Circulating Small Noncoding RNAs Have Specific Expression Patterns in Plasma and Extracellular Vesicles in Myelodysplastic Syndromes and Are Predictive of Patient Outcome

 , ,
, ,
Abstract
1. Introduction
2. Results
2.1. Patient Characteristics
2.2. Characterization of Extracellular Vesicles in MDS Plasma
2.3. General Overview of Circulating sncRNAs of MDS Patients
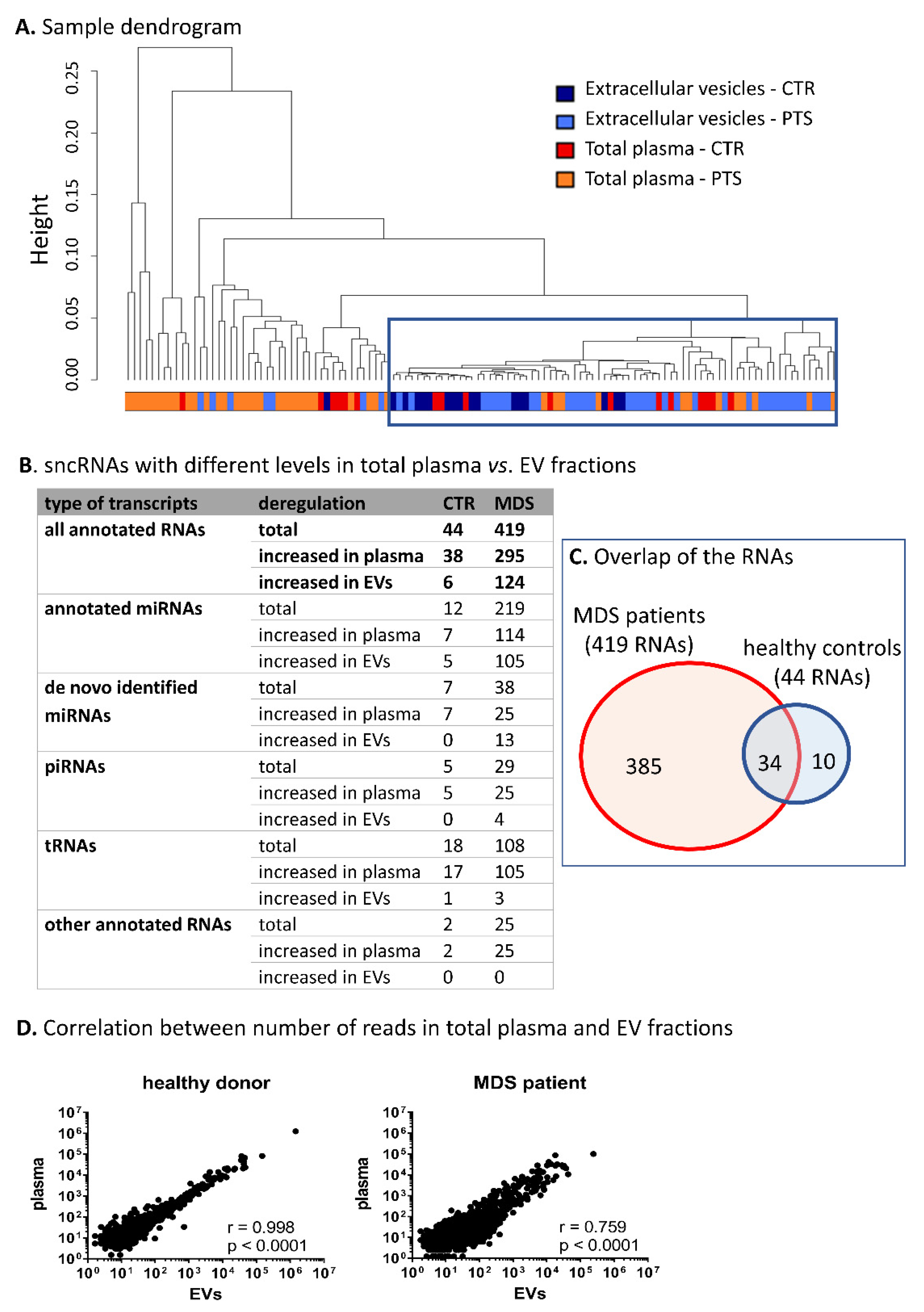
2.4. Comparison of sncRNAs Circulating in Total Plasma vs. Encapsulated in EVs
2.5. SncRNAs Differentially Expressed in MDS
2.6. SncRNAs Differentially Expressed in Different Stages of MDS
2.7. Validation of NGS Results
2.8. Pathway Analysis Based on miRNA Profiles Specific for MDS
2.9. Relationship of Somatic Mutations and Levels of Circulating sncRNAs in MDS
2.10. A-to-I Editing of Circulating miRNAs
2.11. Circulating sncRNAs as Prognostic Biomarkers of MDS Survival
2.12. Circulating sncRNAs Predictive of the AZA Response
3. Discussion
4. Methods
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, P.L.; Tuechler, H.; Schanz, J.; Sanz, G.; Garcia-Manero, G.; Solé, F.; Bennett, J.M.; Bowen, D.; Fenaux, P.; Dreyfus, F.; et al. Revised International Prognostic Scoring System for Myelodysplastic Syndromes. Blood 2012, 120, 2454–2465. [Google Scholar] [CrossRef]
- Vickers, K.C.; Palmisano, B.T.; Shoucri, B.M.; Shamburek, R.D.; Remaley, A.T. MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat. Cell. Biol. 2011, 13, 423–433. [Google Scholar] [CrossRef]
- Arroyo, J.D.; Chevillet, J.R.; Kroh, E.M.; Ruf, I.K.; Pritchard, C.C.; Gibson, D.F.; Mitchell, P.S.; Bennett, C.F.; Pogosova-Agadjanyan, E.L.; Stirewalt, D.L.; et al. Argonaute2 complexes carry a population of circulating microRNAs independent of vesicles in human plasma. Proc. Natl. Acad. Sci. USA 2011, 108, 5003–5008. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Zhang, S.; Weber, J.; Baxter, D.; Galas, D.J. Export of microRNAs and microRNA-protective protein by mammalian cells. Nucleic Acids Res. 2010, 38, 7248–7259. [Google Scholar] [CrossRef] [PubMed]
- Diehl, P.; Fricke, A.; Sander, L.; Stamm, J.; Bassler, N.; Htun, N.; Ziemann, M.; Helbing, T.; El-Osta, A.; Jowett, J.B.; et al. Microparticles: Major transport vehicles for distinct microRNAs in circulation. Cardiovasc. Res. 2012, 93, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Hunter, M.P.; Ismail, N.; Zhang, X.; Aguda, B.D.; Lee, E.J.; Yu, L.; Xiao, T.; Schafer, J.; Lee, M.L.; Schmittgen, T.D.; et al. Detection of microRNA Expression in Human Peripheral Blood Microvesicles. PLoS ONE 2008, 3, e3694. [Google Scholar] [CrossRef]
- Fonsato, V.; Collino, F.; Herrera, M.B.; Cavallari, C.; Deregibus, M.C.; Cisterna, B.; Bruno, S.; Romagnoli, R.; Salizzoni, M.; Tetta, C.; et al. Human liver stem cell-derived microvesicles inhibit hepatoma growth in SCID mice by delivering antitumor microRNAs. Stem Cells 2012, 30, 1985–1998. [Google Scholar] [CrossRef]
- Umezu, T.; Ohyashiki, K.; Kuroda, M.; Ohyashiki, J.H. Leukemia cell to endothelial cell communication via exosomal miRNAs. Oncogene 2013, 32, 2747–2755. [Google Scholar] [CrossRef]
- Yuan, A.; Farber, E.L.; Rapoport, A.L.; Tejada, D.; Deniskin, R.; Akhmedov, N.B.; Farber, D.B. Transfer of MicroRNAs by Embryonic Stem Cell Microvesicles. PLoS ONE 2009, 4, e4722. [Google Scholar] [CrossRef]
- Villarroya-Beltri, C.; Gutiérrez-Vázquez, C.; Sánchez-Cabo, F.; Pérez-Hernández, D.; Vázquez, J.; Martin-Cofreces, N.; Martinez-Herrera, D.J.; Pascual-Montano, A.; Mittelbrunn, M.; Sánchez-Madrid, F. Sumoylated hnRNPA2B1 controls the sorting of miRNAs into exosomes through binding to specific motifs. Nat. Commun. 2013, 4, 2980. [Google Scholar] [CrossRef]
- Temoche-Diaz, M.M.; Shurtleff, M.J.; Nottingham, R.M.; Yao, J.; Fadadu, R.P.; Lambowitz, A.M.; Schekman, R. Distinct mechanisms of microRNA sorting into cancer cell-derived extracellular vesicle subtypes. eLife 2019, 8, e47544. [Google Scholar] [CrossRef]
- Bracken, C.P.; Scott, H.S.; Goodall, G.J. A network-biology perspective of microRNA function and dysfunction in cancer. Nature reviews. Genetics 2016, 17, 719–732. [Google Scholar]
- Zuo, Z.; Calin, G.A.; De Paula, H.M.; Medeiros, L.J.; Fernandez, M.H.; Shimizu, M.; Garcia-Manero, G.; Bueso-Ramos, C.E. Circulating microRNAs let-7a and miR-16 predict progression-free survival and overall survival in patients with myelodysplastic syndrome. Blood 2011, 118, 413–415. [Google Scholar] [CrossRef]
- Kim, Y.; Cheong, J.W.; Kim, Y.K.; Eom, J.I.; Jeung, H.K.; Kim, S.J.; Hwang, D.; Kim, J.S.; Kim, H.J.; Min, Y.H. Serum microRNA-21 as a potential biomarker for response to hypomethylating agents in myelodysplastic syndromes. PLoS ONE 2014, 9, e86933. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Z.; Maiti, S.; Hu, S.; Loghavi, S.; Calin, G.A.; Garcia-Manero, G.; Kantarjian, H.M.; Medeiros, L.J.; Cooper, L.J.; Bueso-Ramos, C.E. Plasma circulating-microRNA profiles are useful for assessing prognosis in patients with cytogenetically normal myelodysplastic syndromes. Mod. Pathol. 2015, 28, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Merkerova, M.D.; Hrustincova, A.; Krejcik, Z.; Votavova, H.; Ratajova, E.; Cermak, J.; Belickova, M. Microarray profiling defines circulating microRNAs associated with myelodysplastic syndromes. Neoplasma 2017, 64, 571–578. [Google Scholar]
- Giudice, V.; Banaszak, L.G.; Gutierrez-Rodrigues, F.; Kajigaya, S.; Panjwani, R.; Ibanez, M.D.; Rios, O.; Bleck, C.K.; Stempinski, E.S.; Raffo, D.Q.; et al. Circulating exosomal microRNAs in acquired aplastic anemia and myelodysplastic syndromes. Haematologica 2018, 103, 1150–1159. [Google Scholar] [CrossRef]
- Enjeti, A.K.; Ariyarajah, A.; D’Crus, A.; Riveros, C.; Seldon, M.; Lincz, L.F. Circulating microvesicles are less procoagulant and carry different miRNA cargo in myelodysplasia. Blood Cells Mol. Dis. 2019, 74, 37–43. [Google Scholar] [CrossRef]
- Bobrie, A.; Colombo, M.; Krumeich, S.; Raposo, G.; Théry, C. Diverse subpopulations of vesicles secreted by different intracellular mechanisms are present in exosome preparations obtained by differential ultracentrifugation. J. Extracell. Vesicles 2012, 1, 18397. [Google Scholar] [CrossRef]
- El-Hefnawy, T.; Raja, S.; Kelly, L.; Bigbee, W.L.; Kirkwood, J.M.; Luketich, J.D.; Godfrey, T.E. Characterization of amplifiable, circulating RNA in plasma and its potential as a tool for cancer diagnostics. Clin. Chem. 2004, 50, 564–573. [Google Scholar] [CrossRef] [PubMed]
- Savelyeva, A.V.; Kuligina, E.V.; Bariakin, D.N.; Kozlov, V.V.; Ryabchikova, E.I.; Richter, V.A.; Semenov, D.V. Variety of RNAs in Peripheral Blood Cells, Plasma, and Plasma Fractions. BioMed Res. Int. 2017, 2017, 7404912. [Google Scholar] [CrossRef] [PubMed]
- Freedman, J.E.; Gerstein, M.; Mick, E.; Rozowsky, J.; Levy, D.; Kitchen, R.; Das, S.; Shah, R.; Danielson, K.; Beaulieu, L.; et al. Diverse human extracellular RNAs are widely detected in human plasma. Nat. Commun. 2016, 7, 11106. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Yuan, T.; Tschannen, M.; Sun, Z.; Jacob, H.; Du, M.; Liang, M.; Dittmar, R.L.; Liu, Y.; Liang, M.; et al. Characterization of human plasma-derived exosomal RNAs by deep sequencing. BMC Genomics 2013, 14, 319. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Deng, C.X. Current Progresses of Exosomes as Cancer Diagnostic and Prognostic Biomarkers. Int. J. Biol. Sci. 2019, 15, 1. [Google Scholar] [CrossRef] [PubMed]
- Szczepanski, M.J.; Szajnik, M.; Welsh, A.; Whiteside, T.L.; Boyiadzis, M. Blast-derived microvesicles in sera from patients with acute myeloid leukemia suppress natural killer cell function via membrane-associated transforming growth factor-beta1. Haematologica 2011, 96, 1302–1309. [Google Scholar] [CrossRef] [PubMed]
- Beatty, M.; Guduric-Fuchs, J.; Brown, E.; Bridgett, S.; Chakravarthy, U.; Hogg, R.E.; Simpson, D.A. Small RNAs from plants, bacteria and fungi within the order Hypocreales are ubiquitous in human plasma. BMC Genomics 2014, 15, 933. [Google Scholar] [CrossRef]
- Whittle, E.; Leonard, M.O.; Harrison, R.; Gant, T.W.; Tonge, D.P. Multi-Method Characterization of the Human Circulating Microbiome. Front Microbiol. 2018, 9, 3266. [Google Scholar] [CrossRef]
- Buck, A.H.; Coakley, G.; Simbari, F.; McSorley, H.J.; Quintana, J.F.; Le Bihan, T.; Kumar, S.; Abreu-Goodger, C.; Lear, M.; Harcus, Y.; et al. Exosomes secreted by nematode parasites transfer small RNAs to mammalian cells and modulate innate immunity. Nat. Commun. 2014, 5, 5488. [Google Scholar] [CrossRef]
- Lunavat, T.R.; Cheng, L.; Kim, D.K.; Bhadury, J.; Jang, S.C.; Lässer, C.; Sharples, R.A.; López, M.D.; Nilsson, J.; Gho, Y.S.; et al. Small RNA deep sequencing discriminates subsets of extracellular vesicles released by melanoma cells - Evidence of unique microRNA cargos. RNA Biol. 2015, 12, 810–823. [Google Scholar] [CrossRef]
- Chevillet, J.R.; Kang, Q.; Ruf, I.K.; Briggs, H.A.; Vojtech, L.N.; Hughes, S.M.; Cheng, H.H.; Arroyo, J.D.; Meredith, E.K.; Gallichotte, E.N.; et al. Quantitative and stoichiometric analysis of the microRNA content of exosomes. Proc. Natl. Acad. Sci. USA 2014, 111, 14888–14893. [Google Scholar] [CrossRef]
- Nolte-’t Hoen, E.N.; Buermans, H.P.; Waasdorp, M.; Stoorvogel, W.; Wauben, M.H.; ’t Hoen, P.A. Deep sequencing of RNA from immune cell-derived vesicles uncovers the selective incorporation of small non-coding RNA biotypes with potential regulatory functions. Nucleic. Acids Res. 2012, 40, 9272–9285. [Google Scholar] [CrossRef] [PubMed]
- Baglio, S.R.; Rooijers, K.; Koppers-Lalic, D.; Verweij, F.J.; Lanzón, M.P.; Zini, N.; Naaijkens, B.; Perut, F.; Niessen, H.W.; Baldini, N.; et al. Human bone marrow- and adipose-mesenchymal stem cells secrete exosomes enriched in distinctive miRNA and tRNA species. Stem Cell Res. Ther. 2015, 6, 127. [Google Scholar] [CrossRef]
- Cheng, L.; Sharples, R.A.; Scicluna, B.J.; Hill, A.F. Exosomes provide a protective and enriched source of miRNA for biomarker profiling compared to intracellular and cell-free blood. J. Extracel. Vesicles 2014, 3, 23743. [Google Scholar] [CrossRef] [PubMed]
- Endzeliņš, E.; Berger, A.; Melne, V.; Bajo-Santos, C.; Soboļevska, K.; Ābols, A.; Rodriguez, M.; Šantare, D.; Rudņickiha, A.; Lietuvietis, V.; et al. Detection of circulating miRNAs: Comparative analysis of extracellular vesicle-incorporated miRNAs and cell-free miRNAs in whole plasma of prostate cancer patients. BMC cancer 2017, 17, 730. [Google Scholar]
- Tian, F.; Shen, Y.; Chen, Z.; Li, R.; Ge, Q. No Significant Difference between Plasma miRNAs and Plasma-Derived Exosomal miRNAs from Healthy People. BioMed Res. Int. 2017. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.X.; Fan, X.; Drummond, C.A.; Majumder, R.; Xie, Y.; Chen, T.; Liu, L.; Haller, S.T.; Brewster, P.S.; Dworkin, L.D.; et al. MicroRNA profiling in kidney disease: Plasma versus plasma-derived exosomes. Gene 2017, 627, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Turchinovich, A.; Drapkina, O.; Tonevitsky, A. Transcriptome of Extracellular Vesicles: State-of-the-Art. Front. Immunol. 2019, 10, 202. [Google Scholar] [CrossRef] [PubMed]
- Guduric-Fuchs, J.; O’Connor, A.; Camp, B.; O’Neill, C.L.; Medina, R.J.; Simpson, D.A. Selective extracellular vesicle-mediated export of an overlapping set of microRNAs from multiple cell types. BMC Genomics 2012, 13, 357. [Google Scholar] [CrossRef] [PubMed]
- Albitar, M.; Manshouri, T.; Shen, Y.; Liu, D.; Beran, M.; Kantarjian, H.M.; Rogers, A.; Jilani, I.; Lin, C.W.; Pierce, S.; et al. Myelodysplastic syndrome is not merely “preleukemia”. Blood 2002, 100, 791–798. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhao, L.; Wei, X.; Guo, Q.; Zhu, X.; Wei, R.; Yin, X.; Zhang, Y.; Wang, B.; Li, X. Integrated bioinformatic analysis of microarray data reveals shared gene signature between MDS and AML. Oncol. Lett. 2018, 16, 5147–5159. [Google Scholar] [CrossRef] [PubMed]
- Dostalova Merkerova, M.; Krejcik, Z.; Votavova, H.; Belickova, M.; Vasikova, A.; Cermak, J. Distinctive microRNA expression profiles in CD34+ bone marrow cells from patients with myelodysplastic syndrome. Eur. J. Hum. Genet. 2011, 19, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Bernasconi, P. Molecular pathways in myelodysplastic syndromes and acute myeloid leukemia: Relationships and distinctions-a review. Br. J. Haematol. 2008, 142, 695–708. [Google Scholar] [CrossRef] [PubMed]
- Pons, A.; Nomdedeu, B.; Navarro, A.; Gaya, A.; Gel, B.; Diaz, T.; Valera, S.; Rozman, M.; Belkaid, M.; Montserrat, E.; et al. Hematopoiesis-related microRNA expression in myelodysplastic syndromes. Leuk Lymphoma 2009, 50, 1854–1859. [Google Scholar] [CrossRef]
- Kotaki, R.; Koyama-Nasu, R.; Yamakawa, N.; Kotani, A. miRNAs in Normal and Malignant Hematopoiesis. Int. J. Mol. Sci. 2017, 18, 1495. [Google Scholar] [CrossRef]
- Chang, T.C.; Wentzel, E.A.; Kent, O.A.; Ramachandran, K.; Mullendore, M.; Lee, K.H.; Feldmann, G.; Yamakuchi, M.; Ferlito, M.; Lowenstein, C.J.; et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol. Cell 2007, 26, 745–752. [Google Scholar] [CrossRef]
- Votavova, H.; Grmanova, M.; Merkerova, M.D.; Belickova, M.; Vasikova, A.; Neuwirtova, R.; Cermak, J. Differential expression of microRNAs in CD34+ cells of 5q- syndrome. J. Hematol. Oncol. 2011, 4, 1. [Google Scholar] [CrossRef]
- Merkerova, M.D.; Krejcik, Z.; Belickova, M.; Hrustincova, A.; Klema, J.; Stara, E.; Zemanova, Z.; Michalova, K.; Cermak, J.; Jonasova, A. Genome-wide miRNA profiling in myelodysplastic syndrome with del(5q) treated with lenalidomide. Eur. J. Haematol. 2015, 95, 35–43. [Google Scholar] [CrossRef]
- Shikama, Y.; Cao, M.; Ono, T.; Feng, X.; Noji, H.; Kimura, H.; Ogawa, K.; Suzuki, Y.; Ikeda, K.; Takeishi, Y.; et al. Reduction of c-Fos via Overexpression of miR-34a Results in Enhancement of TNF- Production by LPS in Neutrophils from Myelodysplastic Syndrome Patients. PLoS ONE 2016, 11, e0158527. [Google Scholar] [CrossRef]
- Guo, S.; Lu, J.; Schlanger, R.; Zhang, H.; Wang, J.Y.; Fox, M.C.; Purton, L.E.; Fleming, H.H.; Cobb, B.; Merkenschlager, M.; et al. MicroRNA miR-125a controls hematopoietic stem cell number. Proc. Natl. Acad. Sci. USA 2010, 107, 14229–14234. [Google Scholar] [CrossRef]
- Tatsumi, N.; Hojo, N.; Yamada, O.; Ogawa, M.; Katsura, Y.; Kawata, S.; Morii, E.; Sakamoto, H.; Inaba, R.; Tsuda, A.; et al. Deficiency in WT1-targeting microRNA-125a leads to myeloid malignancies and urogenital abnormalities. Oncogene 2016, 35, 1003–1014. [Google Scholar] [CrossRef]
- Li, Q.; He, X.; Yu, Q.; Wu, Y.; Du, M.; Chen, J.; Peng, F.; Zhang, W.; Chen, J.; Wang, Y.; et al. The Notch signal mediates macrophage polarization by regulating miR-125a/miR-99b expression. Artif. Cells Nanomed. Biotechnol. 2019, 47, 833–843. [Google Scholar] [CrossRef] [PubMed]
- Bi, L.; Sun, L.; Jin, Z.; Zhang, S.; Shen, Z. MicroRNA-10a/b are regulators of myeloid differentiation and acute myeloid leukemia. Oncology Lett. 2018, 15, 5611–5619. [Google Scholar] [CrossRef] [PubMed]
- Dumas, P.Y.; Mansier, O.; Prouzet-Mauleon, V.; Koya, J.; Villacreces, A.; de la Grange, P.B.; Paz, D.L.; Bidet, A.; Pasquet, J.M.; Praloran, V.; et al. MiR-10a and HOXB4 are overexpressed in atypical myeloproliferative neoplasms. BMC Cancer 2018, 18, 1098. [Google Scholar] [CrossRef] [PubMed]
- Felli, N.; Fontana, L.; Pelosi, E.; Botta, R.; Bonci, D.; Facchiano, F.; Liuzzi, F.; Lulli, V.; Morsilli, O.; Santoro, S.; et al. MicroRNAs 221 and 222 inhibit normal erythropoiesis and erythroleukemic cell growth via kit receptor down-modulation. Proc. Natl. Acad. Sci. USA 2005, 102, 18081–18086. [Google Scholar] [CrossRef] [PubMed]
- Rommer, A.; Steinleitner, K.; Hackl, H.; Schneckenleithner, C.; Engelmann, M.; Scheideler, M.; Vlatkovic, I.; Kralovics, R.; Cerny-Reiterer, S.; Valent, P.; et al. Overexpression of primary microRNA 221/222 in acute myeloid leukemia. BMC cancer 2013, 13, 364. [Google Scholar] [CrossRef]
- Xiao, Y.; Su, C.; Deng, T. miR-223 decreases cell proliferation and enhances cell apoptosis in acute myeloid leukemia via targeting FBXW7. Oncol. Lett. 2016, 12, 3531–3536. [Google Scholar] [CrossRef]
- Wang, X.S.; Gong, J.N.; Yu, J.; Wang, F.; Zhang, X.H.; Yin, X.L.; Tan, Z.Q.; Luo, Z.M.; Yang, G.H.; Shen, C.; et al. MicroRNA-29a and microRNA-1423-p are regulators of myeloid differentiation and acute myeloid leukemia. Blood 2012, 119, 4992–5004. [Google Scholar] [CrossRef]
- Han, Y.C.; Park, C.Y.; Bhagat, G.; Zhang, J.; Wang, Y.; Fan, J.B.; Liu, M.; Zou, Y.; Weissman, I.L.; Gu, H. microRNA-29a induces aberrant self-renewal capacity in hematopoietic progenitors, biased myeloid development, and acute myeloid leukemia. J. Exp. Med. 2010, 207, 475–489. [Google Scholar] [CrossRef]
- Zhou, B.; Wang, S.; Mayr, C.; Bartel, D.P.; Lodish, H.F. miR-150, a microRNA expressed in mature B and T cells, blocks early B cell development when expressed prematurely. Proc. Natl. Acad. Sci. USA 2007, 104, 7080–7085. [Google Scholar] [CrossRef]
- Xiao, C.; Calado, D.P.; Galler, G.; Thai, T.H.; Patterson, H.C.; Wang, J.; Rajewsky, N.; Bender, T.P.; Rajewsky, K. MiR-150 controls B cell differentiation by targeting the transcription factor c-Myb. Cell 2007, 131, 146–159. [Google Scholar] [CrossRef] [PubMed]
- Hussein, K.; Theophile, K.; Büsche, G.; Schlegelberger, B.; Göhring, G.; Kreipe, H.; Bock, O. Significant inverse correlation of microRNA-150/MYB and microRNA-222/p27 in myelodysplastic syndrome. Leuk Res 2010, 34, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Theocharis, A.D.; Skandalis, S.S.; Gialeli, C.; Karamanos, N.K. Extracellular matrix structure. Adv. Drug. Deliv. Rev. 2016, 97, 4–27. [Google Scholar] [CrossRef] [PubMed]
- Piperigkou, Z.; Karamanos, N.K. Dynamic Interplay between miRNAs and the Extracellular Matrix Influences the Tumor Microenvironment. Trends Biochem. Sci. 2019, 44, 1076–1088. [Google Scholar] [CrossRef]
- Harada, H.; Harada, Y. Recent advances in myelodysplastic syndromes: Molecular pathogenesis and its implications for targeted therapies. Cancer Sci. 2015, 106, 329–336. [Google Scholar] [CrossRef]
- Nigita, G.; Distefano, R.; Veneziano, D.; Romano, G.; Rahman, M.; Wang, K.; Pass, H.; Croce, C.M.; Acunzo, M.; Nana-Sinkam, P. Tissue and exosomal miRNA editing in Non-Small Cell Lung. Cancer Sci. Rep. 2018, 8, 10222. [Google Scholar] [CrossRef]
- Crews, L.A.; Jiang, Q.; Balaian, L.; Leu, H.; Delos Santos, N.; Court, A.C.; Sadarangani, A.; Morris, S.; Storb, R.F.; Kulidjian, A.; et al. Decoding the RNA Editome of Normal Versus Malignant Progenitor Cell Aging Short Title: RNA Editing in Normal Aging and MDS/AML. Stem Cells. Blood 2016, 128, 1519. [Google Scholar] [CrossRef]
- Diamantidis, M.; Dimoudis, S.; Klonizakis, P.; Badekas, K.; Koutourli, K.; Haralambidou-Vranitsa, S.; Ioannidou-Papagiannaki, E. The role of apoptosis and current therapeutic challenges in myelodysplastic syndromes. Hippokratia 2007, 11, 178–182. [Google Scholar]
- Park, S.J.; Kim, J.M.; Kim, J.; Hur, J.; Park, S.; Kim, K.; Shin, H.J.; Chwae, Y.J. Molecular mechanisms of biogenesis of apoptotic exosome-like vesicles and their roles as damage-associated molecular patterns. PNAS 2018, 115, E11721–E11730. [Google Scholar] [CrossRef]
- Caruso, S.; Poon, I.K. Apoptotic Cell-Derived Extracellular Vesicles: More Than Just Debris. Front. Immunol. 2018, 9, 1486. [Google Scholar] [CrossRef]
- Benetatos, L.; Hatzimichael, E.; Londin, E.; Vartholomatos, G.; Loher, P.; Rigoutsos, I.; Briasoulis, E. The microRNAs within the DLK1-DIO3 genomic region: Involvement in disease pathogenesis. Cell. Mol. Life Sci. 2013, 70, 795–814. [Google Scholar] [CrossRef] [PubMed]
- Krejčík, Z.; Beličková, M.; Hruštincová, A.; Kléma, J.; Zemanová, Z.; Michalová, K.; Čermák, J.; Jonášová, A.; Merkerová, M.D. Aberrant expression of the microRNA cluster in 14q32 is associated with del(5q) myelodysplastic syndrome and lenalidomide treatment. Cancer Genet. 2015, 208, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Merkerova, M.D.; Remesova, H.; Krejcik, Z.; Loudova, N.; Hrustincova, A.; Szikszai, K.; Cermak, J.; Jonasova, A.; Belickova, M. Relationship between Altered miRNA Expression and DNA Methylation of the DLK1-DIO3 Region in Azacitidine-Treated Patients with Myelodysplastic Syndromes and Acute Myeloid Leukemia with Myelodysplasia-Related Changes. Cells 2018, 7, 138. [Google Scholar] [CrossRef]
- Li, Z.; Lu, J.; Sun, M.; Mi, S.; Zhang, H.; Luo, R.T.; Chen, P.; Wang, Y.; Yan, M.; Qian, Z.; et al. Distinct microRNA expression profiles in acute myeloid leukemia with common translocations. Proc. Natl. Acad. Sci. USA 2008, 105, 15535–15540. [Google Scholar] [CrossRef] [PubMed]
- Shibayama, Y.; Kondo, T.; Ohya, H.; Fujisawa, S.; Teshima, T.; Iseki, K. Upregulation of microRNA-126-5p is associated with drug resistance to cytarabine and poor prognosis in AML patients. Oncol. Rep. 2015, 33, 2176–2182. [Google Scholar] [CrossRef]
- Dakir, E.; Mollinedo, F. Genome-wide miRNA profiling and pivotal roles of miRs 125a-5p and 17-92 cluster in human neutrophil maturation and differentiation of acute myeloid leukemia cells. Oncotarget 2019, 10, 5313–5331. [Google Scholar] [CrossRef]
- Alemdehy, M.F.; Haanstra, J.R.; de Looper, H.W.; van Strien, P.M.; Verhagen-Oldenampsen, J.; Caljouw, Y.; Sanders, M.A.; Hoogenboezem, R.; de Ru, A.H.; Janssen, G.M.; et al. ICL-induced miR139-3p and miR199a-3p have opposite roles in hematopoietic cell expansion and leukemic transformation. Blood 2015, 125, 3937–3948. [Google Scholar] [CrossRef]
- Grivna, S.T.; Beyret, E.; Wang, Z.; Lin, H. A novel class of small RNAs in mouse spermatogenic cells. Genes Dev. 2006, 20, 1709–1714. [Google Scholar] [CrossRef]
- Malone, C.D.; Hannon, G.J. Small RNAs as Guardians of the Genome. Cell 2009, 136, 656–668. [Google Scholar] [CrossRef]
- Martinez, V.D.; Vucic, E.A.; Thu, K.L.; Hubaux, R.; Enfield, K.S.; Pikor, L.A.; Becker-Santos, D.D.; Brown, C.J.; Lam, S.; Lam, W.L. Unique somatic and malignant expression patterns implicate PIWI-interacting RNAs in cancer-type specific biology. Sci. Rep. 2015, 5, 10423. [Google Scholar] [CrossRef]
- Gebetsberger, J.; Wyss, L.; Mleczko, A.M.; Reuther, J.; Polacek, N. A tRNA-derived fragment competes with mRNA for ribosome binding and regulates translation during stress. RNA Biol. 2017, 14, 1364–1373. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Silva, M.R.; Cabrera-Cabrera, F.; Güida, M.C.; Cayota, A. Hints of tRNA-Derived Small RNAs Role in RNA Silencing Mechanisms. Genes (Basel) 2012, 3, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Elbarbary, R.A.; Takaku, H.; Uchiumi, N.; Tamiya, H.; Abe, M.; Takahashi, M.; Nishida, H.; Nashimoto, M. Modulation of gene expression by human cytosolic tRNase Z(L) through 5’-half-tRNA. PLoS ONE 2009, 4, e5908. [Google Scholar] [CrossRef] [PubMed]
- Honda, S.; Loher, P.; Shigematsu, M.; Palazzo, J.P.; Suzuki, R.; Imoto, I.; Rigoutsos, I.; Kirino, Y. Sex hormone-dependent tRNA halves enhance cell proliferation in breast and prostate cancers. Proc. Natl. Acad. Sci. USA 2015, 112, 3816. [Google Scholar] [CrossRef]
- Goodarzi, H.; Liu, X.; Nguyen, H.C.B.; Zhang, S.; Fish, L.; Tavazoie, S.F. Endogenous tRNA-Derived Fragments Suppress Breast Cancer Progression via YBX1 Displacement. Cell 2015, 161, 790–802. [Google Scholar] [CrossRef]
- Maute, R.L.; Schneider, C.; Sumazin, P.; Holmes, A.; Califano, A.; Basso, K.; Dalla-Favera, R. tRNA-derived microRNA modulates proliferation and the DNA damage response and is down-regulated in B cell lymphoma. Proc. Natl. Acad. Sci. USA 2013, 110, 1404–1409. [Google Scholar] [CrossRef]
- Guo, Y.; Bosompem, A.; Mohan, S.; Erdogan, B.; Ye, F.; Vickers, K.C.; Sheng, Q.; Zhao, S.; Li, C.I.; Su, P.F.; et al. Transfer RNA detection by small RNA deep sequencing and disease association with myelodysplastic syndromes. BMC Genom. 2015, 16, 727. [Google Scholar] [CrossRef]
- Guo, Y.; Strickland, S.A.; Mohan, S.; Li, S.; Bosompem, A.; Vickers, K.C.; Zhao, S.; Sheng, Q.; Kim, A.S. MicroRNAs and tRNA-derived fragments predict the transformation of myelodysplastic syndromes to acute myeloid leukemia. Leuk Lymphoma 2017, 58, 1–15. [Google Scholar] [CrossRef]
- Hornick, N.I.; Doron, B.; Abdelhamed, S.; Huan, J.; Harrington, C.A.; Shen, R.; Cambronne, X.A.; Verghese, S.C.; Kurre, P. AML suppresses hematopoiesis by releasing exosomes that contain microRNAs targeting c-MYB. Sci. Signal. 2016, 9, ra88. [Google Scholar] [CrossRef]
- Huson, D.H.; Beier, S.; Flade, I.; Górska, A.; El-Hadidi, M.; Mitra, S.; Ruscheweyh, H.J.; Tappu, R. MEGAN Community Edition - Interactive Exploration and Analysis of Large-Scale Microbiome Sequencing Data. PloS Comput. Biol. 2016, 12, e1004957. [Google Scholar] [CrossRef]
- Lu, Y.; Baras, A.S.; Halushka, M.K. miRge 2.0 for comprehensive analysis of microRNA sequencing data. BMC Bioinform. 2018, 19, 275. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Ishwaran, H.; Kogalur, U.B.; Gorodeski, E.Z.; Minn, A.J.; Lauer, M.S. High-dimensional variable selection for survival data. J. Am. Stat. Assoc. 2010, 105, 205–217. [Google Scholar] [CrossRef]
- Vlachos, I.S.; Zagganas, K.; Paraskevopoulou, M.D.; Georgakilas, G.; Karagkouni, D.; Vergoulis, T.; Dalamagas, T.; Hatzigeorgiou, A.G. DIANA-miRPath v3.0: Deciphering microRNA function with experimental support. Nucleic Acids Res. 2015, 43, W460–W466. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| KEGG Pathway | p-Value |
|---|---|
| plasma: MDS vs. CTR | |
| Mucin type O-Glycan biosynthesis | 9.77 × 10−15 |
| Proteoglycans in cancer | 6.05 × 10−9 |
| ErbB signaling pathway | 2.80 × 10−8 |
| Ras signaling pathway | 2.02 × 10−7 |
| Axon guidance | 2.02 × 10−5 |
| Pathways in cancer | 2.02 × 10−5 |
| Rap1 signaling pathway | 3.30 × 10−5 |
| Lysine degradation | 3.33 × 10−5 |
| Glioma | 6.23 × 10−5 |
| Signaling pathways regulating pluripotency of stem cells | 9.51 × 10−5 |
| EVs: MDS vs. CTR | |
| ECM-receptor interaction | 1.16 × 10−26 |
| Fatty acid biosynthesis | 1.41 × 10−8 |
| ErbB signaling pathway | 1.41 × 10−8 |
| Proteoglycans in cancer | 1.57 × 10−8 |
| Axon guidance | 3.54 × 10−8 |
| Glioma | 5.42 × 10−8 |
| Mucin type O-Glycan biosynthesis | 5.18 × 10−6 |
| Estrogen signaling pathway | 3.52 × 10−5 |
| Focal adhesion | 5.71 × 10−5 |
| Signaling pathways regulating pluripotency of stem cells | 5.71 × 10−5 |
| plasma: early vs. advanced MDS | |
| Amphetamine addiction | 1.56 × 10−7 |
| Signaling pathways regulating pluripotency of stem cells | 5.60 × 10−6 |
| Transcriptional misregulation in cancer | 1.35 × 10−5 |
| Gap junction | 2.31 × 10−5 |
| Glioma | 3.38 × 10−5 |
| FoxO signaling pathway | 5.34 × 10−5 |
| Hippo signaling pathway | 1.16 × 10−4 |
| ErbB signaling pathway | 1.72 × 10−4 |
| Proteoglycans in cancer | 1.75 × 10−4 |
| TGF-beta signaling pathway | 2.54 × 10−4 |
| EVs: early vs. advanced MDS | |
| Biotin metabolism | 7.16 × 10−3 |
| Central carbon metabolism in cancer | 7.16 × 10−3 |
| Signaling pathways regulating pluripotency of stem cells | 7.16 × 10−3 |
| Lysine degradation | 9.83 × 10−3 |
| TGF-beta signaling pathway | 9.83 × 10−3 |
| Steroid biosynthesis | 1.29 × 10−2 |
| Glioma | 1.39 × 10−2 |
| RNA transport | 1.41 × 10−2 |
| ErbB signaling pathway | 1.45 × 10−2 |
| Morphine addiction | 1.45 × 10−2 |
| sncRNA | Univariate Cox Regression Analysis | Prediction Model | |||
|---|---|---|---|---|---|
| Univariate Cox Regression, p-Value | Permutation, p-Value | Hazard Ratio | Coefficient | Cross-Validation, p-Value | |
| Total plasma | |||||
| miR-1260b | 0.0007 | 6 × 10−4 | 0.441 | −0.631 | 0.0002 |
| miR-3191-3p | 0.0009 | 9 × 10−4 | 0.338 | n.a. | n.s. |
| miR-328-3p | 0.0009 | 8 × 10−4 | 0.474 | −0.24 | 0.0008 |
| EV fraction | |||||
| miR-1237-3p | 2 × 10−5 | <1 × 10−7 | 20.135 | 0.615 | 5 × 10−7 |
| U33 | 0.0006 | 6 × 10−4 | 2.499 | 0.917 | 0.0002 |
| hsa_piR_019420 | 0.001 | <1 × 10−7 | 20.135 | −0.106 | 0.0008 |
| miR-548av-5p | 0.001 | 0.001 | 0.217 | −1.01 | 0.0009 |
| Variable | HR | 95.0% CI for HR | p | |
|---|---|---|---|---|
| Lower | Upper | |||
| Age | 1.044 | 0.945 | 1.153 | 0.397 |
| Blasts | 0.882 | 0.767 | 1.013 | 0.076 |
| Hemoglobin | 1.001 | 0.961 | 1.043 | 0.948 |
| Neutrophils | 0.778 | 0.577 | 1.048 | 0.099 |
| Platelets | 1.003 | 0.993 | 1.013 | 0.602 |
| IPSS-R score | 1.410 | 0.840 | 2.366 | 0.193 |
| Combined score (total plasma) | 1.764 | 0.666 | 4.677 | 0.254 |
| Combined score (EVs) | 5.866 | 2.262 | 15.210 | <0.001 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hrustincova, A.; Krejcik, Z.; Kundrat, D.; Szikszai, K.; Belickova, M.; Pecherkova, P.; Klema, J.; Vesela, J.; Hruba, M.; Cermak, J.; et al. Circulating Small Noncoding RNAs Have Specific Expression Patterns in Plasma and Extracellular Vesicles in Myelodysplastic Syndromes and Are Predictive of Patient Outcome. Cells 2020, 9, 794. https://doi.org/10.3390/cells9040794
Hrustincova A, Krejcik Z, Kundrat D, Szikszai K, Belickova M, Pecherkova P, Klema J, Vesela J, Hruba M, Cermak J, et al. Circulating Small Noncoding RNAs Have Specific Expression Patterns in Plasma and Extracellular Vesicles in Myelodysplastic Syndromes and Are Predictive of Patient Outcome. Cells. 2020; 9(4):794. https://doi.org/10.3390/cells9040794
Chicago/Turabian StyleHrustincova, Andrea, Zdenek Krejcik, David Kundrat, Katarina Szikszai, Monika Belickova, Pavla Pecherkova, Jiri Klema, Jitka Vesela, Monika Hruba, Jaroslav Cermak, and et al. 2020. "Circulating Small Noncoding RNAs Have Specific Expression Patterns in Plasma and Extracellular Vesicles in Myelodysplastic Syndromes and Are Predictive of Patient Outcome" Cells 9, no. 4: 794. https://doi.org/10.3390/cells9040794
APA StyleHrustincova, A., Krejcik, Z., Kundrat, D., Szikszai, K., Belickova, M., Pecherkova, P., Klema, J., Vesela, J., Hruba, M., Cermak, J., Hrdinova, T., Krijt, M., Valka, J., Jonasova, A., & Dostalova Merkerova, M. (2020). Circulating Small Noncoding RNAs Have Specific Expression Patterns in Plasma and Extracellular Vesicles in Myelodysplastic Syndromes and Are Predictive of Patient Outcome. Cells, 9(4), 794. https://doi.org/10.3390/cells9040794