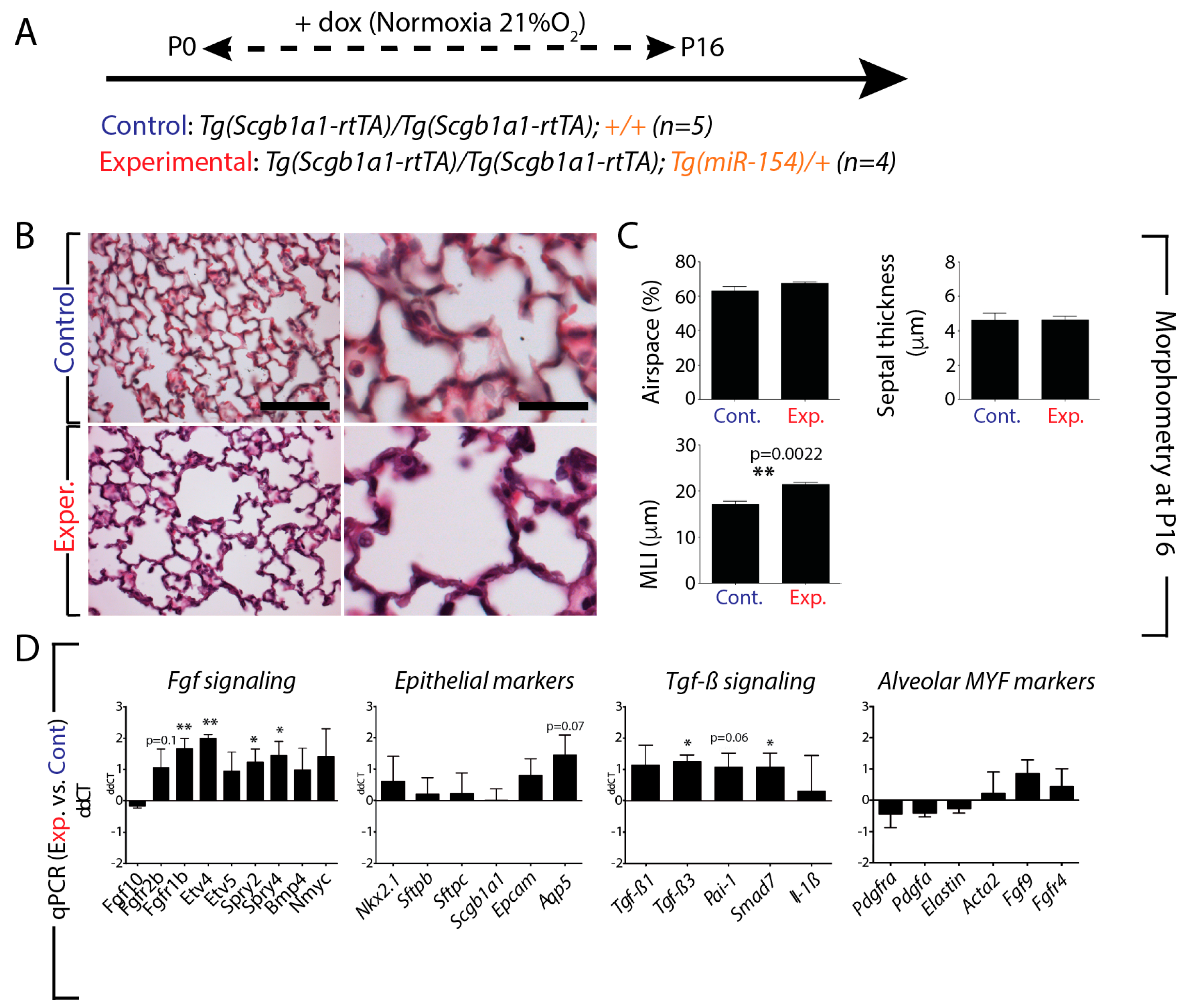
3.4. miR-154 Overexpression in the Lung Epithelium Postnatally under Normoxic Conditions is Sufficient to Impair Alveologenesis
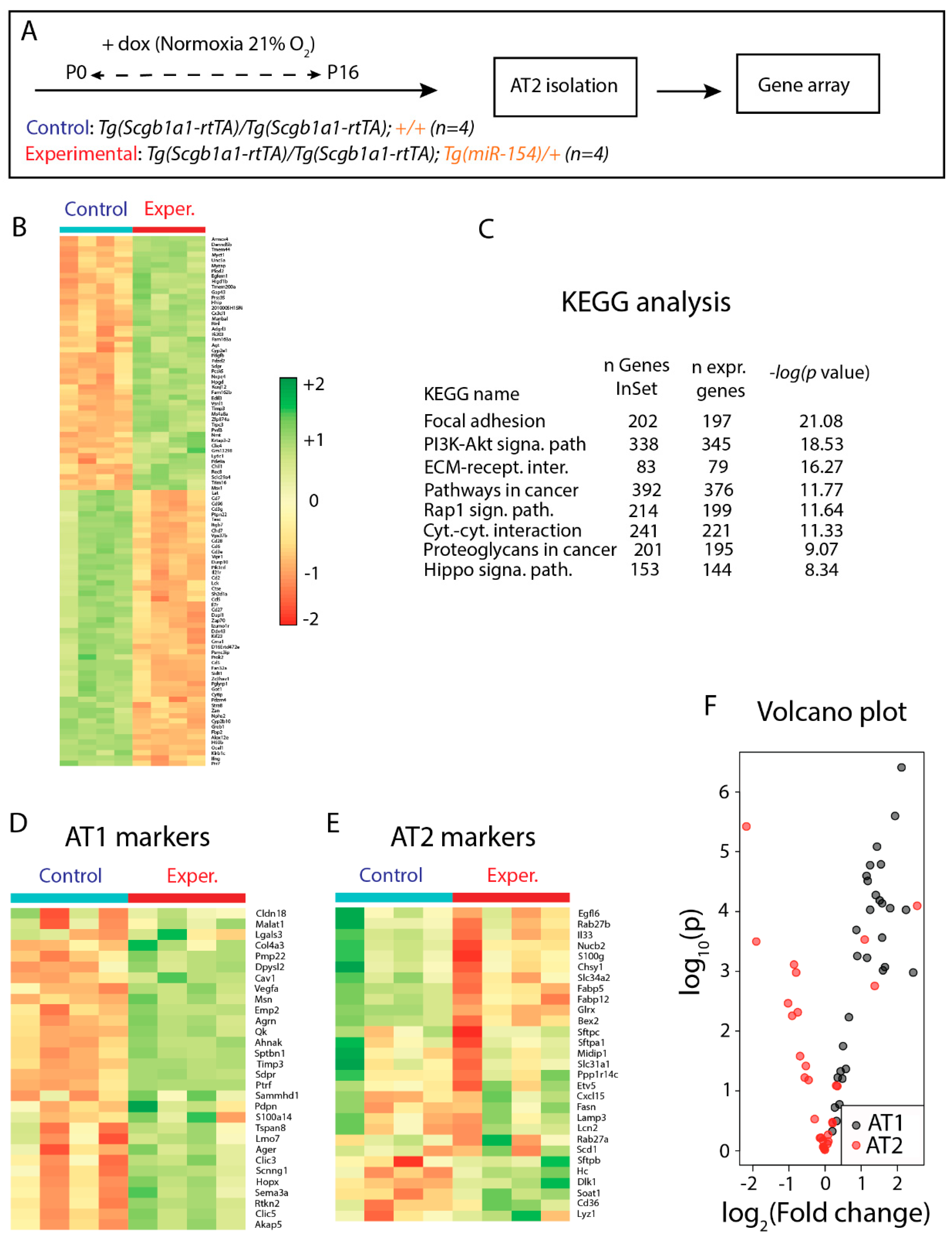
3.5. Identification of Genes Regulated in AT2 Cells from Experimental Versus Control Lungs and Analysis of AT2 Cell Differentiation
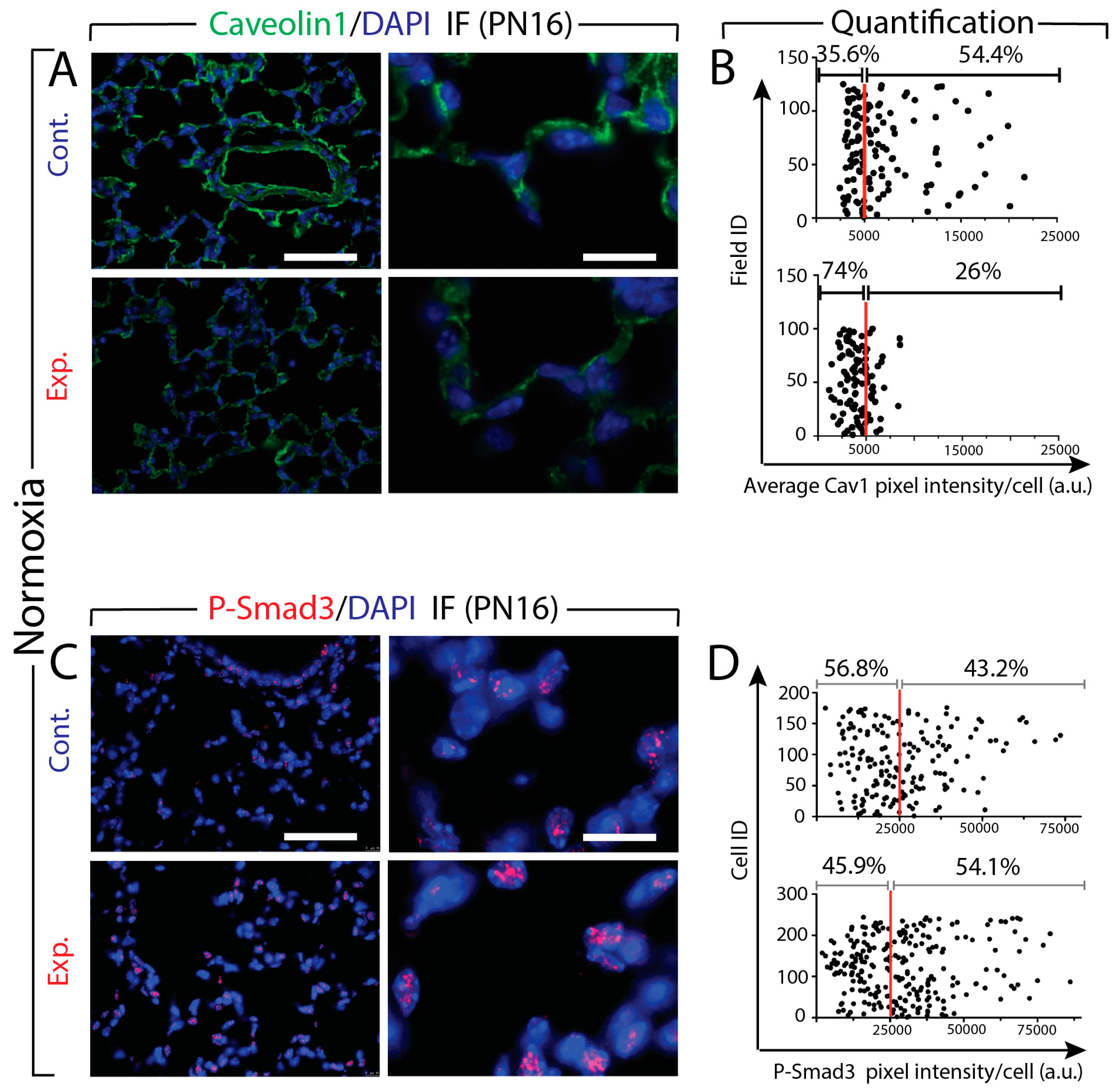
3.6. Increased Tgf-β Signaling in Experimental Versus Control Lungs in Normoxia
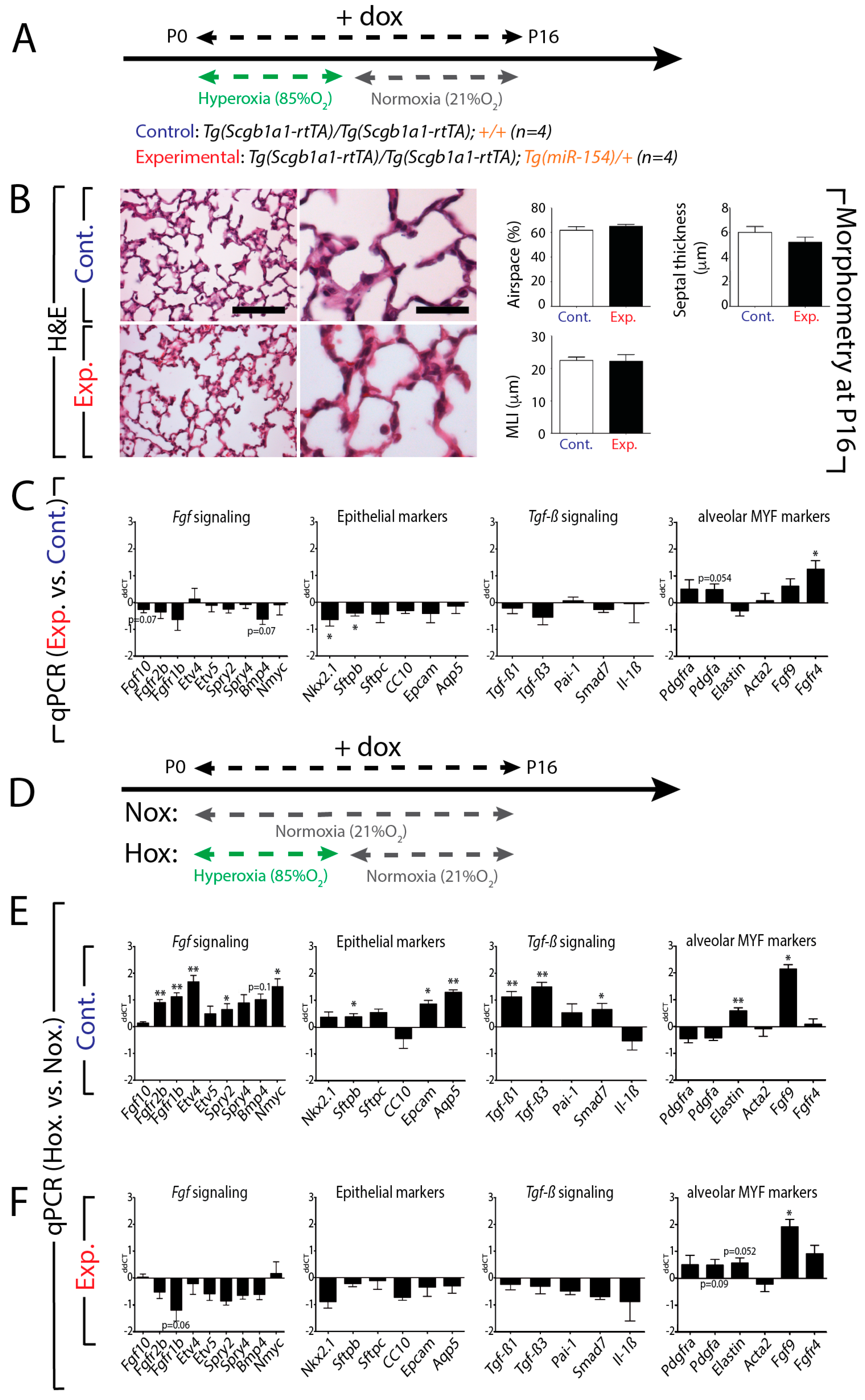
3.7. Upon Hyperoxia Injury, Key Signaling Pathways Were Differentially Affected between the miR-154 Overexpressing Lungs Versus Control Lungs, But the Alveolar Simplification Phenotype in Both Cases Was Morphologically Indistinguishable
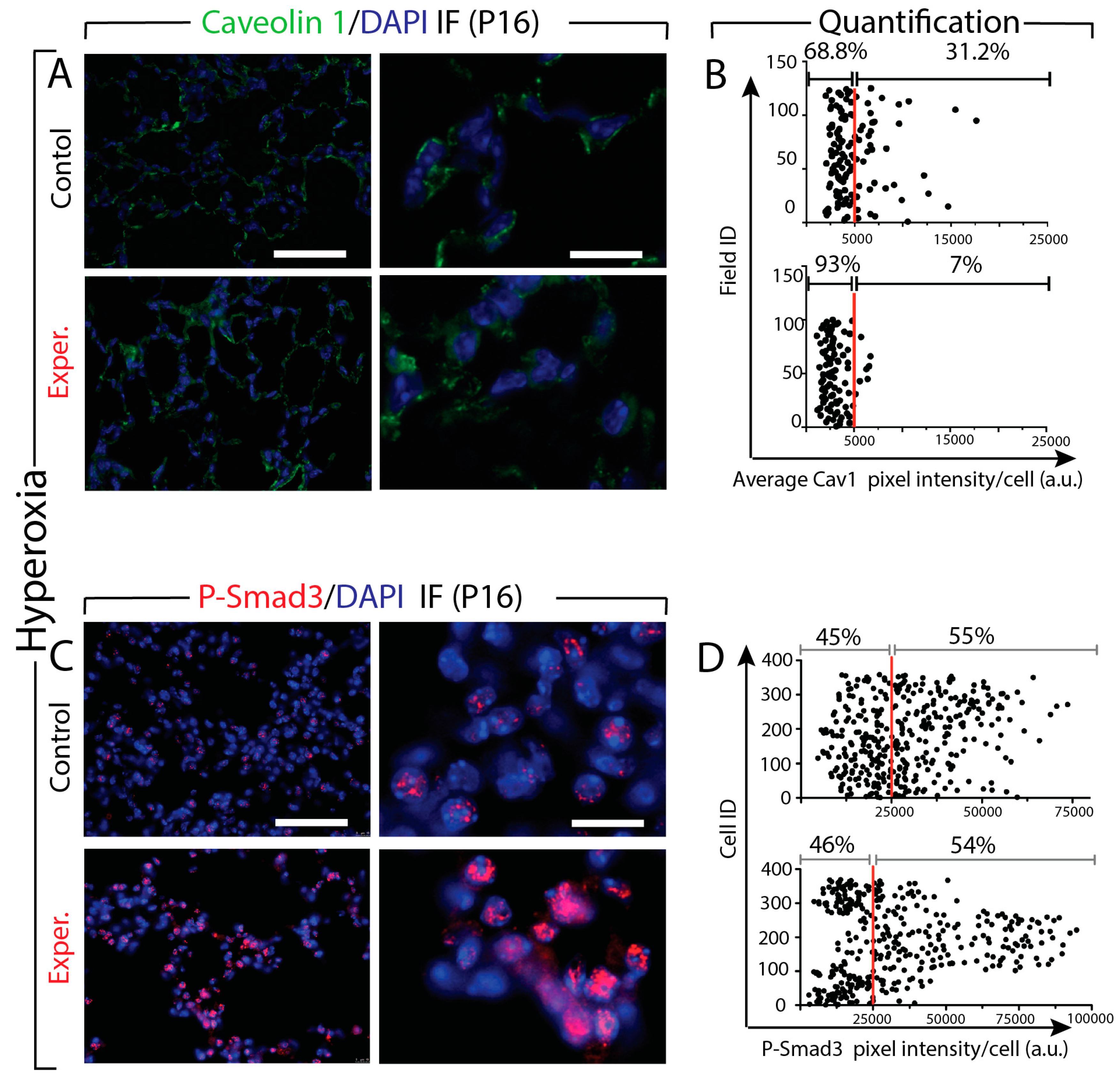
3.8. Increased Tgf-β Signaling in miR-154 Experimental Versus Control Lungs in Hyperoxia
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kugler, M.C.; Loomis, C.A.; Zhao, Z.; Cushman, J.C.; Liu, L.; Munger, J.S. Sonic Hedgehog Signaling Regulates Myofibroblast Function during Alveolar Septum Formation in Murine Postnatal Lung. Am. J. Respir. Cell Mol. Biol. 2017, 57, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Bostrom, H.; Willetts, K.; Pekny, M.; Leveen, P.; Lindahl, P.; Hedstrand, H.; Pekna, M.; Hellstrom, M.; Gebre-Medhin, S.; Schalling, M.; et al. PDGF-A signaling is a critical event in lung alveolar myofibroblast development and alveogenesis. Cell 1996, 85, 863–873. [Google Scholar] [CrossRef]
- Popova, A.P.; Bentley, J.K.; Cui, T.X.; Richardson, M.N.; Linn, M.J.; Lei, J.; Chen, Q.; Goldsmith, A.M.; Pryhuber, G.S.; Hershenson, M.B. Reduced platelet-derived growth factor receptor expression is a primary feature of human bronchopulmonary dysplasia. Am. J. Physiol. Lung Cell Mol. Physiol. 2014, 307, L231–L239. [Google Scholar] [CrossRef] [PubMed]
- Perl, A.K.; Gale, E. FGF signaling is required for myofibroblast differentiation during alveolar regeneration. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 297, L299–L308. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, M.; Xu, X.; Ohyama, K.; Deng, C.X. FGFR-3 and FGFR-4 function cooperatively to direct alveogenesis in the murine lung. Development 1998, 125, 3615–3623. [Google Scholar] [PubMed]
- De Langhe, S.P.; Carraro, G.; Warburton, D.; Hajihosseini, M.K.; Bellusci, S. Levels of mesenchymal FGFR2 signaling modulate smooth muscle progenitor cell commitment in the lung. Dev. Biol. 2006, 299, 52–62. [Google Scholar] [CrossRef]
- Baraldi, E.; Filippone, M. Chronic lung disease after premature birth. N Engl. J. Med. 2007, 357, 1946–1955. [Google Scholar] [CrossRef]
- Nardiello, C.; Morty, R.E. MicroRNA in late lung development and bronchopulmonary dysplasia: The need to demonstrate causality. Mol. Cell Pediatr. 2016, 3, 19. [Google Scholar] [CrossRef]
- Chao, C.M.; El Agha, E.; Tiozzo, C.; Minoo, P.; Bellusci, S. A breath of fresh air on the mesenchyme: Impact of impaired mesenchymal development on the pathogenesis of bronchopulmonary dysplasia. Front. Med. (Lausanne) 2015, 2, 27. [Google Scholar] [CrossRef]
- Northway, W.H., Jr.; Rosan, R.C.; Porter, D.Y. Pulmonary disease following respirator therapy of hyaline-membrane disease. Bronchopulmonary dysplasia. N Engl. J. Med. 1967, 276, 357–368. [Google Scholar] [CrossRef]
- Voynow, J.A. “New” bronchopulmonary dysplasia and chronic lung disease. Paediatr. Respir. Rev. 2017, 24, 17–18. [Google Scholar] [CrossRef] [PubMed]
- Owen, L.S.; Manley, B.J.; Davis, P.G.; Doyle, L.W. The evolution of modern respiratory care for preterm infants. Lancet 2017, 389, 1649–1659. [Google Scholar] [CrossRef]
- Xing, Y.; Fu, J.; Yang, H.; Yao, L.; Qiao, L.; Du, Y.; Xue, X. MicroRNA expression profiles and target prediction in neonatal Wistar rat lungs during the development of bronchopulmonary dysplasia. Int. J. Mol. Med. 2015, 36, 1253–1263. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Silva, D.M.; Nardiello, C.; Pozarska, A.; Morty, R.E. Recent advances in the mechanisms of lung alveolarization and the pathogenesis of bronchopulmonary dysplasia. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 309, L1239–L1272. [Google Scholar] [CrossRef] [PubMed]
- Jonas, S.; Izaurralde, E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat. Rev. Genet. 2015, 16, 421–433. [Google Scholar] [CrossRef]
- Krol, J.; Loedige, I.; Filipowicz, W. The widespread regulation of microRNA biogenesis, function and decay. Nat. Rev. Genet. 2010, 11, 597–610. [Google Scholar] [CrossRef]
- Sessa, R.; Hata, A. Role of microRNAs in lung development and pulmonary diseases. Pulm. Circ. 2013, 3, 315–328. [Google Scholar] [CrossRef]
- Benetatos, L.; Hatzimichael, E.; Londin, E.; Vartholomatos, G.; Loher, P.; Rigoutsos, I.; Briasoulis, E. The microRNAs within the DLK1-DIO3 genomic region: Involvement in disease pathogenesis. Cell Mol. Life Sci. 2013, 70, 795–814. [Google Scholar] [CrossRef]
- Williams, A.E.; Moschos, S.A.; Perry, M.M.; Barnes, P.J.; Lindsay, M.A. Maternally imprinted microRNAs are differentially expressed during mouse and human lung development. Dev. Dyn. 2007, 236, 572–580. [Google Scholar] [CrossRef]
- Seitz, H.; Royo, H.; Bortolin, M.L.; Lin, S.P.; Ferguson-Smith, A.C.; Cavaille, J. A large imprinted microRNA gene cluster at the mouse Dlk1-Gtl2 domain. Genome Res. 2004, 14, 1741–1748. [Google Scholar] [CrossRef]
- Dixon-McIver, A.; East, P.; Mein, C.A.; Cazier, J.B.; Molloy, G.; Chaplin, T.; Andrew Lister, T.; Young, B.D.; Debernardi, S. Distinctive patterns of microRNA expression associated with karyotype in acute myeloid leukaemia. PloS ONE 2008, 3, e2141. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.P.; Youngson, N.; Takada, S.; Seitz, H.; Reik, W.; Paulsen, M.; Cavaille, J.; Ferguson-Smith, A.C. Asymmetric regulation of imprinting on the maternal and paternal chromosomes at the Dlk1-Gtl2 imprinted cluster on mouse chromosome 12. Nat. Genet. 2003, 35, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Kagami, M.; O’Sullivan, M.J.; Green, A.J.; Watabe, Y.; Arisaka, O.; Masawa, N.; Matsuoka, K.; Fukami, M.; Matsubara, K.; Kato, F.; et al. The IG-DMR and the MEG3-DMR at human chromosome 14q32.2: Hierarchical interaction and distinct functional properties as imprinting control centers. PloS Genet. 2010, 6, e1000992. [Google Scholar] [CrossRef] [PubMed]
- Available online: http://www.mirbase.org/ (accessed on 1 December 2019).
- Parsa, S.; Ramasamy, S.K.; De Langhe, S.; Gupte, V.V.; Haigh, J.J.; Medina, D.; Bellusci, S. Terminal end bud maintenance in mammary gland is dependent upon FGFR2b signaling. Dev. Biol. 2008, 317, 121–131. [Google Scholar] [CrossRef] [PubMed]
- McGrath-Morrow, S.A.; Cho, C.; Soutiere, S.; Mitzner, W.; Tuder, R. The effect of neonatal hyperoxia on the lung of p21Waf1/Cip1/Sdi1-deficient mice. Am. J. Respir. Cell Mol. Biol. 2004, 30, 635–640. [Google Scholar] [CrossRef]
- Woyda, K.; Koebrich, S.; Reiss, I.; Rudloff, S.; Pullamsetti, S.S.; Ruhlmann, A.; Weissmann, N.; Ghofrani, H.A.; Gunther, A.; Seeger, W.; et al. Inhibition of phosphodiesterase 4 enhances lung alveolarisation in neonatal mice exposed to hyperoxia. Eur. Respir. J. 2009, 33, 861–870. [Google Scholar] [CrossRef]
- Seimetz, M.; Parajuli, N.; Pichl, A.; Veit, F.; Kwapiszewska, G.; Weisel, F.C.; Milger, K.; Egemnazarov, B.; Turowska, A.; Fuchs, B.; et al. Inducible NOS inhibition reverses tobacco-smoke-induced emphysema and pulmonary hypertension in mice. Cell 2011, 147, 293–305. [Google Scholar] [CrossRef]
- Gentleman, R.C.; Carey, V.J.; Bates, D.M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; et al. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004, 5, R80. [Google Scholar] [CrossRef]
- Smyth, G.K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004, 3, 3. [Google Scholar] [CrossRef]
- McQualter, J.L.; Yuen, K.; Williams, B.; Bertoncello, I. Evidence of an epithelial stem/progenitor cell hierarchy in the adult mouse lung. Proc. Natl. Acad. Sci. USA 2010, 107, 1414–1419. [Google Scholar] [CrossRef]
- Sanders, Y.Y.; Cui, Z.; Le Saux, C.J.; Horowitz, J.C.; Rangarajan, S.; Kurundkar, A.; Antony, V.B.; Thannickal, V.J. SMAD-independent down-regulation of caveolin-1 by TGF-beta: Effects on proliferation and survival of myofibroblasts. PloS ONE 2015, 10, e0116995. [Google Scholar] [CrossRef] [PubMed]
- Al Alam, D.; Danopoulos, S.; Schall, K.; Sala, F.G.; Almohazey, D.; Fernandez, G.E.; Georgia, S.; Frey, M.R.; Ford, H.R.; Grikscheit, T.; et al. Fibroblast growth factor 10 alters the balance between goblet and Paneth cells in the adult mouse small intestine. Am. J. Physiol. Gastrointest Liver Physiol. 2015, 308, G678–G690. [Google Scholar] [CrossRef] [PubMed]
- Treutlein, B.; Brownfield, D.G.; Wu, A.R.; Neff, N.F.; Mantalas, G.L.; Espinoza, F.H.; Desai, T.J.; Krasnow, M.A.; Quake, S.R. Reconstructing lineage hierarchies of the distal lung epithelium using single-cell RNA-seq. Nature 2014, 509, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kang, Y.; Kojima, Y.; Lighthouse, J.K.; Hu, X.; Aldred, M.A.; McLean, D.L.; Park, H.; Comhair, S.A.; Greif, D.M.; et al. An endothelial apelin-FGF link mediated by miR-424 and miR-503 is disrupted in pulmonary arterial hypertension. Nat. Med. 2013, 19, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Al Alam, D.; El Agha, E.; Sakurai, R.; Kheirollahi, V.; Moiseenko, A.; Danopoulos, S.; Shrestha, A.; Schmoldt, C.; Quantius, J.; Herold, S.; et al. Evidence for the involvement of fibroblast growth factor 10 in lipofibroblast formation during embryonic lung development. Development 2015, 142, 4139–4150. [Google Scholar] [CrossRef]
- de Maximy, A.A.; Nakatake, Y.; Moncada, S.; Itoh, N.; Thiery, J.P.; Bellusci, S. Cloning and expression pattern of a mouse homologue of drosophila sprouty in the mouse embryo. Mech. Dev. 1999, 81, 213–216. [Google Scholar] [CrossRef]
- Jones, M.R.; Lingampally, A.; Dilai, S.; Shrestha, A.; Stripp, B.; Helmbacher, F.; Chen, C.; Chao, C.M.; Bellusci, S. Characterization of Tg(Etv4-GFP) and Etv5 (RFP) Reporter Lines in the Context of Fibroblast Growth Factor 10 Signaling During Mouse Embryonic Lung Development. Front. Genet. 2019, 10, 178. [Google Scholar] [CrossRef]
- Hwangbo, C.; Tae, N.; Lee, S.; Kim, O.; Park, O.K.; Kim, J.; Kwon, S.H.; Lee, J.H. Syntenin regulates TGF-beta1-induced Smad activation and the epithelial-to-mesenchymal transition by inhibiting caveolin-mediated TGF-beta type I receptor internalization. Oncogene 2016, 35, 389–401. [Google Scholar] [CrossRef]
- Gauldie, J.; Galt, T.; Bonniaud, P.; Robbins, C.; Kelly, M.; Warburton, D. Transfer of the active form of transforming growth factor-beta 1 gene to newborn rat lung induces changes consistent with bronchopulmonary dysplasia. Am. J. Pathol. 2003, 163, 2575–2584. [Google Scholar] [CrossRef]
- Vicencio, A.G.; Lee, C.G.; Cho, S.J.; Eickelberg, O.; Chuu, Y.; Haddad, G.G.; Elias, J.A. Conditional overexpression of bioactive transforming growth factor-beta1 in neonatal mouse lung: A new model for bronchopulmonary dysplasia? Am. J. Respir. Cell Mol. Biol. 2004, 31, 650–656. [Google Scholar] [CrossRef]
- Alejandre-Alcazar, M.A.; Kwapiszewska, G.; Reiss, I.; Amarie, O.V.; Marsh, L.M.; Sevilla-Perez, J.; Wygrecka, M.; Eul, B.; Kobrich, S.; Hesse, M.; et al. Hyperoxia modulates TGF-beta/BMP signaling in a mouse model of bronchopulmonary dysplasia. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 292, L537–L549. [Google Scholar] [CrossRef] [PubMed]
- Milosevic, J.; Pandit, K.; Magister, M.; Rabinovich, E.; Ellwanger, D.C.; Yu, G.; Vuga, L.J.; Weksler, B.; Benos, P.V.; Gibson, K.F.; et al. Profibrotic role of miR-154 in pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2012, 47, 879–887. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Frank, D.B.; Morley, M.P.; Zhou, S.; Wang, X.; Lu, M.M.; Lazar, M.A.; Morrisey, E.E. HDAC3-Dependent Epigenetic Pathway Controls Lung Alveolar Epithelial Cell Remodeling and Spreading via miR-17-92 and TGF-beta Signaling Regulation. Dev. Cell 2016, 36, 303–315. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, A.; Carraro, G.; El Agha, E.; Mukhametshina, R.; Chao, C.M.; Rizvanov, A.; Barreto, G.; Bellusci, S. Generation and Validation of miR-142 Knock Out Mice. PloS ONE 2015, 10, e0136913. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chao, C.-M.; Carraro, G.; Rako, Z.A.; Kolck, J.; Sedighi, J.; Zimmermann, V.; Moiseenko, A.; Wilhelm, J.; Young, B.M.; Chong, L.; et al. Failure to Down-Regulate miR-154 Expression in Early Postnatal Mouse Lung Epithelium Suppresses Alveologenesis, with Changes in Tgf-β Signaling Similar to those Induced by Exposure to Hyperoxia. Cells 2020, 9, 859. https://doi.org/10.3390/cells9040859
Chao C-M, Carraro G, Rako ZA, Kolck J, Sedighi J, Zimmermann V, Moiseenko A, Wilhelm J, Young BM, Chong L, et al. Failure to Down-Regulate miR-154 Expression in Early Postnatal Mouse Lung Epithelium Suppresses Alveologenesis, with Changes in Tgf-β Signaling Similar to those Induced by Exposure to Hyperoxia. Cells. 2020; 9(4):859. https://doi.org/10.3390/cells9040859
Chicago/Turabian StyleChao, Cho-Ming, Gianni Carraro, Zvonimir A. Rako, Johannes Kolck, Jamschid Sedighi, Volker Zimmermann, Alena Moiseenko, Jochen Wilhelm, Brittany M. Young, Lei Chong, and et al. 2020. "Failure to Down-Regulate miR-154 Expression in Early Postnatal Mouse Lung Epithelium Suppresses Alveologenesis, with Changes in Tgf-β Signaling Similar to those Induced by Exposure to Hyperoxia" Cells 9, no. 4: 859. https://doi.org/10.3390/cells9040859
APA StyleChao, C.-M., Carraro, G., Rako, Z. A., Kolck, J., Sedighi, J., Zimmermann, V., Moiseenko, A., Wilhelm, J., Young, B. M., Chong, L., Wu, J., Contreras, A., Minoo, P., Barreto, G., Warburton, D., & Bellusci, S. (2020). Failure to Down-Regulate miR-154 Expression in Early Postnatal Mouse Lung Epithelium Suppresses Alveologenesis, with Changes in Tgf-β Signaling Similar to those Induced by Exposure to Hyperoxia. Cells, 9(4), 859. https://doi.org/10.3390/cells9040859