Transcription Factors in Cancer: When Alternative Splicing Determines Opposite Cell Fates




Abstract
1. Introduction
1.1. Alternative Splicing
1.2. Transcription Factors
1.3. Deregulation of Transcription Factors in Cancer
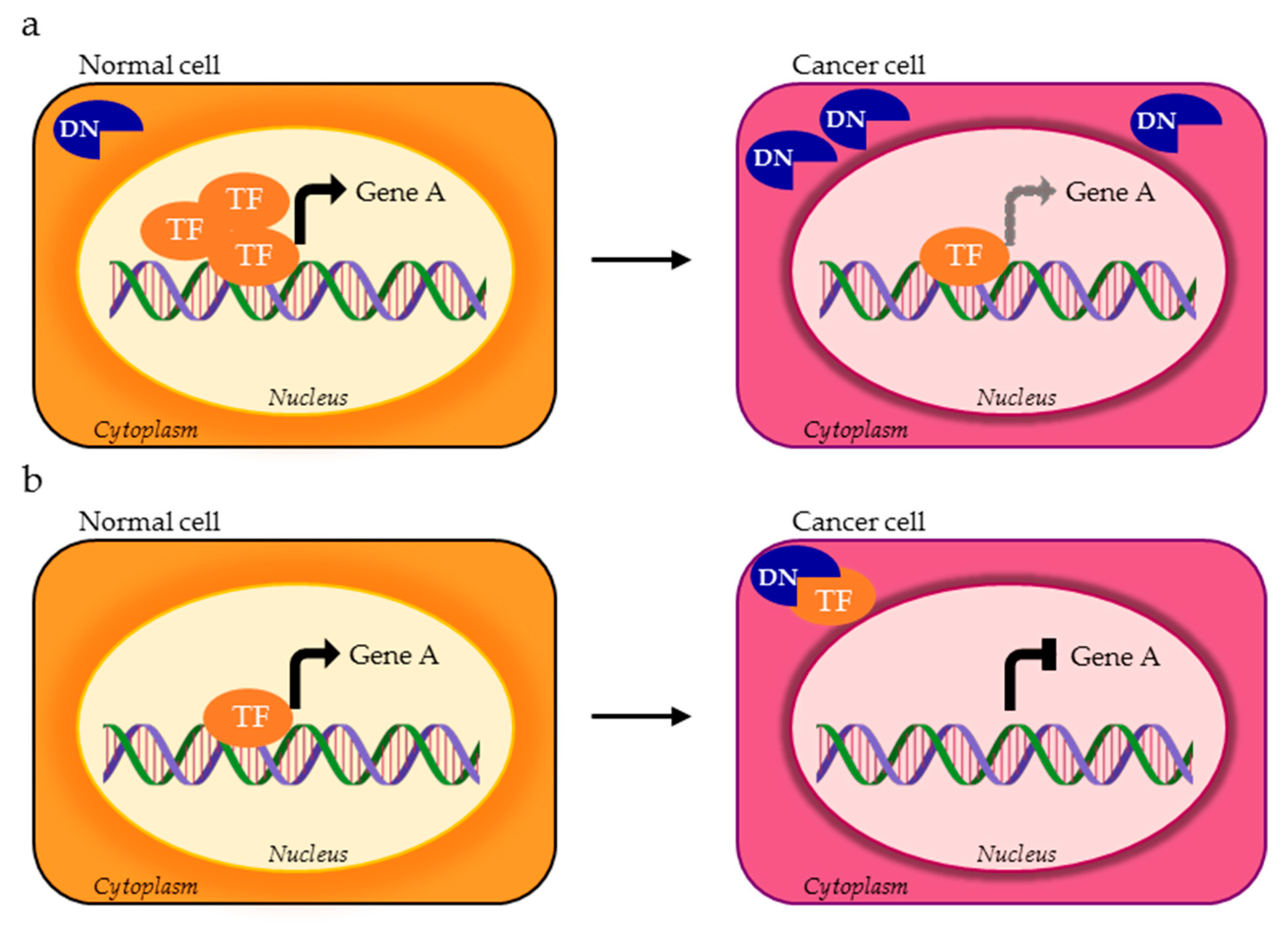
2. TFs Splice Variants that Directly Control Different Transcriptional Programs
2.1. Nuclear Transcription Factor Y (NF-Y)
2.2. Signal Transducer and Activator of Transcription 3 (STAT3)
2.3. T Cell Factor 4 (TCF4)
2.4. Wilm’s Tumor 1 (WT1)
3. TFs Splice Variants that Differently Control the Same set of Genes
3.1. Myocyte Enhancer Factor-2C (MEF2C)
3.2. Melanocyte Transcription Factor (MITF)
3.3. Nuclear Factor of Activated T Cells 1 (NFAT2/ NFATC1)
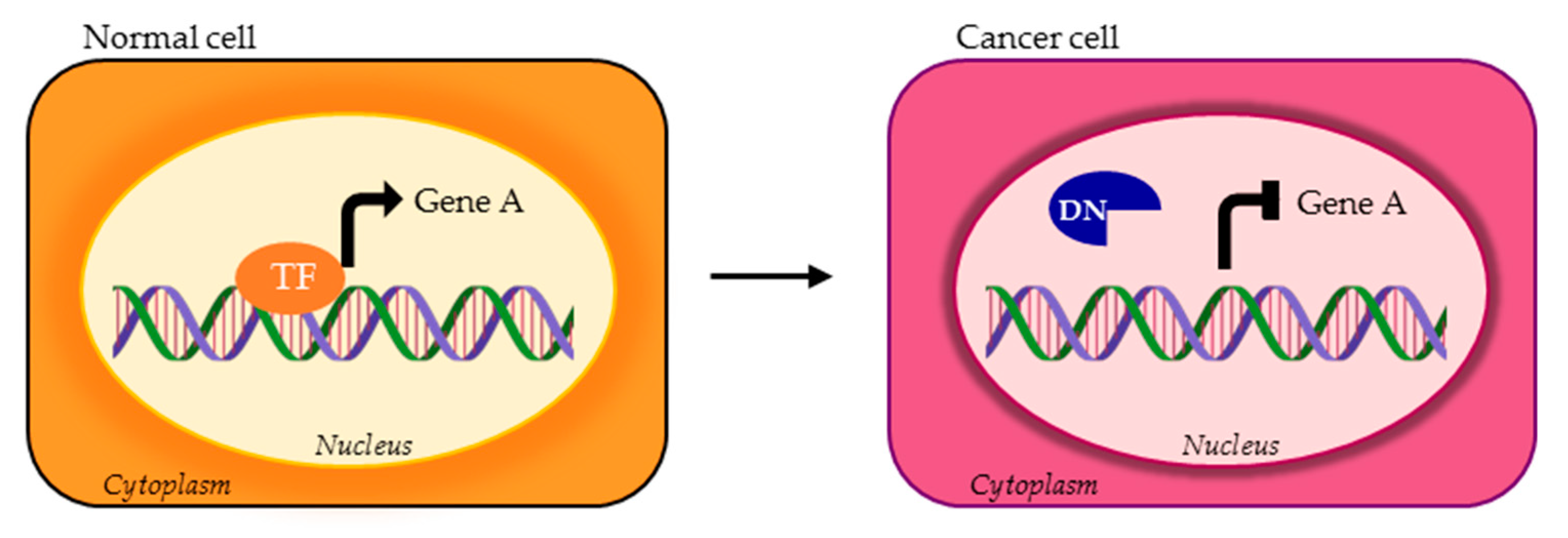
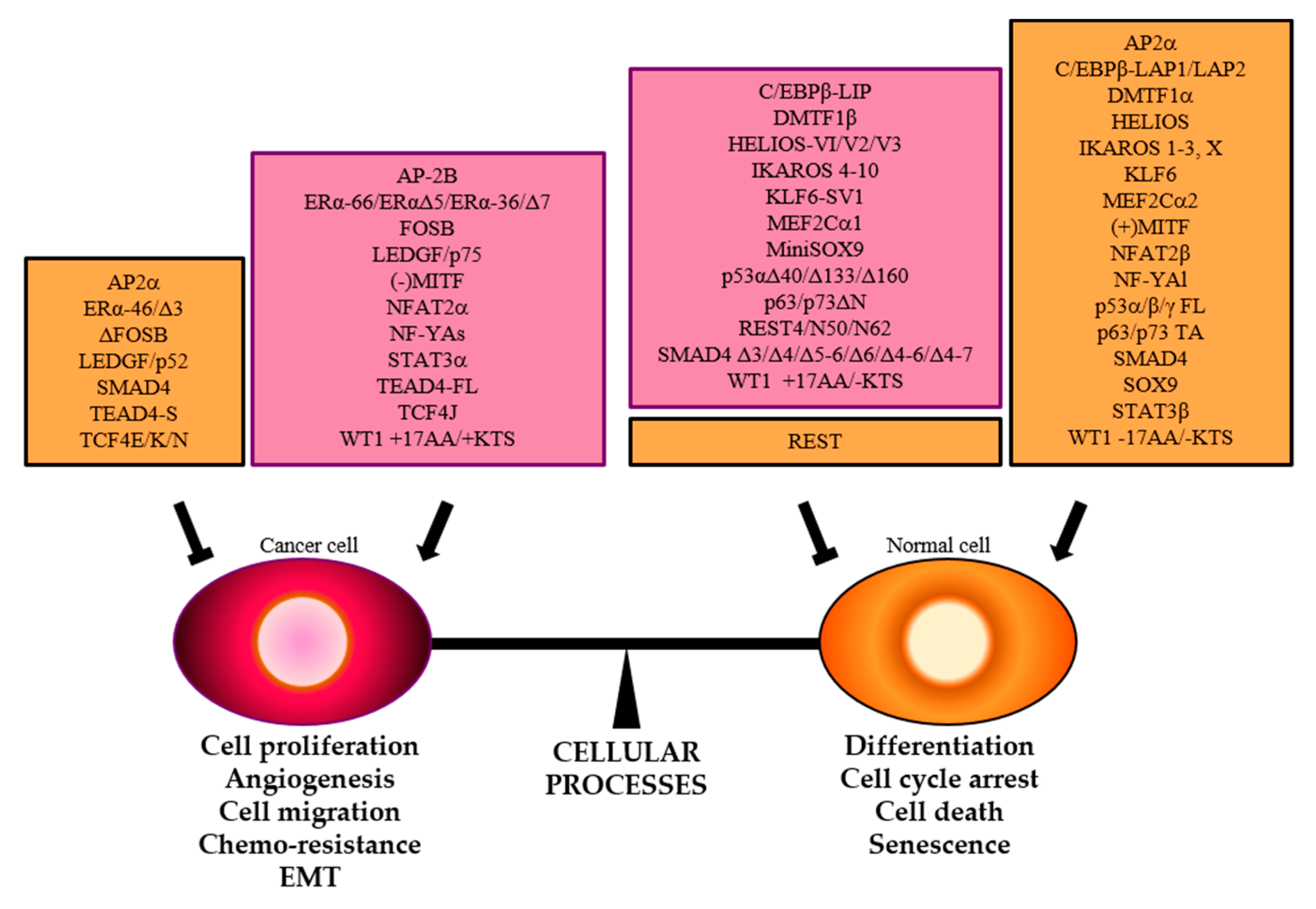
4. TFs Splice Variants with Dominant Negative Activity that Hamper TF Physiological Function
4.1. DNs with Cellular Mislocalization
4.1.1. Ikaros Family Zinc Finger Protein HELIOS
4.1.2. Krüppel-like Factor 6 (KLF6)
4.2. DNs Impaired in DNA Binding Ability
4.2.1. Ikaros Family Zinc Finger Protein 1 (IKAROS)
4.2.2. TEA Domain Family Member 4 (TEAD4)
4.3. DNs with Altered Regultory Ability
4.3.1. CCAAT-Enhancer Binding Protein β (C/EBPβ)
4.3.2. Lens Epithelium-Derived Growth Factor (LEDGF)
4.3.3. RE1 Silencing Transcription Factor (REST)
4.3.4. SMAD Family Member 4 (SMAD4)
4.3.5. Estrogen Receptor Alpha (ERα)
5. Conclusions and Prospects
Author Contributions
Funding
Conflicts of Interest
References
- Park, E.; Pan, Z.; Zhang, Z.; Lin, L.; Xing, Y. The Expanding Landscape of Alternative Splicing Variation in Human Populations. Am. J. Hum. Genet. 2018, 102, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yan, L.; Zeng, J.; Zhou, H.; Liu, H.; Yu, G.; Yao, W.; Chen, K.; Ye, Z.; Xu, H. Pan-cancer analysis of clinical relevance of alternative splicing events in 31 human cancers. Oncogene 2019, 38, 6678–6695. [Google Scholar] [CrossRef] [PubMed]
- El Marabti, E.; Younis, I. The cancer spliceome: Reprograming of alternative splicing in cancer. Front. Mol. Biosci. 2018, 5, 80. [Google Scholar] [CrossRef] [PubMed]
- Escobar-Hoyos, L.; Knorr, K.; Abdel-Wahab, O. Aberrant RNA Splicing in Cancer. Annu. Rev. Cancer Biol. 2019, 3, 167–185. [Google Scholar] [CrossRef]
- Faustino, N.A.; Cooper, T.A. Pre-mRNA splicing and human disease. Genes Dev. 2003, 17, 419–437. [Google Scholar] [CrossRef]
- Pajares, M.J.; Ezponda, T.; Catena, R.; Calvo, A.; Pio, R.; Montuenga, L.M. Alternative splicing: An emerging topic in molecular and clinical oncology. Lancet Oncol. 2007, 8, 349–357. [Google Scholar] [CrossRef]
- Luscombe, N.M.; Austin, S.E.; Berman, H.M.; Thornton, J.M. An overview of the structures of protein-DNA complexes. Genome Biol. 2000. [Google Scholar] [CrossRef]
- Lambert, S.A.; Jolma, A.; Campitelli, L.F.; Das, P.K.; Yin, Y.; Albu, M.; Chen, X.; Taipale, J.; Hughes, T.R.; Weirauch, M.T. The Human Transcription Factors. Cell 2018, 172, 650–665. [Google Scholar] [CrossRef]
- Cusanovich, D.A.; Pavlovic, B.; Pritchard, J.K.; Gilad, Y. The Functional Consequences of Variation in Transcription Factor Binding. PLoS Genet. 2014, 10, e1004226. [Google Scholar] [CrossRef]
- Spitz, F.; Furlong, E.E.M. Transcription factors: From enhancer binding to developmental control. Nat. Rev. Genet. 2012, 13, 613–626. [Google Scholar] [CrossRef]
- Lee, T.I.; Young, R.A. Transcriptional regulation and its misregulation in disease. Cell 2013, 152, 1237–1251. [Google Scholar] [CrossRef] [PubMed]
- Levine, M.; Manley, J.L. Transcriptional repression of eukaryotic promoters. Cell 1989, 59, 405–408. [Google Scholar] [CrossRef]
- Payankaulam, S.; Li, L.M.; Arnosti, D.N. Transcriptional repression: Conserved and evolved features. Curr. Biol. 2010, 20, R764–R771. [Google Scholar] [CrossRef]
- Reiter, F.; Wienerroither, S.; Stark, A. Combinatorial function of transcription factors and cofactors. Curr. Opin. Genet. Dev. 2017, 43, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Bradner, J.E.; Hnisz, D.; Young, R.A. Transcriptional Addiction in Cancer. Cell 2017, 168, 629–643. [Google Scholar] [CrossRef]
- Garraway, L.A.; Lander, E.S. Lessons from the cancer genome. Cell 2013, 153, 17–37. [Google Scholar] [CrossRef]
- Dong, X.; Chen, R. Understanding aberrant RNA splicing to facilitate cancer diagnosis and therapy. Oncogene 2019, 39, 2231–2242. [Google Scholar] [CrossRef]
- Sveen, A.; Kilpinen, S.; Ruusulehto, A.; Lothe, R.A.; Skotheim, R.I. Aberrant RNA splicing in cancer; Expression changes and driver mutations of splicing factor genes. Oncogene 2016, 35, 2413–2427. [Google Scholar] [CrossRef]
- Biamonti, G.; Infantino, L.; Gaglio, D.; Amato, A. An Intricate Connection between Alternative Splicing and Phenotypic Plasticity in Development and Cancer. Cells 2019, 9, 34. [Google Scholar] [CrossRef]
- David, C.J.; Manley, J.L. Alternative pre-mRNA splicing regulation in cancer: Pathways and programs unhinged. Genes Dev. 2010, 24, 2343–2364. [Google Scholar] [CrossRef]
- Danan-Gotthold, M.; Golan-Gerstl, R.; Eisenberg, E.; Meir, K.; Karni, R.; Levanon, E.Y. Identification of recurrent regulated alternative splicing events across human solid tumors. Nucleic Acids Res. 2015, 43, 5130–5144. [Google Scholar] [CrossRef] [PubMed]
- Gurtner, A.; Manni, I.; Piaggio, G. NF-Y in cancer: Impact on cell transformation of a gene essential for proliferation. Biochim. Biophys. Acta Gene Regul. Mech. 2017, 1860, 604–616. [Google Scholar] [CrossRef] [PubMed]
- Goodarzi, H.; Elemento, O.; Tavazoie, S. Revealing Global Regulatory Perturbations across Human Cancers. Mol. Cell 2009, 36, 900–911. [Google Scholar] [CrossRef] [PubMed]
- Dolfini, D.; Minuzzo, M.; Pavesi, G.; Mantovani, R. The Short Isoform of NF-YA Belongs to the Embryonic Stem Cell Transcription Factor Circuitry. Stem Cells 2012, 30, 2450–2459. [Google Scholar] [CrossRef]
- Basile, V.; Baruffaldi, F.; Dolfini, D.; Belluti, S.; Benatti, P.; Ricci, L.; Artusi, V.; Tagliafico, E.; Mantovani, R.; Molinari, S.; et al. NF-YA splice variants have different roles on muscle differentiation. Biochim. Biophys. Acta Gene Regul. Mech. 2016, 1859, 627–638. [Google Scholar] [CrossRef]
- Cicchillitti, L.; Corrado, G.; Carosi, M.; Dabrowska, M.E.; Loria, R.; Falcioni, R.; Cutillo, G.; Piaggio, G.; Vizza, E. Prognostic role of NF-YA splicing isoforms and Lamin A status in low grade endometrial cancer. Oncotarget 2017, 8, 7935–7945. [Google Scholar] [CrossRef]
- Dolfini, D.; Andrioletti, V.; Mantovani, R. Overexpression and alternative splicing of NF-YA in breast cancer. Sci. Rep. 2019, 9, 12955. [Google Scholar] [CrossRef]
- Bezzecchi, E.; Ronzio, M.; Dolfini, D.; Mantovani, R. NF-YA overexpression in lung cancer: LUSC. Genes 2019, 10, 937. [Google Scholar] [CrossRef]
- Bezzecchi, E.; Ronzio, M.; Semeghini, V.; Andrioletti, V.; Mantovani, R.; Dolfini, D. NF-YA Overexpression in Lung Cancer: LUAD. Genes 2020, 11, 198. [Google Scholar] [CrossRef]
- Inghirami, G.; Chiarle, R.; Simmons, W.J.; Piva, R.; Schlessinger, K.; Levy, D.E. New and old functions of STAT3: A pivotal target for individualized treatment of cancer. Cell Cycle 2005, 4, 1131–1133. [Google Scholar] [CrossRef]
- Maritano, D.; Sugrue, M.L.; Tininini, S.; Dewilde, S.; Strobl, B.; Fu, X.P.; Murray-Tait, V.; Chiarle, R.; Poli, V. The STAT3 isoforms α and β have unique and specific functions. Nat. Immunol. 2004, 5, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Ng, I.H.W.; Ng, D.C.H.; Jans, D.A.; Bogoyevitch, M.A. Selective STAT3-α or -β expression reveals spliceform-specific phosphorylation kinetics, nuclear retention and distinct gene expression outcomes. Biochem. J. 2012, 447, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Park, O.K.; Schaefer, L.K.; Wang, W.; Schaefer, T.S. Dimer stability as a determinant of differential DNA binding activity of Stat3 isoforms. J. Biol. Chem. 2000, 275, 32244–32249. [Google Scholar] [CrossRef]
- Aziz, M.H.; Hafeez, B.B.; Sand, J.M.; Pierce, D.B.; Aziz, S.W.; Dreckschmidt, N.E.; Verma, A.K. Protein kinase C mediates Stat3Ser727 phosphorylation, Stat3-regulated gene expression, and cell invasion in various human cancer cell lines through integration with MAPK cascade (RAF-1, MEK1/2, and ERK1/2). Oncogene 2010, 29, 3100–3109. [Google Scholar] [CrossRef]
- Tang, J.-Z.; Kong, X.-J.; Banerjee, A.; Muniraj, N.; Pandey, V.; Steiner, M.; Perry, J.K.; Zhu, T.; Liu, D.-X.; Lobie, P.E. STAT3α Is Oncogenic for Endometrial Carcinoma Cells and Mediates the Oncogenic Effects of Autocrine Human Growth Hormone. Endocrinology 2010, 151, 4133–4145. [Google Scholar] [CrossRef] [PubMed]
- Sheng, W.J.; Jiang, H.; Wu, D.L.; Zheng, J.H. Early responses of the STAT3 pathway to platinum drugs are associated with cisplatin resistance in epithelial ovarian cancer. Brazilian J. Med. Biol. Res. 2013, 46, 650–658. [Google Scholar] [CrossRef]
- Schaefer, L.K.; Ren, Z.; Fuller, G.N.; Schaefer, T.S. Constitutive of Stat3α in brain tumors: Localization to tumor endothelial cells and activation by the endothelial tyrosine kinase receptor (VEGFR-2). Oncogene 2002, 21, 2058–2065. [Google Scholar] [CrossRef]
- Chakraborty, A.; White, S.M.; Schaefer, T.S.; Ball, E.D.; Dyer, K.F.; Tweardy, D.J. Granulocyte colony-stimulating factor activation of Stat3α and Stat3β in immature normal and leukemic human myeloid cells. Blood 1996, 88, 2442–2449. [Google Scholar] [CrossRef]
- Carpenter, R.L.; Lo, H.W. STAT3 target genes relevant to human cancers. Cancers 2014, 6, 897–925. [Google Scholar] [CrossRef]
- Ivanov, V.N.; Zhou, H.; Partridge, M.A.; Hei, T.K. Inhibition of ataxia telangiectasia mutated kinase activity enhances TRAIL-mediated apoptosis in human melanoma cells. Cancer Res. 2009, 69, 3510–3519. [Google Scholar] [CrossRef]
- Niu, G.; Shain, K.H.; Huang, M.; Ravi, R.; Bedi, A.; Dalton, W.S.; Jove, R.; Yu, H. Overexpression of a Dominant-Negative Signal Transducer and Activator of Transcription 3 Variant in Tumor Cells Leads to Production of SolubleFactors That Induce Apoptosis and Cell Cycle Arrest. Cancer Res. 2001, 61, 3276–3280. [Google Scholar] [PubMed]
- Ivanov, V.N.; Bhoumik, A.; Krasilnikov, M.; Raz, R.; Owen-Schaub, L.B.; Levy, D.; Horvath, C.M.; Ronai, Z. Cooperation between STAT3 and c-Jun suppresses Fas transcription. Mol. Cell 2001, 7, 517–528. [Google Scholar] [CrossRef]
- Aigner, P.; Mizutani, T.; Horvath, J.; Eder, T.; Heber, S.; Lind, K.; Just, V.; Moll, H.P.; Yeroslaviz, A.; Fischer, M.J.; et al. STAT3b is a tumor suppressor in acute myeloid leukemia. Blood Adv. 2019, 3, 1989–2002. [Google Scholar] [CrossRef] [PubMed]
- Weise, A.; Bruser, K.; Elfert, S.; Wallmen, B.; Wittel, Y.; Wöhrle, S.; Hecht, A. Alternative splicing of Tcf7l2 transcripts generates protein variants with differential promoter-binding and transcriptional activation properties at Wnt/β-catenin targets. Nucleic Acids Res. 2009, 38, 1964–1981. [Google Scholar] [CrossRef]
- Fan, Y.C.; Min, L.; Chen, H.; Liu, Y.L. Alternative splicing isoform of T cell factor 4K suppresses the proliferation and metastasis of non-small cell lung cancer cells. Genet. Mol. Res. 2015, 14, 14009–14018. [Google Scholar] [CrossRef]
- Tsedensodnom, O.; Koga, H.; Rosenberg, S.A.; Nambotin, S.B.; Carroll, J.J.; Wands, J.R.; Kim, M. Identification of T-cell factor-4 isoforms that contribute to the malignant phenotype of hepatocellular carcinoma cells. Exp. Cell Res. 2011, 317, 920–931. [Google Scholar] [CrossRef]
- Liu, Z.; Song, J.; Wu, Y.; Yao, S.; Xu, G.Z.; Diao, B. Expression and functional analysis of TCF4 isoformsin human glioma cells. Mol. Med. Rep. 2018, 17, 6023–6027. [Google Scholar]
- He, G.; Guan, X.; Chen, X.; Wang, Y.; Luo, C.; Zhang, B. Expression and splice variant analysis of human TCF4 transcription factor in esophageal cancer. J. Cancer 2015, 6, 333–341. [Google Scholar] [CrossRef]
- Valenta, T.; Lukas, J.; Korinek, V. HMG box transcription factor TCF-4’s interaction with CtBP1 controls the expression of the Wnt target Axin2/Conductin in human embryonic kidney cells. Nucleic Acids Res. 2003, 31, 2369–2380. [Google Scholar] [CrossRef]
- Koga, H.; Tsedensodnom, O.; Tomimaru, Y.; Walker, E.J.; Lee, H.C.; Kim, K.M.; Yano, H.; Wands, J.R.; Kim, M. Loss of the SxxSS Motif in a Human T-Cell Factor-4 Isoform Confers Hypoxia Resistance to Liver Cancer: An Oncogenic Switch in Wnt Signaling. PLoS ONE 2012, 7, e39981. [Google Scholar] [CrossRef]
- Kennell, J.A.; O’Leary, E.E.; Gummow, B.M.; Hammer, G.D.; MacDougald, O.A. T-Cell Factor 4N (TCF-4N), a Novel Isoform of Mouse TCF-4, Synergizes with -Catenin To Coactivate C/EBP and Steroidogenic Factor 1 Transcription Factors. Mol. Cell. Biol. 2003, 23, 5366–5375. [Google Scholar] [CrossRef] [PubMed]
- Yamanouchi, K.; Ohta, T.; Liu, Z.; Oji, Y.; Sugiyama, H.; Shridhar, V.; Matsumura, S.; Takahashi, T.; Takahashi, K.; Kurachi, H. The Wilms’ tumor gene WT1 −17AA/−KTS splice variant increases tumorigenic activity through up-regulation of vascular endothelial growth factor in an in vivo ovarian cancer model. Transl. Oncol. 2014, 7, 580–589. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Han, Y.; Saiz, S.; Minden, M.D. Review a tumor suppressor and oncogene: The WT1 story. Leukemia 2007, 21, 868–876. [Google Scholar] [CrossRef] [PubMed]
- Hammes, A.; Guo, J.K.; Lutsch, G.; Leheste, J.R.; Landrock, D.; Ziegler, U.; Gubler, M.C.; Schedl, A. Two splice variants of the wilms’ tumor 1 gene have distinct functions during sex determination and nephron formation. Cell 2001, 106, 319–329. [Google Scholar] [CrossRef]
- Baudry, D.; Hamelin, M.; Cabanis, M.-O.; Fournet, J.-C.; Tournade, M.-F.; Sarnacki, S.; Junien, C.; Jeanpierre, C. WT1 Splicing Alterations in Wilms’ Tumors. Clin. Cancer Res. 2000, 6, 3957–3965. [Google Scholar] [PubMed]
- Baudry, D.; Faussillon, M.; Cabanis, M.O.; Rigolet, M.; Zucker, J.M.; Patte, C.; Sarnacki, S.; Boccon-Gibod, L.; Junien, C.; Jeanpierre, C. Changes in WT1 splicing are associated with a specific gene expression profile in Wilms’ tumour. Oncogene 2002, 21, 5566–5573. [Google Scholar] [CrossRef]
- Kramarzova, K.; Stuchly, J.; Willasch, A.; Gruhn, B.; Schwarz, J.; Cermak, J.; MacHova-Polakova, K.; Fuchs, O.; Stary, J.; Trka, J.; et al. Real-time PCR quantification of major Wilms tumor gene 1 (WT1) isoforms in acute myeloid leukemia, their characteristic expression patterns and possible functional consequences. Leukemia 2012, 26, 2086–2095. [Google Scholar] [CrossRef][Green Version]
- Siehl, J.M.; Reinwald, M.; Heufelder, K.; Menssen, H.D.; Keilholz, U.; Thiel, E. Expression of Wilms’ tumor gene 1 at different stages of acute myeloid leukemia and analysis of its major splice variants. Ann. Hematol. 2004, 83, 745–750. [Google Scholar] [CrossRef]
- Burwell, E.A.; McCarty, G.P.; Simpson, L.A.; Thompson, K.A.; Loeb, D.M. Isoforms of Wilms’ tumor suppressor gene (WT1) have distinct effects on mammary epithelial cells. Oncogene 2007, 26, 3423–3430. [Google Scholar] [CrossRef][Green Version]
- Loeb, D.M.; Evron, E.; Patel, C.B.; Sharma, P.M.; Niranjan, B.; Buluwela, L.; Weitzman, S.A.; Korz, D.; Sukumar, S. Wilms’ tumor suppressor gene (WT1) is expressed in primary breast tumors despite tumor-specific promoter methylation. Cancer Res. 2001, 61, 921–925. [Google Scholar]
- Potthoff, M.J.; Olson, E.N. MEF2: A central regulator of diverse developmental programs. Development 2007, 134, 4131–4140. [Google Scholar] [CrossRef] [PubMed]
- Di Giorgio, E.; Hancock, W.W.; Brancolini, C. MEF2 and the tumorigenic process, hic sunt leones. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Infantino, V.; Convertini, P.; Menga, A.; Iacobazzi, V. MEF2C exon α: Role in gene activation and differentiation. Gene 2013, 531, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Sebastian, S.; Faralli, H.; Yao, Z.; Rakopoulos, P.; Palii, C.; Cao, Y.; Singh, K.; Liu, Q.C.; Chu, A.; Aziz, A.; et al. Tissue-specific splicing of a ubiquitously expressed transcription factor is essential for muscle differentiation. Genes Dev. 2013, 27, 1247–1259. [Google Scholar] [CrossRef]
- Zhang, M.; Zhu, B.; Davie, J. Alternative splicing of MEF2C pre-mRNA controls its activity in normal myogenesis and promotes tumorigenicity in rhabdomyosarcoma cells. J. Biol. Chem. 2015, 290, 310–324. [Google Scholar] [CrossRef]
- Baruffaldi, F.; Montarras, D.; Basile, V.; de Feo, L.; Badodi, S.; Ganassi, M.; Battini, R.; Nicoletti, C.; Imbriano, C.; Musarò, A.; et al. Dynamic phosphorylation of the myocyte enhancer factor 2Cα1 splice variant promotes skeletal muscle regeneration and hypertrophy. Stem Cells 2017, 35, 725–738. [Google Scholar] [CrossRef]
- Goding, C.R. Mitf from neural crest to melanoma: Signal transduction and transcription in the melanocyte lineage. Genes Dev. 2000, 14, 1712–1728. [Google Scholar]
- Bismuth, K.; Maric, D.; Arnheiter, H. MITF and cell proliferation: The role of alternative splice forms. Pigment Cell Res. 2005, 18, 349–359. [Google Scholar] [CrossRef]
- Salti, G.I.; Manougian, T.; Farolan, M.; Shilkaitis, A.; Majumdar, D.; Das Gupta, T.K. Micropthalmia transcription factor: A new prognostic marker in intermediate-thickness cutaneous malignant melanoma. Cancer Res. 2000, 60, 5012–5016. [Google Scholar]
- Pogenberg, V.; Ögmundsdóttir, M.H.; Bergsteinsdóttir, K.; Schepsky, A.; Phung, B.; Deineko, V.; Milewski, M.; Steingrímsson, E.; Wilmanns, M. Restricted leucine zipper dimerization and specificity of DNA recognition of the melanocyte master regulator MITF. Genes Dev. 2012, 26, 2647–2658. [Google Scholar] [CrossRef]
- Carreira, S.; Goodall, J.; Aksan, I.; La Rocca, S.A.; Galibert, M.D.; Denat, L.; Larue, L.; Goding, C.R. Mitf cooperates with Rb1 and activates p21 Cip1 expression to regulate cell cycle progression. Nature 2005, 433, 764–769. [Google Scholar] [CrossRef] [PubMed]
- Loercher, A.E.; Tank, E.M.H.; Delston, R.B.; Harbour, J.W. MITF links differentiation with cell cycle arrest in melanocytes by transcriptional activation of INK4A. J. Cell Biol. 2005, 168, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Primot, A.; Mogha, A.; Corre, S.; Roberts, K.; Debbache, J.; Adamski, H.; Dreno, B.; Khammari, A.; Lesimple, T.; Mereau, A.; et al. ERK-regulated differential expression of the Mitf 6a/b splicing isoforms in melanoma. Pigment Cell Melanoma Res. 2010, 23, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Rengarajan, J.; Mittelstadt, P.R.; Mages, H.W.; Gerth, A.J.; Kroczek, R.A.; Ashwell, J.D.; Glimcher, L.H. Sequential involvement of NFAT and Egr transcription factors in FasL regulation. Immunity 2000, 12, 293–300. [Google Scholar] [CrossRef]
- Caetano, M.S.; Vieira-de-Abreu, A.; Teixeira, L.K.; Werneck, M.B.F.; Barcinski, M.A.; Viola, J.P.B. NFATC2 transcription factor regulates cell cycle progression during lymphocyte activation: Evidence of its involvement in the control of cyclin gene expression. FASEB J. 2002, 16, 1940–1942. [Google Scholar] [CrossRef]
- Ranger, A.M.; Gerstenfeld, L.C.; Wang, J.; Kon, T.; Bae, H.; Gravallese, E.M.; Glimcher, M.J.; Glimcher, L.H. The nuclear factor of activated T cells (NFAT) transcription factor NFATp (NFATc2) is a repressor of chondrogenesis. J. Exp. Med. 2000, 191, 9–22. [Google Scholar] [CrossRef]
- Chuvpilo, S.; Jankevics, E.; Tyrsin, D.; Akimzhanov, A.; Moroz, D.; Jha, M.K.; Schulze-Luehrmann, J.; Santner-Nanan, B.; Feoktistova, E.; König, T.; et al. Autoregulation of NFATc1/A expression facilitates effector T cells to escape from rapid apoptosis. Immunity 2002, 16, 881–895. [Google Scholar] [CrossRef]
- Robbs, B.K.; Cruz, A.L.S.; Werneck, M.B.F.; Mognol, G.P.; Viola, J.P.B. Dual Roles for NFAT Transcription Factor Genes as Oncogenes and Tumor Suppressors. Mol. Cell. Biol. 2008, 28, 7168–7181. [Google Scholar] [CrossRef]
- Levin-Gromiko, U.; Koshelev, V.; Kushnir, P.; Fedida-Metula, S.; Voronov, E.; Fishman, D. Amplified lipid rafts of malignant cells constitute a target for inhibition of aberrantly active NFAT and melanoma tumor growth by the aminobisphosphonate zoledronic acid. Carcinogenesis 2014, 35, 2555–2566. [Google Scholar] [CrossRef]
- Tripathi, M.K.; Deane, N.G.; Zhu, J.; An, H.; Mima, S.; Wang, X.; Padmanabhan, S.; Shi, Z.; Prodduturi, N.; Ciombor, K.K.; et al. Nuclear factor of activated T-cell activity is associated with metastatic capacity in colon cancer. Cancer Res. 2014, 74, 6947–6957. [Google Scholar] [CrossRef]
- Viola, J.P.B.; Carvalho, L.D.S.; Fonseca, B.P.F.; Teixeira, L.K. NFAT transcription factors: From cell cycle to tumor development. Brazilian J. Med. Biol. Res. 2005, 38, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Lucena, P.I.; Faget, D.V.; Pachulec, E.; Robaina, M.C.; Klumb, C.E.; Robbs, B.K.; Viola, J.P.B. NFAT2 isoforms differentially regulate gene expression, cell death and transformation through alternative N-terminal domains. Mol. Cell. Biol. 2015, 36, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Tabayashi, T.; Ishimaru, F.; Takata, M.; Kataoka, I.; Nakase, K.; Kozuka, T.; Tanimoto, M. Characterization of the short isoform of Helios overexpressed in patients with T-cell malignancies. Cancer Sci. 2007, 98, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Liu, W.; Li, Y.; Liu, P.; Li, S.; Dou, D.; Wang, Y.; Yang, R.; Xiang, R.; Liu, F. Alternative Splice Variants Modulates Dominant-Negative Function of Helios in T-Cell Leukemia. PLoS ONE 2016, 11, e0163328. [Google Scholar] [CrossRef][Green Version]
- DiFeo, A.; Martignetti, J.A.; Narla, G. The role of KLF6 and its splice variants in cancer therapy. Drug Resist. Updat. 2009, 12, 1–7. [Google Scholar] [CrossRef]
- Li, D.; Yea, S.; Li, S.; Chen, Z.; Narla, G.; Banck, M.; Laborda, J.; Tan, S.; Friedman, J.M.; Friedman, S.L.; et al. Krüppel-like factor-6 promotes preadipocyte differentiation through histone deacetylase 3-dependent repression of DLK1. J. Biol. Chem. 2005, 280, 26941–26952. [Google Scholar] [CrossRef]
- Ito, G.; Uchiyama, M.; Kondo, M.; Mori, S.; Usami, N.; Maeda, O.; Kawabe, T.; Hasegawa, Y.; Shimokata, K.; Sekido, Y. Krüppel-like factor 6 is frequently down-regulated and induces apoptosis in non-small cell lung cancer cells. Cancer Res. 2004, 64, 3838–3843. [Google Scholar] [CrossRef]
- Narla, G.; Heath, K.E.; Reeves, H.L.; Li, D.; Giono, L.E.; Kimmelman, A.C.; Glucksman, M.J.; Narla, J.; Eng, F.J.; Chan, A.M.; et al. KLF6, a candidate tumor suppressor gene mutated in prostate cancer. Science 2001, 294, 2563–2566. [Google Scholar] [CrossRef]
- Narla, G.; Kremer-Tal, S.; Matsumoto, N.; Zhao, X.; Yao, S.; Kelley, K.; Tarocchi, M.; Friedman, S.L. In vivo regulation of p21 by the Kruppel-like factor 6 tumor-suppressor gene in mouse liver and human hepatocellular carcinoma. Oncogene 2007, 26, 4428–4434. [Google Scholar] [CrossRef]
- Slavin, D.A.; Koritschoner, N.P.; Prieto, C.C.; López-Díaz, F.J.; Chatton, B.; Bocco, J.L. A new role for the Kruppel-like transcription factor KLF6 as an inhibitor of c-Jun proto-oncoprotein function. Oncogene 2004, 23, 8196–8205. [Google Scholar] [CrossRef]
- Benzeno, S.; Narla, G.; Allina, J.; Cheng, G.Z.; Reeves, H.L.; Banck, M.S.; Odin, J.A.; Diehl, J.A.; Germain, D.; Friedman, S.L. Cyclin-dependent kinase inhibition by the KLF6 tumor suppressor protein through interaction with cyclin D1. Cancer Res. 2004, 64, 3885–3891. [Google Scholar] [CrossRef] [PubMed]
- Narla, G.; DiFeo, A.; Reeves, H.L.; Schaid, D.J.; Hirshfeld, J.; Hod, E.; Katz, A.; Isaacs, W.B.; Hebbring, S.; Komiya, A.; et al. A germline DNA polymorphism enhances alternative splicing of the KLF6 tumor suppressor gene and is associated with increased prostate cancer risk. Cancer Res. 2005, 65, 1213–1222. [Google Scholar] [CrossRef] [PubMed]
- DiFeo, A.; Feld, L.; Rodriguez, E.; Wang, C.; Beer, D.G.; Martignetti, J.A.; Narla, G. A functional role for KLF6-SV1 in lung adenocarcinoma prognosis and chemotherapy response. Cancer Res. 2008, 68, 965–970. [Google Scholar] [CrossRef] [PubMed]
- Narla, G.; Difeo, A.; Yao, S.; Banno, A.; Hod, E.; Reeves, H.L.; Qiao, R.F.; Camacho-Vanegas, O.; Levine, A.; Kirschenbaum, A.; et al. Targeted inhibition of the KLF6 splice variant, KLF6 SV1, suppresses prostate cancer cell growth and spread. Cancer Res. 2005, 65, 5761–5768. [Google Scholar] [CrossRef]
- Difeo, A.; Huang, F.; Sangodkar, J.; Terzo, E.A.; Leake, D.; Narla, G.; Martignetti, J.A. KLF6-SV1 is a novel antiapoptotic protein that targets the BH3-only protein NOXA for degradation and whose inhibition extends survival in an ovarian cancer model. Cancer Res. 2009, 69, 4733–4741. [Google Scholar] [CrossRef]
- McConnell, B.B.; Yang, V.W. Mammalian Krüppel-Like factors in health and diseases. Physiol. Rev. 2010, 90, 1337–1381. [Google Scholar] [CrossRef]
- Hatami, R.; Sieuwerts, A.M.; Izadmehr, S.; Yao, Z.; Qiao, R.F.; Papa, L.; Look, M.P.; Smid, M.; Ohlssen, J.; Levine, A.C.; et al. KLF6-SV1 Drives Breast Cancer Metastasis and Is Associated with Poor Survival. Sci. Transl. Med. 2013, 5, 169ra12. [Google Scholar] [CrossRef]
- Narla, G.; DiFeo, A.; Fernandez, Y.; Dhanasekaran, S.; Huang, F.; Sangodkar, J.; Hod, E.; Leake, D.; Friedman, S.L.; Hall, S.J.; et al. KLF6-SV1 overexpression accelerates human and mouse prostate cancer progression and metastasis. J. Clin. Investig. 2008, 118, 2711–2721. [Google Scholar] [CrossRef]
- Jimenez, M.; Arechederra, M.; Ávila, M.A.; Berasain, C. Splicing alterations contributing to cancer hallmarks in the liver: Central role of dedifferentiation and genome instability. Transl. Gastroenterol. Hepatol. 2018, 3, 84. [Google Scholar] [CrossRef]
- DiFeo, A.; Narla, G.; Martignetti, J.A. Emerging roles of Kruppel-like factor 6 and Kruppel-like factor 6 splice variant 1 in ovarian cancer progression and treatment. Mt. Sinai J. Med. 2009, 76, 557–566. [Google Scholar] [CrossRef]
- Hilger-Eversheim, K.; Moser, M.; Schorle, H.; Buettner, R. Regulatory roles of AP-2 transcription factors in vertebrate development, apoptosis and cell-cycle control. Gene 2000, 260, 1–12. [Google Scholar] [CrossRef]
- Jean, D.; Gershenwald, J.E.; Huang, S.; Luca, M.; Hudson, M.J.; Tainsky, M.A.; Bar-Eli, M. Loss of AP-2 results in up-regulation of MCAM/MUC18 and an increase in tumor growth and metastasis of human melanoma cells. J. Biol. Chem. 1998, 273, 16501–16508. [Google Scholar] [CrossRef] [PubMed]
- Gershenwald, J.E.; Sumner, W.; Calderone, T.; Wang, Z.; Huang, S.; Bar-Eli, M. Dominant-negative transcription factor AP-2 augments SB-2 melanoma tumor growth in vivo. Oncogene 2001, 20, 3363–3375. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Buettner, R.; Kannan, P.; Imhof, A.; Bauer, R.; Yim, S.O.; Glockshuber, R.; Van Dyke, M.W.; Tainsky, M.A. An alternatively spliced mRNA from the AP-2 gene encodes a negative regulator of transcriptional activation by AP-2. Mol. Cell. Biol. 1993, 13, 4174–4185. [Google Scholar] [CrossRef][Green Version]
- Fan, Y.; Lu, D. The Ikaros family of zinc-finger proteins. Acta Pharm. Sin. B 2016, 6, 513–521. [Google Scholar] [CrossRef]
- Vshyukova, V.; Valochnik, A.; Meleshko, A. Expression of aberrantly spliced oncogenic Ikaros isoforms coupled with clonal IKZF1 deletions and chimeric oncogenes in acute lymphoblastic leukemia. Blood Cells Mol. Dis. 2018, 71, 29–38. [Google Scholar] [CrossRef]
- Nakase, K.; Ishimaru, F.; Avitahl, N.; Dansako, H.; Matsuo, K.; Fujii, K.; Sezaki, N.; Nakayama, H.; Yano, T.; Fukuda, S.; et al. Dominant Negative Isoform of the Ikaros Gene in Patients with Adult B-Cell Acute Lymphoblastic Leukemia. Cancer Res. 2000, 60, 4062–4065. [Google Scholar]
- Yagi, T.; Kano, G.; Imamura, T. High frequency of Ikaros isoform 6 expression in acute myelomonocytic and monocytic leukemias: Implications for up-regulation of the antiapoptotic protein Bcl-XL in leukemogenesis. Blood 2011, 99, 1350–1355. [Google Scholar] [CrossRef]
- Shi, Z.; He, F.; Chen, M.; Hua, L.; Wang, W.; Jiao, S.; Zhou, Z. DNA-binding mechanism of the Hippo pathway transcription factor TEAD4. Oncogene 2017, 36, 4362–4369. [Google Scholar] [CrossRef]
- Tang, J.-Y.; Yu, C.-Y.; Bao, Y.-J.; Chen, L.; Chen, J.; Yang, S.-L.; Chen, H.-Y.; Hong, J.; Fang, J.-Y. Cell Cycle TEAD4 promotes colorectal tumorigenesis via transcriptionally targeting YAP1 TEAD4 promotes colorectal tumorigenesis via transcriptionally targeting YAP1. Cell Cycle 2018, 17, 102–109. [Google Scholar] [CrossRef]
- Huh, H.D.; Kim, D.H.; Jeong, H.-S.; Park, H.W. Regulation of TEAD Transcription Factors in Cancer Biology. Cells 2019, 8, 600. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Ren, F.; Zhang, Q.; Chen, Y.; Wang, B.; Jiang, J. The TEAD/TEF family of transcription factor Scalloped mediates Hippo signaling in organ size control. Dev. Cell 2008, 14, 377–387. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Yuan, L.; Sun, Y.; Wang, P.; Zhang, H.; Feng, X.; Wang, Z.; Zhang, W.; Yang, C.; Zeng, Y.A.; et al. Glucocorticoid receptor signaling activates TEAD4 to promote breast cancer progression. Cancer Res. 2019, 79, 4399–4411. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Zeng, Z.; Wei, H.; Wang, Z. Alternative splicing in cancers: From aberrant regulation to new therapeutics. Semin. Cell Dev. Biol. 2018, 75, 13–22. [Google Scholar] [CrossRef]
- Qi, Y.; Yu, J.; Han, W.; Fan, X.; Qian, H.; Wei, H.; Tsai, Y.H.S.; Zhao, J.; Zhang, W.; Liu, Q.; et al. A splicing isoform of TEAD4 attenuates the Hippo-YAP signalling to inhibit tumour proliferation. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef]
- Vieler, M.; Sanyal, S. P53 isoforms and their implications in cancer. Cancers 2018, 10, 288. [Google Scholar] [CrossRef]
- Surget, S.; Khoury, M.P.; Bourdon, J.C. Uncovering the role of p53 splice variants in human malignancy: A clinical perspective. Onco Targets. Ther. 2013, 7, 57–67. [Google Scholar]
- Harms, K.; Nozell, S.; Chen, X. The common and distinct target genes of the p53 family transcription factors. C Cell. Mol. Life Sci 2004, 61, 822–842. [Google Scholar] [CrossRef]
- Maglic, D.; Stovall, D.B.; Cline, J.M.; Fry, E.A.; Mallakin, A.; Taneja, P.; Caudell, D.L.; Willingham, M.C.; Sui, G.; Inoue, K. DMP1β, a splice isoform of the tumour suppressor DMP1 locus, induces proliferation and progression of breast cancer. J. Pathol. 2015, 236, 90–102. [Google Scholar] [CrossRef]
- Tian, N.; Li, J.; Shi, J.; Sui, G. From General Aberrant Alternative Splicing in Cancers and Its Therapeutic Application to the Discovery of an Oncogenic DMTF1 Isoform. Int. J. Mol. Sci. 2017, 18, 191. [Google Scholar] [CrossRef]
- Inoue, K.; Mallakin, A.; Frazier, D.P. Dmp1 and tumor suppression. Oncogene 2007, 26, 4329–4335. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Samad, R.; Zalzali, H.; Rammah, C.; Giraud, J.; Naudin, C.; Dupasquier, S.; Poulat, F.; Boizet-Bonhoure, B.; Lumbroso, S.; Mouzat, K.; et al. MiniSOX9, a dominant-negative variant in colon cancer cells. Oncogene 2011, 30, 2493–2503. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nestler, E.J. δfosB: A transcriptional regulator of stress and antidepressant responses. Eur. J. Pharmacol. 2015, 753, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Luft, F.C. C/EBPβ LIP induces a tumor menagerie making it an oncogene. J. Mol. Med. 2014, 93, 39–49. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nerlov, C. Transcriptional and translational control of C/EBPs: The case for “deep” genetics to understand physiological function. BioEssays 2010, 32, 680–686. [Google Scholar] [CrossRef] [PubMed]
- Robinson, G.W.; Johnson, P.F.; Hennighausen, L.; Sterneck, E. The C/EBPβ transcription factor regulates epithelial cell proliferation and differentiation in the mammary gland. Genes Dev. 1998, 12, 1907–1916. [Google Scholar] [CrossRef]
- Seagroves, T.N.; Krnacik, S.; Raught, B.; Gay, J.; Burgess-Beusse, B.; Darlington, G.J.; Rosen, J.M. C/EBPβ, but not C/EBPα, is essential for ductal morphogenesis, lobuloalveolar proliferation, and functional differentiation in the mouse mammary gland. Genes Dev. 1998, 12, 1917–1928. [Google Scholar] [CrossRef]
- Screpanti, I.; Romani, L.; Musiani, P.; Modesti, A.; Fattori, E.; Lazzaro, D.; Sellitto, C.; Scarpa, S.; Bellavia, D.; Lattanzio, G. Lymphoproliferative disorder and imbalanced T-helper response in C/EBP beta-deficient mice. EMBO J. 1995, 14, 1932–1941. [Google Scholar] [CrossRef]
- Chen, X.; Lu, J.; Zhao, X.; Chen, C.; Qiao, D.; Wang, H.; Yue, X. Role of C/EBP-β in Methamphetamine-Mediated Microglial Apoptosis. Front. Cell. Neurosci. 2019, 13, 366. [Google Scholar] [CrossRef]
- Kajimura, S.; Seale, P.; Kubota, K.; Lunsford, E.; Frangioni, J.V.; Gygi, S.P.; Spiegelman, B.M. Initiation of myoblast to brown fat switch by a PRDM16-C/EBP-β transcriptional complex. Nature 2009, 460, 1154–1158. [Google Scholar] [CrossRef]
- Zahnow, C.A. CCAAT/enhancer-binding protein β: Its role in breast cancer and associations with receptor tyrosine kinases. Expert Rev. Mol. Med. 2009, 11, e12. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.-H.; Wang, Y.; Qiao, S.; Wang, M.-J.; Chen, F.; Zi, X.-Y.; Li, J.-X.; Zhang, H.-B.; Yu, B.; Hu, Y.-P. Liver-enriched activator protein 1 as an isoform of CCAAT/enhancer-binding protein beta suppresses stem cell features of hepatocellular carcinoma. Cancer Manag. Res. 2018, 10, 873–885. [Google Scholar] [CrossRef] [PubMed]
- Anand, S.; Ebner, J.; Warren, C.B.; Raam, M.S.; Piliang, M.; Billings, S.D.; Maytin, E. V C/EBP transcription factors in human squamous cell carcinoma: Selective changes in expression of isoforms correlate with the neoplastic state. PLoS ONE 2014, 9, e112073. [Google Scholar] [CrossRef] [PubMed]
- Milde-Langosch, K.; Löning, T.; Bamberger, A.M. Expression of the CCAAT/enhancer-binding proteins C/EBPα, C/EBPβ and C/EBPδ in breast cancer: Correlations with clinicopathologic parameters and cell-cycle regulatory proteins. Breast Cancer Res. Treat. 2003, 79, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Kurzejamska, E.; Johansson, J.; Jirström, K.; Prakash, V.; Ananthaseshan, S.; Boon, L.; Fuxe, J.; Religa, P. C/EBPβ expression is an independent predictor of overall survival in breast cancer patients by MHCII/CD4-dependent mechanism of metastasis formation. Oncogenesis 2014, 3, e125. [Google Scholar] [CrossRef] [PubMed]
- Fang, T.; Cui, M.; Sun, J.; Ge, C.; Zhao, F.; Zhang, L.; Tian, H.; Zhang, L.; Chen, T.; Jiang, G.; et al. Orosomucoid 2 inhibits tumor metastasis and is upregulated by CCAAT/enhancer binding protein β in hepatocellular carcinomas. Oncotarget 2015, 6, 16106–16119. [Google Scholar] [CrossRef]
- Brown-Bryan, T.A.; Leoh, L.S.; Ganapathy, V.; Pacheco, F.J.; Mediavilla-Varela, M.; Filippova, M.; Linkhart, T.A.; Gijsbers, R.; Debyser, Z.; Casiano, C.A. Alternative splicing and caspase-mediated cleavage generate antagonistic variants of the stress oncoprotein LEDGF/p75. Mol. Cancer Res. 2008, 6, 1293–1307. [Google Scholar] [CrossRef]
- Turlure, F.; Maertens, G.; Rahman, S.; Cherepanov, P.; Engelman, A. A tripartite DNA-binding element, comprised of the nuclear localization signal and two AT-hook motifs, mediates the association of LEDGF/p75 with chromatin in vivo. Nucleic Acids Res. 2006, 34, 1653–1665. [Google Scholar] [CrossRef]
- Pradeepa, M.M.; Sutherland, H.G.; Ule, J.; Grimes, G.R.; Bickmore, W.A. Psip1/Ledgf p52 Binds Methylated Histone H3K36 and Splicing Factors and Contributes to the Regulation of Alternative Splicing. PLoS Genet. 2012, 8, e1002717. [Google Scholar] [CrossRef]
- Basu, A.; Sanchez, T.W.; Casiano, C.A. DFS70/LEDGFp75: An Enigmatic Autoantigen at the Interface between Autoimmunity, AIDS, and Cancer. Front. Immunol. 2015, 6, 116. [Google Scholar] [CrossRef]
- Blokken, J.; De Rijck, J.; Christ, F.; Debyser, Z. Protein–protein and protein–chromatin interactions of LEDGF/p75 as novel drug targets. Drug Discov. Today Technol. 2017, 24, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.P.; Fatma, N.; Kimura, A.; Chylack, L.T.; Shinohara, T. LEDGF binds to heat shock and stress-related element to activate the expression of stress-related genes. Biochem. Biophys. Res. Commun. 2001, 283, 943–955. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Rojas, H.; Banerjee, H.; Cabrera, I.B.; Perez, K.Y.; De León, M.; Casiano, C.A. Expression of the Stress Response Oncoprotein LEDGF/p75 in Human Cancer: A Study of 21 Tumor Types. PLoS ONE 2012, 7, e30132. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.P.; Ohguro, N.; Kikuchi, T.; Sueno, T.; Reddy, V.N.; Yuge, K.; Chylack, L.T.; Shinohara, T. Lens epithelium-derived growth factor: Effects on growth and survival of lens epithelial cells, keratinocytes, and fibroblasts. Biochem. Biophys. Res. Commun. 2000, 267, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.S.; Myklebust, L.M.; Kjarland, E.; Gjertsen, B.T.; Pendino, F.; Bruserud, Ø.; Døskeland, S.O.; Lillehaug, J.R. LEDGF/p75 has increased expression in blasts from chemotherapy-resistant human acute myelogenic leukemia patients and protects leukemia cells from apoptosis in vitro. Mol. Cancer 2007, 6, 31. [Google Scholar] [CrossRef]
- Leoh, L.S.; Van Heertum, B.; De Rijck, J.; Filippova, M.; Rios-Colon, L.; Basu, A.; Martinez, S.R.; Tungteakkhun, S.S.; Filippov, V.; Christ, F.; et al. The stress oncoprotein LEDGF/p75 interacts with the methyl CpG binding protein MeCP2 and influences its transcriptional activity. Mol. Cancer Res. 2012, 10, 378–391. [Google Scholar] [CrossRef]
- Yokoyama, A.; Cleary, M.L. Menin Critically Links MLL Proteins with LEDGF on Cancer-Associated Target Genes. Cancer Cell 2008, 14, 36–46. [Google Scholar] [CrossRef]
- Singh, D.K.; Gholamalamdari, O.; Jadaliha, M.; Li, X.L.; Lin, Y.-C.; Zhang, Y.; Guang, S.; Hashemikhabir, S.; Tiwari, S.; Zhu, Y.J.; et al. PSIP1/p75 promotes tumorigenicity in breast cancer cells by promoting the transcription of cell cycle genes. Carcinogenesis 2017, 38, 966–975. [Google Scholar] [CrossRef]
- Daugaard, M.; Kirkegaard-Sørensen, T.; Ostenfeld, M.S.; Aaboe, M.; Høyer-Hansen, M.; Ørntoft, T.F.; Rohde, M.; Jäättelä, M. Lens Epithelium-Derived Growth Factor Is an Hsp70-2 Regulated Guardian of Lysosomal Stability in Human Cancer. Cancer Res. 2007, 67, 2559–2567. [Google Scholar] [CrossRef]
- Bruce, A.W.; Donaldson, I.J.; Wood, I.C.; Yerbury, S.A.; Sadowski, M.I.; Chapman, M.; Gottgens, B.; Buckley, N.J. Genome-wide analysis of repressor element 1 silencing transcription factor/neuron-restrictive silencing factor (REST/NRSF) target genes. Proc. Natl. Acad. Sci. USA 2004, 101, 10458–10463. [Google Scholar] [CrossRef]
- Deng, P.; Zuo, Y.; Feng, S.; Li, Z.; Chen, W.; Li, H.; Wang, X. Knockdown of NRSF inhibits cell proliferation of ovarian cancer via activating Hippo pathway. Life Sci. 2018, 215, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Liu, P.; Kong, L.; Fei, X.; Gao, Y.; Wu, T.; Sun, D.; Tan, X. Identification of RE1-Silencing Transcription Factor as a Promoter of Metastasis in Pancreatic Cancer. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef]
- Wagoner, M.P.; Gunsalus, K.T.W.; Schoenike, B.; Richardson, A.L.; Friedl, A.; Roopra, A. The transcription factor REST is lost in aggressive breast cancer. PLoS Genet. 2010, 6, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Majumder, S. REST in good times and bad: Roles in tumor suppressor and oncogenic activities. Cell Cycle 2006, 5, 1929–1935. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wang, Z.; Tang, X.; Zeng, L.; Fan, X.; Li, Z. Molecular mechanisms and potential prognostic effects of REST and REST4 in glioma (Review). Mol. Med. Rep. 2017, 16, 3707–3712. [Google Scholar] [CrossRef] [PubMed]
- Coulson, J.M.; Edgson, J.L.; Woll, P.J.; Quinn, J.P. A splice variant of the neuron-restrictive silencer factor repressor is expressed in small cell lung cancer: A potential role in derepression of neuroendocrine genes and a useful clinical marker. Cancer Res. 2000, 60, 1840–1844. [Google Scholar]
- Chen, G.-L.; Miller, G.M. Alternative REST Splicing Underappreciated. eNeuro 2018, 5. [Google Scholar] [CrossRef]
- Chen, G.-L.; Miller, G.M. Extensive Alternative Splicing of the Repressor Element Silencing Transcription Factor Linked to Cancer. PLoS ONE 2013, 8, e62217. [Google Scholar] [CrossRef]
- Lee, A.R.; Che, N.; Lovnicki, J.M.; Dong, X. Development of neuroendocrine prostate cancers by the Ser/Arg repetitive matrix 4-mediated RNA splicing network. Front. Oncol. 2018, 8, 93. [Google Scholar] [CrossRef]
- Palm, K.; Metsis, M.; Timmusk, T. Neuron-specific splicing of zinc finger transcription factor REST/NRSF/XBR is frequent in neuroblastomas and conserved in human, mouse and rat. Mol. Brain Res. 1999, 72, 30–39. [Google Scholar] [CrossRef]
- Morikawa, M.; Koinuma, D.; Miyazono, K.; Heldin, C.H. Genome-wide mechanisms of Smad binding. Oncogene 2013, 32, 1609–1615. [Google Scholar] [CrossRef] [PubMed]
- Ullah, I.; Sun, W.; Tang, L.; Feng, J. Roles of SmAds family and alternative splicing variants of Smad4 in different cancers. J. Cancer 2018, 9, 4018–4028. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Wu, C.-J.; Chu, G.C.; Xiao, Y.; Ho, D.; Zhang, J.; Perry, S.R.; Labrot, E.S.; Wu, X.; Lis, R.; et al. SMAD4-dependent barrier constrains prostate cancer growth and metastatic progression. Nature 2011, 470, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Haeger, S.M.; Thompson, J.J.; Kalra, S.; Cleaver, T.G.; Merrick, D.; Wang, X.-J.; Malkoski, S.P. Smad4 loss promotes lung cancer formation but increases sensitivity to DNA topoisomerase inhibitors. Oncogene 2016, 35, 577–586. [Google Scholar] [CrossRef]
- Zhao, M.; Mishra, L.; Deng, C.-X. The role of TGF-β/SMAD4 signaling in cancer. Int. J. Biol. Sci. 2018, 14, 111–123. [Google Scholar] [CrossRef]
- Yang, J.; Wang, Y.; Zeng, Z.; Qiao, L.; Zhuang, L.; Gao, Q.; Ma, D.; Huang, X. Smad4 deletion in blood vessel endothelial cells promotes ovarian cancer metastasis. Int. J. Oncol. 2017, 50, 1693–1700. [Google Scholar] [CrossRef][Green Version]
- Yang, L.; Mao, C.; Teng, Y.; Li, W.; Zhang, J.; Cheng, X.; Li, X.; Han, X.; Xia, Z.; Deng, H.; et al. Targeted disruption of Smad4 in mouse epidermis results in failure of hair follicle cycling and formation of skin tumors. Cancer Res. 2005, 65, 8671–8678. [Google Scholar] [CrossRef]
- Pierreux, C.E.; Nicolas, F.J.; Hill, C.S. Transforming Growth Factor beta -Independent Shuttling of Smad4 between the Cytoplasm and Nucleus. Mol. Cell. Biol. 2000, 20, 9041–9054. [Google Scholar] [CrossRef]
- Lazzereschi, D.; Nardi, F.; Turco, A.; Ottini, L.; D’Amico, C.; Mariani-Costantini, R.; Gulino, A.; Coppa, A. A complex pattern of mutations and abnormal splicing of Smad4 is present in thyroid tumours. Oncogene 2005, 24, 5344–5354. [Google Scholar] [CrossRef]
- Kageyama, H.; Seki, N.; Yamada, S.; Sakiyama, S.; Nakagawara, A. DPC4 splice variants in neuroblastoma. Cancer Lett. 1998, 122, 187–193. [Google Scholar] [CrossRef]
- De Winter, J.P.; Roelen, B.A.J.; Ten Dijke, P.; Van Der Burg, B.; Van Den Eijnden-Van Raaij, A.J.M. DPC4 (SMAD4) mediates transforming growth factar-β1 (TGF-β1) induced growth inhibition and transcriptional response in breast tumour cells. Oncogene 1997, 14, 1891–1899. [Google Scholar] [CrossRef] [PubMed]
- Chantalat, E.; Boudou, F.; Laurell, H.; Palierne, G.; Houtman, R.; Melchers, D.; Rochaix, P.; Filleron, T.; Stella, A.; Burlet-Schiltz, O.; et al. The AF-1-deficient estrogen receptor ERα46 isoform is frequently expressed in human breast tumors. Breast Cancer Res. 2016, 18, 123. [Google Scholar] [CrossRef] [PubMed]
- Herynk, M.H.; Fuqua, S.A.W. Estrogen receptor mutations in human disease. Endocr. Rev. 2004, 25, 869–898. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ren, J.; Wei, J.; Chong, C.C.N.; Yang, D.; He, Y.; Chen, G.G.; Lai, P.B.S. Alternative splicing of estrogen receptor alpha in hepatocellular carcinoma. BMC Cancer 2016, 16, 926. [Google Scholar] [CrossRef] [PubMed]
- Villa, E.; Colantoni, A.; Cammà, C.; Grottola, A.; Buttafoco, P.; Gelmini, R.; Ferretti, I.; Manenti, F. Estrogen receptor classification for hepatocellular carcinoma: Comparison with clinical staging systems. J. Clin. Oncol. 2003, 21, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Bushweller, J.H. Targeting transcription factors in cancer—From undruggable to reality. Nat. Rev. Cancer 2019, 19, 611–624. [Google Scholar] [CrossRef]
- Lambert, M.; Jambon, S.; Depauw, S.; David-Cordonnier, M.H. Targeting transcription factors for cancer treatment. Molecules 2018, 23, 1479. [Google Scholar] [CrossRef]
- Brinkman, B.M.N. Splice variants as cancer biomarkers. Clin. Biochem. 2004, 37, 584–594. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Lu, C.; Luber, B.; Wang, H.; Chen, Y.; Zhu, Y.; Silberstein, J.L.; Taylor, M.N.; Maughan, B.L.; Denmeade, S.R.; et al. Clinical significance of androgen receptor splice variant-7 mRNA detection in circulating tumor cells of men with metastatic castration-resistant prostate cancer treated with first & second-line Abiraterone & Enzalutamide. J. Clin. Oncol. 2017, 35, 2149–2156. [Google Scholar]
- Wang, Y.; Bernhardy, A.J.; Cruz, C.; Krais, J.J.; Nacson, J.; Nicolas, E.; Peri, S.; Van Der Gulden, H.; Van Der Heijden, I.; O’Brien, S.W.; et al. The BRCA1-Δ11q alternative splice isoform bypasses germline mutations and promotes therapeutic resistance to PARP inhibition and cisplatin. Cancer Res. 2016, 76, 2778–2790. [Google Scholar] [CrossRef]
- Liu, S.S.; Chan, K.Y.K.; Leung, R.C.Y.; Law, H.K.W.; Leung, T.W.; Ngan, H.Y.S. Enhancement of the radiosensitivity of cervical cancer cells by overexpressing p73α. Mol. Cancer Ther. 2006, 5, 1209–1215. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pfeifer, D.; Wallin, Å.; Holmlund, B.; Sun, X.F. Protein expression following γ-irradiation relevant to growth arrest and apoptosis in colon cancer cells. J. Cancer Res. Clin. Oncol. 2009, 135, 1583–1592. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.; Schleithoff, E.S.; Stremmel, W.; Melino, G.; Krammer, P.H.; Schilling, T. One, two, three-p53, p63, p73 and chemosensitivity. Drug Resist. Updat. 2006, 9, 288–306. [Google Scholar] [CrossRef] [PubMed]
- Havens, M.A.; Hastings, M.L. SURVEY AND SUMMARY Splice-switching antisense oligonucleotides as therapeutic drugs. Nucleic Acids Res. 2016, 44, 6549–6563. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TF | DNA Binding Motif | Physiological Regulated Processes | Splice Variants | TP Splice Variants | AS Domains in TP Splice Variants | Ref. |
|---|---|---|---|---|---|---|
| NF-YA | CCAAT | Cell proliferation and differentiation, metabolism, cell death | NF-YAs NF-YAl | NF-YAs | TAD | [26,27,28,29] |
| STAT3 | TTCC(G=C)GGAA | Cell proliferation, cell death | STAT3α STAT3β | STAT3α | TAD | [31,32,33,34,35,36,37,38,39,40,41,42,43] |
| TCF4 | (A/T)(A/T)CAAAG | Cell proliferation, apoptosis | TCF4 TCF4N/E/M/B/S/K/J | TCF4J | SxxSS motif | [44,45,46,47,48,49,50,51] |
| WT1 | GCGTGGGAGT | Cell proliferation and differentiation, metabolism, apoptosis | -17AA/-KTS -17AA/+KTS +17AA/-KTS +17AA/+KTS | -17AA* +17AA* +17AA/+KTS | TAD DBD | [54,55,56,57,58,59,60] |
| TF | DNA Binding Motif | Physiological Regulated Processes | Splice Variants | TP Splice Variants | AS Domains in TP Splice Variants | Ref. |
|---|---|---|---|---|---|---|
| MEF2C | YTA(A/T)4TAR | Muscle cell proliferation and differentiation | Mef2Cα1 Mef2Cα2 | Mef2Cα1 | Adjacent to DBD | [65] |
| MITF | TCATGTGCT | Melanocyte proliferation and differentiation | (+) MITF (−) MITF | (−) MITF | Adjacent to DBD | [68,69,70,71,72,73] |
| NFAT2 | (A/T)GGAAA | Cell proliferation and differentiation, apoptosis, inflammatory response | NFAT2α NFAT2β | NFAT2α | TAD | [78,82] |
| TF | DNA Binding Motif | Physiological Regulated Processes | Splice Variants | TP Splice Variants | AS Domains in TP Splice Variants | Ref. |
|---|---|---|---|---|---|---|
| AP-2α | GCCNNNGGC | Development, cell growth and differentiation, apoptosis | AP-2α AP-2B | AP-2B | Dimerization domain | [103,104] |
| CEBPβ | T(TG)NNGNAA (TG) | Cell cycle, differentiation, apoptosis and senescence | LAP1 LAP2 LIP | LIP | TAD | [131,132,133,134,135,136] |
| DMTF1 | CCCG(G/T)ATGT | Cell proliferation, apoptosis | DMTF1α/β/γ | DMTF1β | TAD DBD | [119,120,121] |
| ERα | AGGTCANNNTGACCT | Cell proliferation, apoptosis, inflammation | ERα (ERα-66) ERαΔ1 (ERα-46) ERαΔ2/Δ3/Δ4/ Δ5/Δ7 ERα−36 ERα−30 | ERα-66 ERα−36 ERαΔ3/Δ5/Δ7 | TAD DBD LBD | [172,173,174,175] |
| FOSB | TGAC/GTCA | Cell proliferation and differentiation, apoptosis, stress response | FOSB ΔFOSB | FOSB | TAD Degron domain | [123] |
| HELIOS | GGGAA | T-lineage differentiation | HELIOS HELIOS-V1/V2/V3 | HELIOS-V1/V2/V3 | NLS DBD | [84] |
| IKAROS | GGAAA | Hematopoiesis, myelopoiesis, lymphopoiesis | IK1-10 IKX | IK4-10 | DBD | [105,106,107,108] |
| KLF6 | GC box CACC box | Cell proliferation and differentiation, adhesion, tissue repair | KLF6 KLF6-SV1/SV2/SV3 | KLF6-SV1 | DBD | [88,92,93,94,95,96,97,98,99,100] |
| LEDGF | NGAAN T/AGGGG | Neuroepithelial stem cell differentiation and neurogenesis, stress-induced apoptosis, lens epithelial cell growth and differentiation, host-virus interaction | LEDGF/p52 LEDGF/p75 | LEDGF/p75 | IBD CTD | [140,143,144,145,146,147,148,149] |
| p53/p63/p73 | RRRC(A/T)(A/T)GYYY | Cell cycle arrest, cell death, genome stability, cell differentiation, development | p53α/β/γ Δ40/Δ133/ Δ160 p53α/β/γ TA/ΔN p63 TA/ΔN p73 | Δ40p53α Δ133p53α Δ160p53α ΔN p63/p73 | TAD DBD | [116,117,118] |
| REST/NRSF | NT(T/C)AG(A/C)(A/G)CCNN(A/G)G(A/C)(G/S)AG | Cell differentiation | REST REST1/4/5 REST-N50/N62 REST-5FΔ | REST4 REST-N50 REST-N62 | DBD NLS | [153,155,156,157,158,159,160] |
| SMAD4 | GTCTAGAC | Cell proliferation and differentiation | SMAD4 SMAD4 Δ3/ Δ4/Δ5-6/Δ6/ Δ4-6/Δ4-7 | SMAD4 Δ3/ Δ4/Δ5-6/ Δ6/Δ4-6/Δ4-7 | Linker domain TAD | [168,169,170,171] |
| SOX9 | (A/T)(A/T)CAA(A/T)G | Stem cell maintenance and commitment, differentiation, matrix deposition | SOX9 MiniSOX9 | MiniSOX9 | TAD | [122] |
| TEAD4 | CATTCCA | Cell proliferation and differentiation, apoptosis | TEAD-FL TEAD-S | TEAD-FL | DBD | [111,113,114,115] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belluti, S.; Rigillo, G.; Imbriano, C. Transcription Factors in Cancer: When Alternative Splicing Determines Opposite Cell Fates. Cells 2020, 9, 760. https://doi.org/10.3390/cells9030760
Belluti S, Rigillo G, Imbriano C. Transcription Factors in Cancer: When Alternative Splicing Determines Opposite Cell Fates. Cells. 2020; 9(3):760. https://doi.org/10.3390/cells9030760
Chicago/Turabian StyleBelluti, Silvia, Giovanna Rigillo, and Carol Imbriano. 2020. "Transcription Factors in Cancer: When Alternative Splicing Determines Opposite Cell Fates" Cells 9, no. 3: 760. https://doi.org/10.3390/cells9030760
APA StyleBelluti, S., Rigillo, G., & Imbriano, C. (2020). Transcription Factors in Cancer: When Alternative Splicing Determines Opposite Cell Fates. Cells, 9(3), 760. https://doi.org/10.3390/cells9030760