Validation of Reference Genes for Gene Expression Studies by RT-qPCR in HepaRG Cells during Toxicity Testing and Disease Modelling

 ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Viability Assays
2.3. Total RNA Isolation
2.4. Real-Time Polymerase Chain Reaction (RT-qPCR)
2.5. Identification of Suitable Reference Genes
2.6. Quality Control on Post-RT-qPCR Data
2.7. RT-qPCR Efficiency Evaluation
2.7.1. Determination of Reference Gene Expression Stability
2.7.2. Assessment of Positive and Negative Controls
2.7.3. Deviating Sample Normalisation Factors
2.8. Statistical Analysis
3. Results
3.1. Cell Viability: Total Adenosine Triphosphate (ATP) and PrestoBlue Assays
3.2. RT-qPCR Efficiency
3.3. RNA Transcription Levels of Putative Reference Genes in Various Experimental Settings
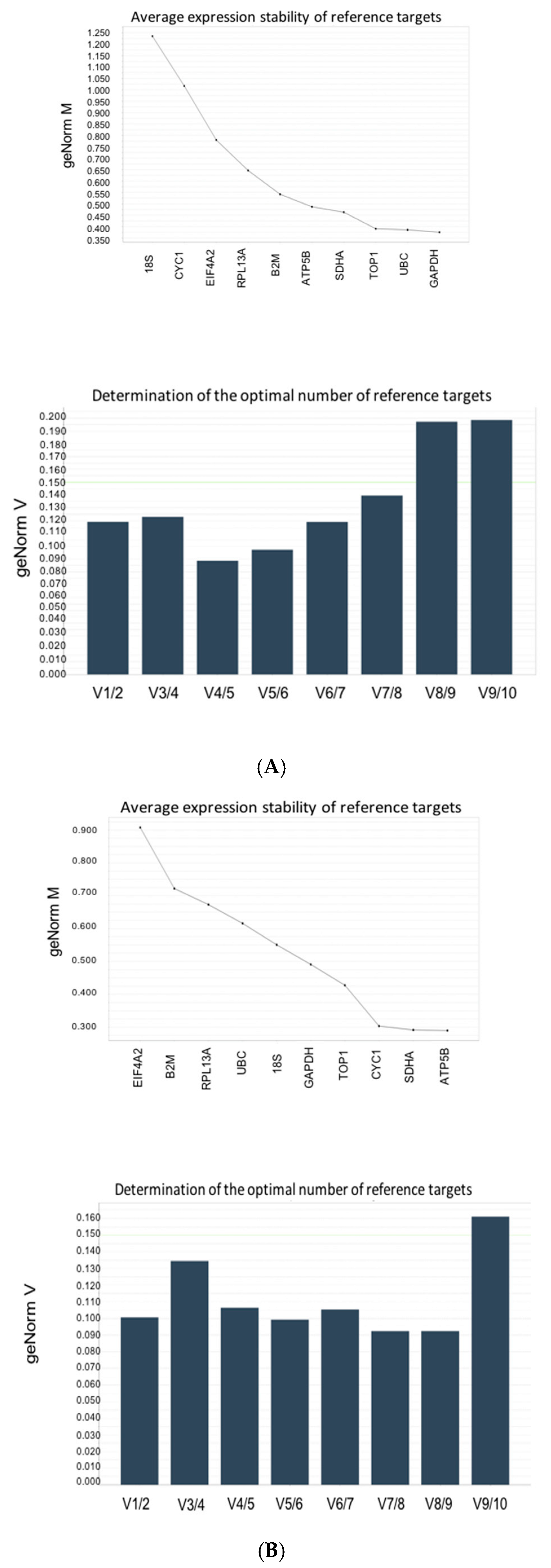
3.4. Stability Analysis Per Experimental Condition
3.5. Stability of RNA Transcription Following CPZ and APAP Stimulation (Data from CYP3A4 Analysis)
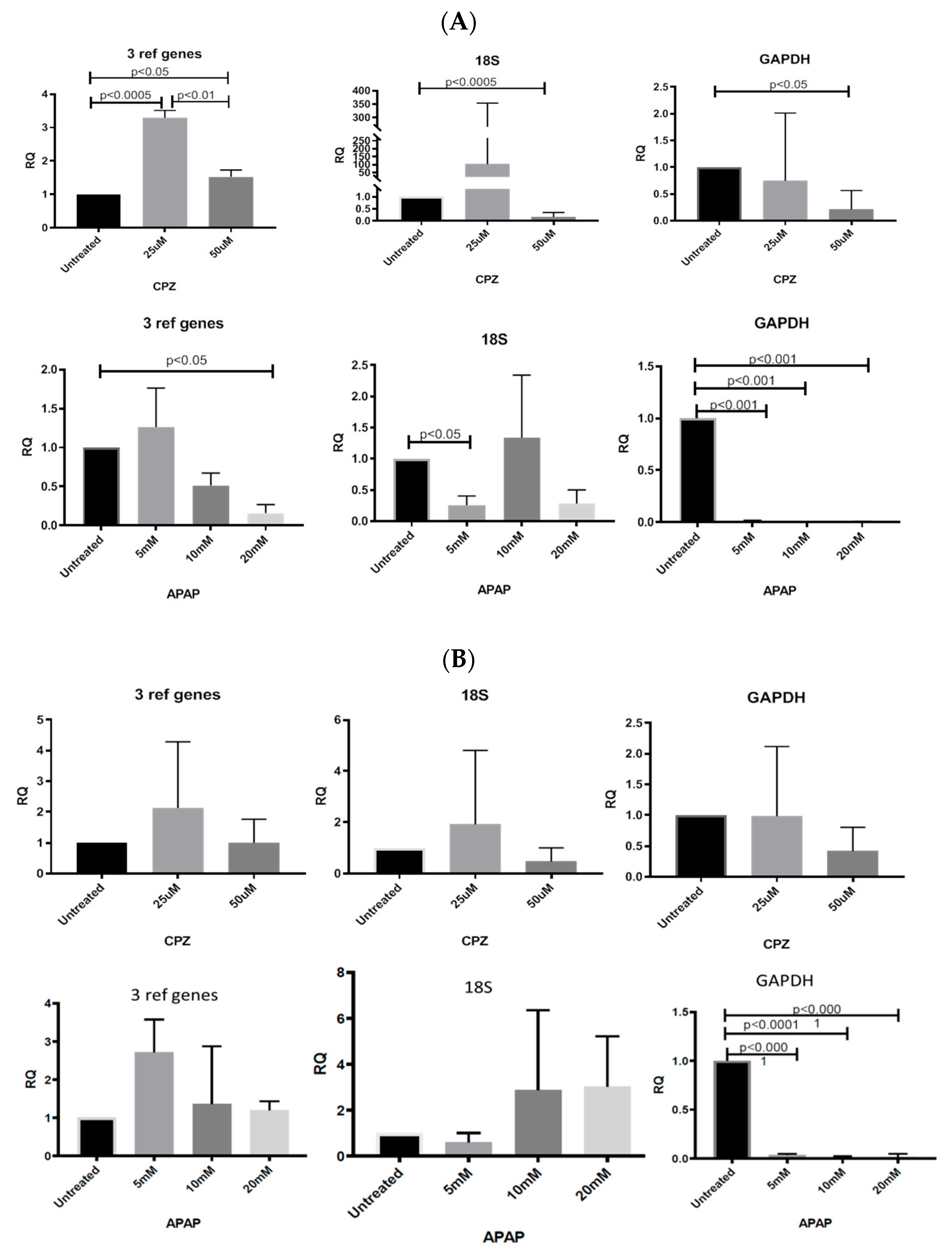
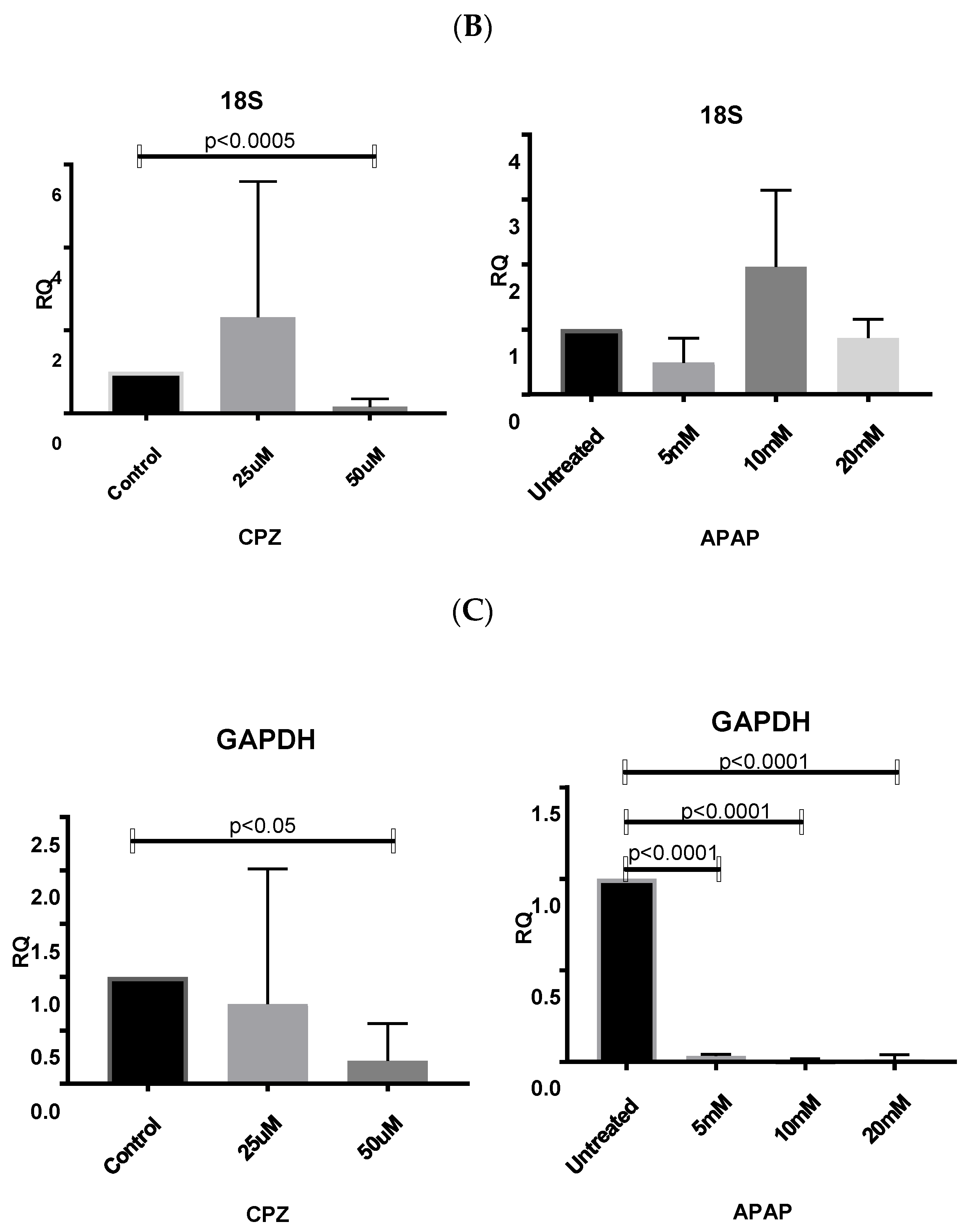
3.6. Application of Identified Reference Genes
3.6.1. Normalizing Data from CPZ-Treated HepaRG Cells
3.6.2. Normalising Data for APAP-Treated HepaRG Cells
3.6.3. Normalising Data for Low and Highly Abundant Liver Gene
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE guidelines: Minimum information for publication of quantitative real-time RT-QPCR experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Bonefeld, B.E.; Elfving, B.; Wegener, G. Reference genes for normalization: A study of rat brain tissue. Synapse 2008, 62, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Dheda, K.; Huggett, J.F.; Chang, J.S.; Kim, L.U.; Bustin, S.A.; Johnson, M.A.; Rook, G.A.; Zumla, A. The implications of using an inappropriate reference gene for real-time reverse transcription RT-QPCR data normalization. Anal. Biochem. 2005, 344, 141–143. [Google Scholar] [CrossRef]
- Thellin, O.; Zorzi, W.; Lakaye, B.; De Borman, B.; Coumans, B.; Hennen, G.; Grisar, T.; Igout, A.; Heinen, E. Housekeeping genes as internal standards: Use and limits. J. Biotechnol. 1999, 75, 291–295. [Google Scholar] [CrossRef]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-RT-QPCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3, RESEARCH0034. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W.; Tichopad, A.; Prgomet, C.; Neuvians, T.P. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper--Excel-based tool using pair-wise correlations. Biotechnol. Lett. 2004, 26, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Kubista, M.; Andrade, J.M.; Bengtsson, M.; Forootan, A.; Jonák, J.; Lind, K.; Sindelkae, R.; Sjöbacka, R.; Sjögreend, B.; Strömbom, L.; et al. The real-time polymerase chain reaction. Mol. Asp. Med. 2006, 27, 95–125. [Google Scholar] [CrossRef]
- Parent, R.; Marion, M.J.; Furio, L.; Trépo, C.; Petit, M.A. Origin and characterization of a human bipotent liver progenitor cell line. Gastroenterology 2004, 126, 1147–1156. [Google Scholar] [CrossRef]
- Cerec, V.; Glaise, D.; Garnier, D.; Morosan, S.; Turlin, B.; Drenou, B.; Gripon, P.; Kremsdorf, D.; Guguen-Guillouzo, C.; Corlu, A. Transdifferentiation of hepatocyte-like cells from the human hepatoma HepaRG cell line through bipotent progenitor. Hepatology 2007, 45, 957–967. [Google Scholar] [CrossRef]
- Gripon, P.; Rumin, S.; Urban, S.; Le Seyec, J.; Glaise, D.; Cannie, I.; Guyomard, C.; Lucas, J.; Trepo, C.; Guguen-Guillouzo, C. Infection of a human hepatoma cell line by hepatitis B virus. Proc. Natl. Acad. Sci. USA 2002, 99, 15655–15660. [Google Scholar] [CrossRef]
- Guillouzo, A.; Corlu, A.; Aninat, C.; Glaise, D.; Morel, F.; Guguen-Guillouzo, C. The human hepatoma HepaRG cells: A highly differentiated model for studies of liver metabolism and toxicity of xenobiotics. Chem. Biol. Interact. 2007, 168, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Ceelen, L.; De Spiegelaere, W.; David, M.; De Craene, J.; Vinken, M.; Vanhaecke, T.; Rogiers, V. Critical selection of reliable reference genes for gene expression study in the HepaRG cell line. Biochem. Pharmacol. 2011, 81, 1255–1261. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ceelen, L.; De Craene, J.; De Spiegelaere, W. Evaluation of normalization strategies used in real-time quantitative RT-QPCR experiments in HepaRG cell line studies. Clin. Chem. 2014, 60, 451–454. [Google Scholar] [CrossRef]
- Prinz, F.; Schlange, T.; Asadullah, K. Believe it or not: How much can we rely on published data on potential drug targets? Nat. Rev. Drug Discov. 2011, 10, 712–713. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Brzeszczynska, J.; Samuel, K.; Black, J.; Palakkan, A.; Anderson, R.; Gallagher, R.; Ross, J.A. Efficient episomal reprogramming of blood mononuclear cells and differentiation to hepatocytes with functional drug metabolism. Experimental Cell Research. 2015, 338, 203–213. [Google Scholar] [CrossRef]
- Bustin, S.A.; Benes, V.; Garson, J.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.; et al. The need for transparency and good practices in the qRT-QPCR literature. Nat. Methods. 2013, 10, 1063–1067. [Google Scholar] [CrossRef]
- Gamal, W.; Treskes, P.; Samuel, K.; Sullivan, G.J.; Siller, R.; Srsen, V.; Morgan, K.; Bryans, A.; Kozlowska, A.; Koulovasilopoulos, A.; et al. Low-dose acetaminophen induces early disruption of cell-cell tight junctions in human hepatic cells and mouse liver. Sci. Rep. 2017, 7, 37541. [Google Scholar] [CrossRef]
- Larrey, D.; Ripault, M.P. Hepatotoxicity of Psychotropic Drugs and Drugs of Abuse. In Drug-Induced Liver Disease; Kaplowitz, N., DeLeve, L.D., Eds.; Academic Press: San Diego, CA, USA, 2013; pp. 443–462. [Google Scholar]
- Morgan, K.; Martucci, N.; Kozlowska, A.; Gamal, W.; Brzeszczyński, F.; Treskes, P.; Samuel, K.; Hayes, P.; Nelson, L.; Bagnaninchi, P.; et al. Chlorpromazine toxicity is associated with disruption of cell membrane integrity and initiation of a pro-inflammatory response in the HepaRG hepatic cell line. Biomed. Pharmacother. 2019, 111, 1408–1416. [Google Scholar] [CrossRef]
- Brzeszczyńska, J.; Meyer, A.; McGregor, R.; Schilb, A.; Degen, S.; Tadini, V.; Johns, N.; Langen, R.; Schols, A.; Glass, D.J.; et al. Alterations in the in vitro and in vivo regulation of muscle regeneration in healthy ageing and the influence of sarcopenia. J. Cachexia. Sarcop. Muscle 2017, 9, 93–105. [Google Scholar] [CrossRef]
- Radonić, A.S.; Thulke, I.M.; Mackay, O.; Landt, W.; Siegert, A. Guideline to reference gene selection for quantitative real-time PCR. Biochem. Biophys. Res. Commun. 2004, 313, 856–862. [Google Scholar] [CrossRef]
- Nicholson, J.P.; Wolmarans, M.R.; Park, G.R. The role of albumin in critical illness. Br. J. Anaesth. 2000, 85, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Derveaux, S.; Vandesompele, J.; Hellemans, J. How to do successful gene expression analysis using real-time RT-QPCR. Methods 2010, 50, 227–230. [Google Scholar] [CrossRef]
- Taylor, S.; Wakem, M.; Dijkman, G.; Alsarraj, M.; Nguyen, M.A. Practical approach to RT-qRT-QPCR-Publishing data that conform to the MIQE guidelines. Methods 2010, 50, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Lockman, K.A.; Baren, J.P.; Pemberton, C.J.; Baghdadi, H.; Burgess, K.E.; Plevris-Papaioannou, N.; Lee, P.; Howie, F.; Beckett, G.; Pryde, A.; et al. Oxidative stress rather than triglyceride accumulation is a determinant of mitochondrial dysfunction in in vitro models of hepatic cellular steatosis. Liver Int. 2012, 32, 1079–1092. [Google Scholar] [CrossRef] [PubMed]
- Lockman, K.A.; Htun, V.; Sinha, R.; Treskes, P.; Nelson, L.J.; Martin, S.F.; Rogers, S.M.; Le Bihan, T.; Hayes, P.C.; Plevris, J.N. Proteomic profiling of cellular steatosis with concomitant oxidative stress in vitro. Lipid. Health Dis. 2016, 15, 114. [Google Scholar] [CrossRef]
- Lambert, C.B.; Spire, C.; Claude, N.; Guillouzo, A. Dose- and time-dependent effects of phenobarbital on gene expression profiling in human hepatoma HepaRG cells. Toxicol. Appl. Pharmacol. 2009, 234, 345–360. [Google Scholar] [CrossRef]
- Antherieu, C.; Chesne, R.; Li, S.; Camus, A.; Lahoz, L.; Picazo, M.; Turpeinen, M.; Tolonen, A.; Uusitalo, J.; Guguen-Guillouzo, C.; et al. Stable expression, activity, and inducibility of cytochromes P450 in differentiated HepaRG cells. Drug Metab. Dispos. 2010, 38, 516–525. [Google Scholar] [CrossRef]
- Chapman, J.R.; Waldenström, J. With Reference to Reference Genes: A Systematic Review of Endogenous Controls in Gene Expression Studies. PLoS ONE 2015, 10, e0141853. [Google Scholar] [CrossRef]
- Bu, H.Z.A. literature review of enzyme kinetic parameters for CYP3A4-mediated metabolic reactions of 113 drugs in human liver microsomes: Structure-kinetics relationship assessment. Curr. Drug Metab. 2006, 7, 231–249. [Google Scholar] [CrossRef]
- Liu, Y.; Flynn, T.J.; Xia, M.; Wiesenfeld, P.L.; Ferguson, M.S. Evaluation of CYP3A4 inhibition and hepatotoxicity using DMSO-treated human hepatoma HuH-7 cells. Cell Biol. Toxicol. 2015, 31, 221–230. [Google Scholar] [CrossRef]
- Liu, Y.T.; Hao, H.P.; Liu, C.X.; Wang, G.J.; Xie, H.G. Drugs as CYP3A probes, inducers, and inhibitors. Drug Metab. Rev. 2007, 39, 699–721. [Google Scholar] [CrossRef]
- Bachour-El Azzi, P.; Sharanek, A.; Abdel-Razzak, Z.; Antherieu, S.; Al-Attrache, H.; Savary, C.C.; Lepage, S.; Morel, I.; Labbe, G.; Guguen-Guillouzo, C.; et al. Impact of inflammation on chlorpromazine-induced cytotoxicity and cholestatic features in HepaRG cells. Drug Metab. Dispos. 1556, 42, 1556–1566. [Google Scholar] [CrossRef] [PubMed]
- Jones, B.C.; Rollison, H.; Johansson, S.; Kanebratt, K.P.; Lambert, C.; Vishwanathan, K.; Andersson, T.B. Managing the Risk of CYP3A Induction in Drug Development: A Strategic Approach. Drug Metab. Dispos. 2017, 45, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Borkowska, P.; Zielińska, A.; Paul-Samojedny, M.; Stojko, R.; Kowalski, J. Evaluation of reference genes for quantitative real-time PCR in Wharton’s Jelly-derived mesenchymal stem cells after lentiviral transduction and differentiation. Mol. Biol. Rep. 2020, 47, 1107–1115. [Google Scholar] [CrossRef] [PubMed]
- Ayanoğlu, F.B.; Elçin, E.A.; Elçin, Y.M. Evaluation of the stability of standard reference genes of adipose-derived mesenchymal stem cells during in vitro proliferation and differentiation. Mol. Biol. Rep. 2020, 47, 2109–2122. [Google Scholar] [CrossRef] [PubMed]
- Zárybnický, T.; Matoušková, P.; Ambrož, M.; Šubrt, Z.; Skálová, L.; Boušová, I. The Selection and Validation of Reference Genes for mRNA and microRNA Expression Studies in Human Liver Slices Using RT-qPCR. Genes 2019, 10, 763. [Google Scholar] [CrossRef]
- Araujo, L.C.C.; Bordin, S.; Carvalho, C.R.O. Reference Gene and Protein Expression Levels in Two Different NAFLD Mouse Models. Gastroenterol. Res. Prac. 2020, 2020, 1–7. [Google Scholar] [CrossRef]
- Nygard, A.; Jørgensen, C.B.; Cirera, S.; Fredholm, M. Selection of reference genes for gene expression studies in pig tissues using SYBR green qPCR. BMC Mol. Biol. 2007, 8, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Lyu, S.; Yu, Y.; Xu, S.; Cai, W.; Chen, G.; Chen, J.; Pan, D.; She, W. Identification of Appropriate Reference Genes for Normalizing miRNA Expression in Citrus Infected by Xanthomonas citri subsp. Citri. Genes 2020, 11, 17. [Google Scholar] [CrossRef]
- Congiu, M.; Slavin, J.L.; Desmond, P.V. Expression of common housekeeping genes is affected by disease in human hepatitis C virus-infected liver. Liver International 2011, 31, 386–390. [Google Scholar] [CrossRef]
- Boujedidi, H.; Bouchet-Delbos, L.; Cassard-Doulcieret, A.M. Housekeeping gene variability in the liver of alcoholic patients. Alcohol. Clin. Exper. Res. 2012, 36, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Sandercock, D.A.; Coe, J.C.; Giminiani, P.D.; Edwards, S.A. Determination of stable reference genes for RT-qPCR expression data in mechanistic pain studies on pig dorsal root ganglia and spinal cord. Res. Vet. Sci. 2017, 114, 493–501. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Gene Name | Forward Primer | Reverse Primer | Amplicon |
|---|---|---|---|---|
| ALB | Albumin | TGACAAATCACTTCATACCCTTTTT | GCATTCATTTCTCTCAGGTTCTTG | 118 |
| CYP3A4 | Cytochrome P450 family 3 subfamily A member 4 | ACCGTAAGTGGAGCCTGAAT | AAGTAATTTGAGGTCTCTGGTGTT | 90 |
| HNF4α | Hepatocyte nuclear factor 4α | GACCTCTACTGCCTTGGACAA | GATGAAGTCGGGGGTTGGA | 87 |
| CDKNA | Cycli-dependent kinase inhibitor 2A | GGAAGGTCCCTCAGACATCC | CTTCGGTGACTGATGATCTAAGTT | 84 |
| Gene Name | Gene Function |
|---|---|
| GAPDH (glyceralde- hyde-3-phosphate dehydrogenase) | Product catalyses a step during carbohydrate metabolism, also has uracil DNA glycosylase activity in the nucleus, and contains peptide involved in antimicrobial activity. |
| RPL13a (ribosomal protein L13a) | Eukaryotic cytoplasmic ribosomal subunit. |
| 18S (18S ribosomal RNA subunit) | Eukaryotic cytoplasmic ribosomal subunit. |
| TOP1 (topoisomerase 1) | Encodes a DNA topoisomerase, an enzyme that controls and alters the orientation of DNA during transcription. |
| ATP5b (ATP synthase subunit 5b) | Encodes a subunit of mitochondrial ATP synthase. |
| UBC (ubiquitin C) | Encodes a polyubiquitin precursor. |
| SDHA (succinate dehydrogenase complex, subunit A) | Encodes major catalytic subunit of the mitochondrial respiratory chain. |
| B2M (beta-2 microglobulin) | Encodes a serum protein found on the surface of most nucleated cells. |
| β-Actin (beta-actin) | Encodes one of six different actin proteins, involved in cell motility, integrity, structure, and intercellular signalling. |
| CYC1 (cyclin D1) | Role in cell proliferation. |
| EiIF4a2 (eukaryotic initiation factor 4a2) | Regulates lipid metabolism, and translation factors. |
| YWHAZ (phospholipase A2) | Encodes mediator of signal transduction. |
| Experiment | All Conditions | APAP | CPZ |
|---|---|---|---|
| Samples Types | Number of samples | ||
| Treated | 5 | 6 | 6 |
| Untreated | 5 | 6 | 6 |
| Reference Genes | |||
| Gene | M-value | ||
| UBC | 2.262 | 0.6125 | 0.387 |
| YWHA | |||
| SDHA | 1.9 | 0.287 | 0.462 |
| GAPDH | 1.50 | 0.487 | 0.375 |
| EIF4A1 | 1.362 | 0.912 | 0.775 |
| B2M | 1.65 | 0.725 | 0.567 |
| TOP1 | 1.012 | 0.425 | 0.387 |
| RPL13A | 2.062 | 0.675 | 0.65 |
| ATP5B | 0.8 | 0.287 | 0.487 |
| CYC1 | 0.912 | 0.3 | 1.012 |
| 18S | 1.25 | 0.55 | 1.137 |
| ACTB | |||
| Average M | 1.0672 | 0.291333 | 0.383 |
| CV | 0.19604 | 0.021035 | 0.01477 |
| Required number of genes | 5 | 3 | 3 |
| Average Cq Values | |||
| Untreated | GAPDH,UBC,TOP1 | GAPDH | 18S |
| 32.4 | 31.2 | 24.4 | |
| 31.2 | 31.5 | 22.7 | |
| 31.1 | 33.7 | 25.8 | |
| average | 31.6 | 32.1 | 24.4 |
| st.dev | 0.6 | 1.1 | 1.1 |
| 25 μM CPZ | |||
| 33.5 | 31.10 | 22.9 | |
| 35.3 | nd | 24.1 | |
| 34.0 | 33.45 | 27.0 | |
| 35.3 | 22.63 | 10.4 | |
| nd | 22.29 | 11.5 | |
| 30.2 | 22.19 | 36.8 | |
| average | 33.6 | 26.3 | 22.1 |
| st.dev | 1.9 | 4.9 | 9.1 |
| 50 μM CPZ | |||
| 34.0 | 33.1 | 24.4 | |
| 34.5 | 30.8 | 24.2 | |
| 34.9 | 22.2 | ||
| 34.9 | 26.1 | 10.9 | |
| 28.5 | 28.3 | 10.8 | |
| 30.2 | 29.0 | 14.7 | |
| average | 32.8 | 29.4 | 17.9 |
| st.dev | 0.4 | 2.3 | 5.9 |
| Average Cq Values | |||
| Untreated | ATP,SDHA,CYC1 | GAPDH | 18S |
| 26.7 | 31.2 | 22.7 | |
| 26.4 | 31.5 | 25.8 | |
| 27.3 | 33.7 | 24.8 | |
| average | 26.8 | 32.1 | 24.4 |
| st.dev | 0.4 | 1.1 | 1.3 |
| 5 mM APAP | |||
| 31.6 | 26.5 | 21.5 | |
| 32.2 | 27.6 | 23.9 | |
| 32.4 | 27.9 | 24.6 | |
| average | 32.1 | 27.3 | 23.3 |
| st.dev | 0.4 | 0.6 | 1.4 |
| 10 mM APAP | |||
| 31.6 | 27.1 | 24.9 | |
| 32.2 | 27.1 | 27.4 | |
| 31.4 | 26.7 | 27.8 | |
| average | 31.7 | 27.0 | 26.7 |
| st.dev | 0.3 | 0.2 | 1.3 |
| 20 mM APAP | 31.6 | 28.9 | 24.9 |
| 32.2 | 24.0 | 27.4 | |
| 31.4 | 23.7 | 27.8 | |
| average | 31.7 | 25.6 | 26.7 |
| st.dev | 0.3 | 2.4 | 1.3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brzeszczyńska, J.; Brzeszczyński, F.; Samuel, K.; Morgan, K.; Morley, S.D.; Plevris, J.N.; Hayes, P.C. Validation of Reference Genes for Gene Expression Studies by RT-qPCR in HepaRG Cells during Toxicity Testing and Disease Modelling. Cells 2020, 9, 770. https://doi.org/10.3390/cells9030770
Brzeszczyńska J, Brzeszczyński F, Samuel K, Morgan K, Morley SD, Plevris JN, Hayes PC. Validation of Reference Genes for Gene Expression Studies by RT-qPCR in HepaRG Cells during Toxicity Testing and Disease Modelling. Cells. 2020; 9(3):770. https://doi.org/10.3390/cells9030770
Chicago/Turabian StyleBrzeszczyńska, Joanna, Filip Brzeszczyński, Kay Samuel, Katie Morgan, Steven D. Morley, John N. Plevris, and Peter C. Hayes. 2020. "Validation of Reference Genes for Gene Expression Studies by RT-qPCR in HepaRG Cells during Toxicity Testing and Disease Modelling" Cells 9, no. 3: 770. https://doi.org/10.3390/cells9030770
APA StyleBrzeszczyńska, J., Brzeszczyński, F., Samuel, K., Morgan, K., Morley, S. D., Plevris, J. N., & Hayes, P. C. (2020). Validation of Reference Genes for Gene Expression Studies by RT-qPCR in HepaRG Cells during Toxicity Testing and Disease Modelling. Cells, 9(3), 770. https://doi.org/10.3390/cells9030770