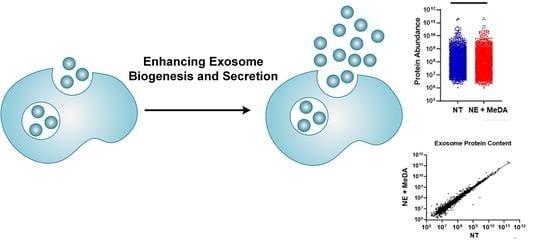
Boosting the Biogenesis and Secretion of Mesenchymal Stem Cell-Derived Exosomes


Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Extracellular Vesicle Isolation
2.3. Exosome Characterization by Nanoparticle Tracking Analysis (NTA)
2.4. Exosome Characterization by Transmission Electron Microscopy (TEM)
2.5. Western Blotting
2.6. MTT Assay
2.7. Collagen Expression Assay
2.8. Angiogenesis Assay
2.9. Bone Marrow-Derived Macrophage (BMDM) Isolation and Culture
2.10. Macrophage Polarization Assay
2.11. Mechanism Study
2.12. Mass Spectrometric Analysis of Exosomes
2.13. Bioinformatic Data Analysis
3. Results
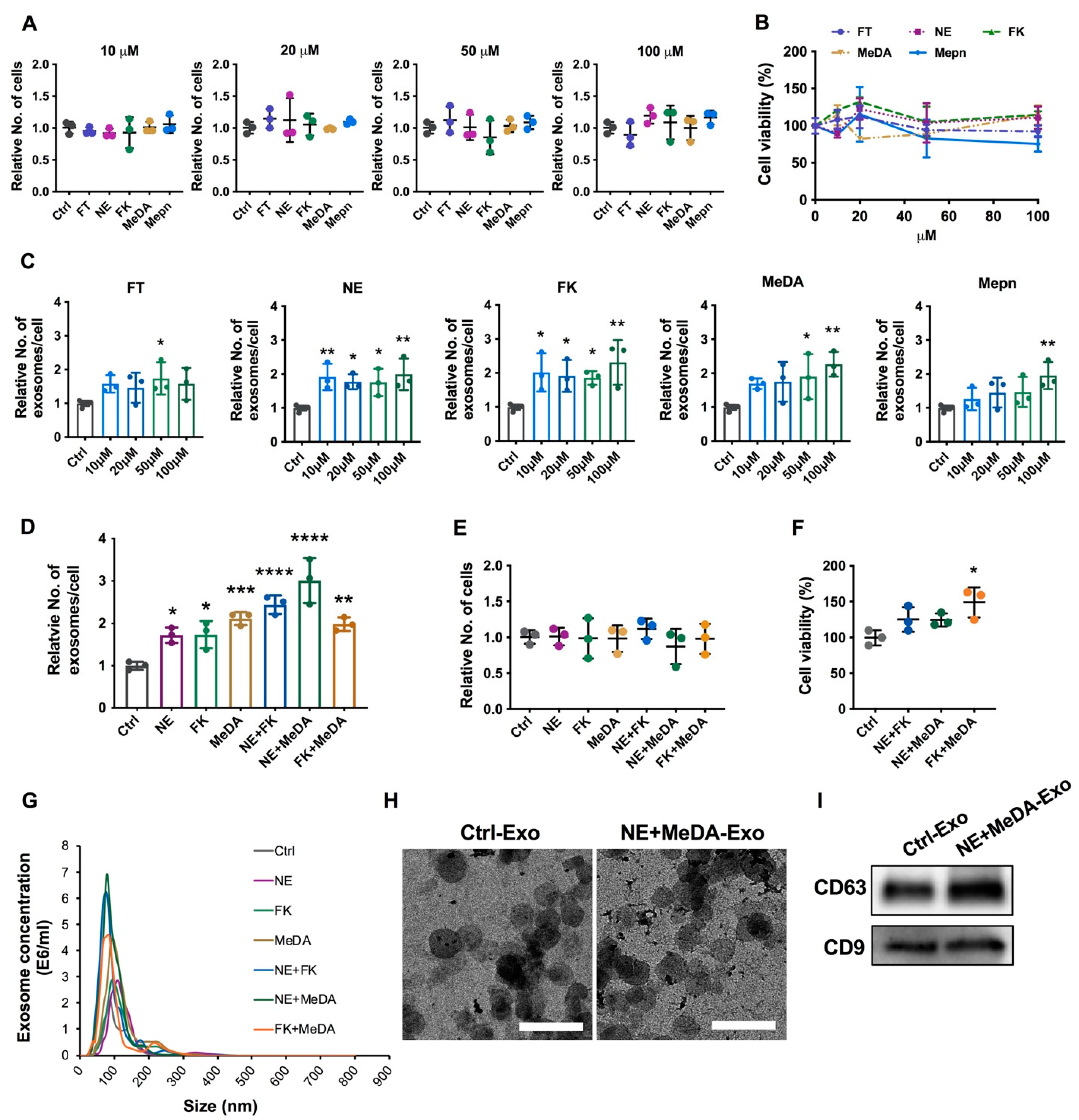
3.1. Effects of Small Molecules on Exosomal Production Efficiency
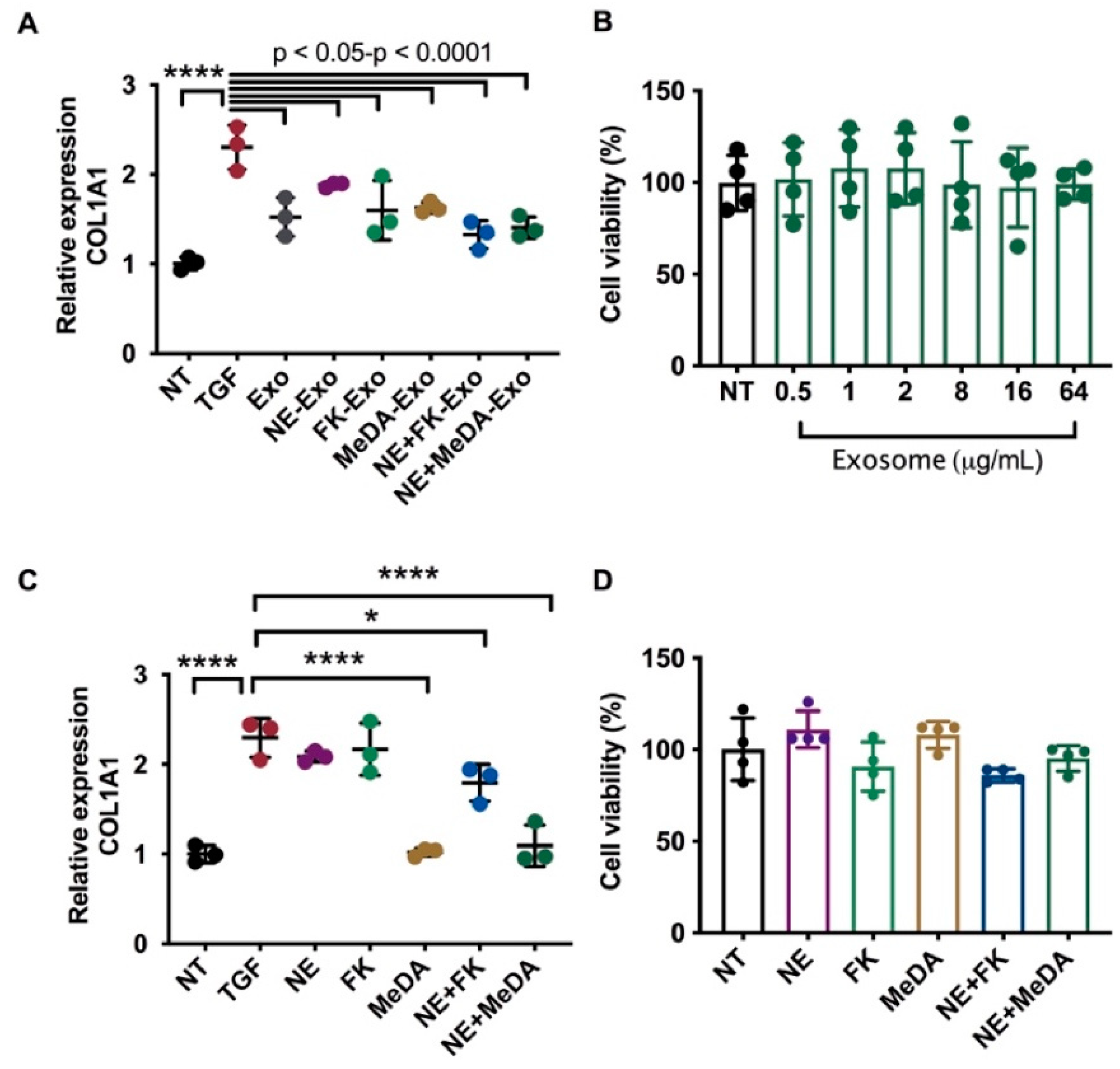
3.2. Downregulation of Collagen Expression
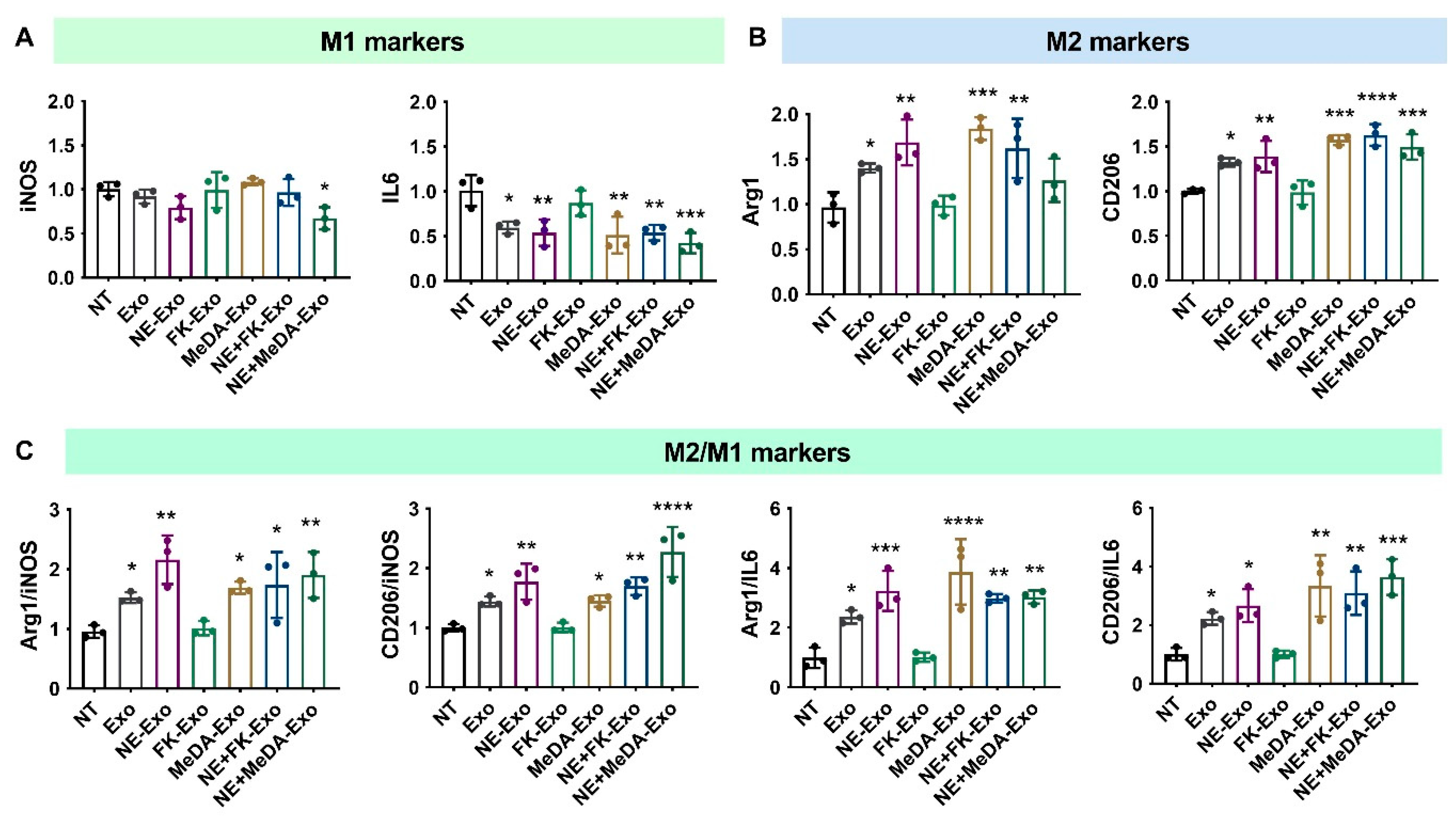
3.3. Macrophage Polarization
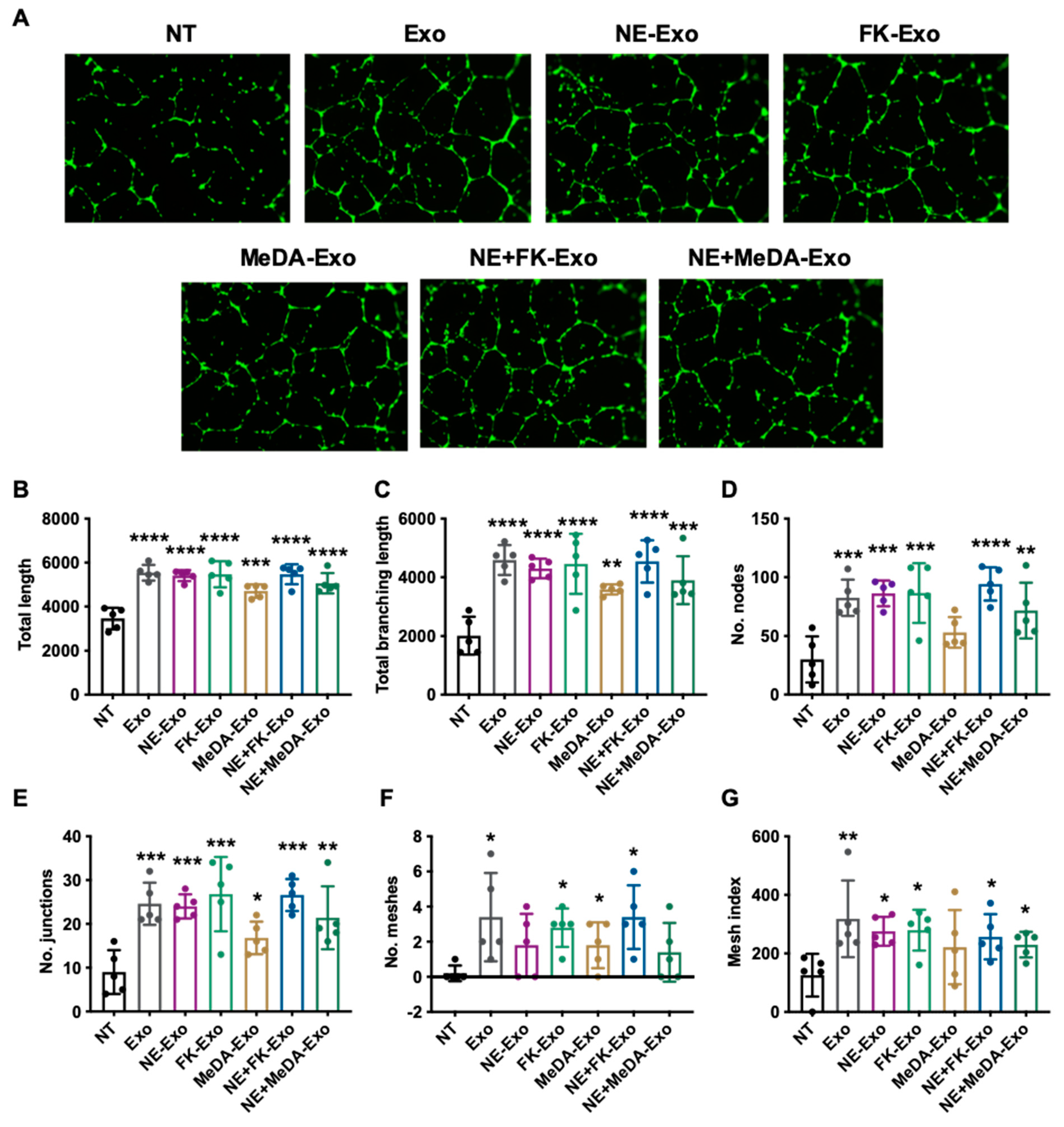
3.4. Angiogenesis Effect
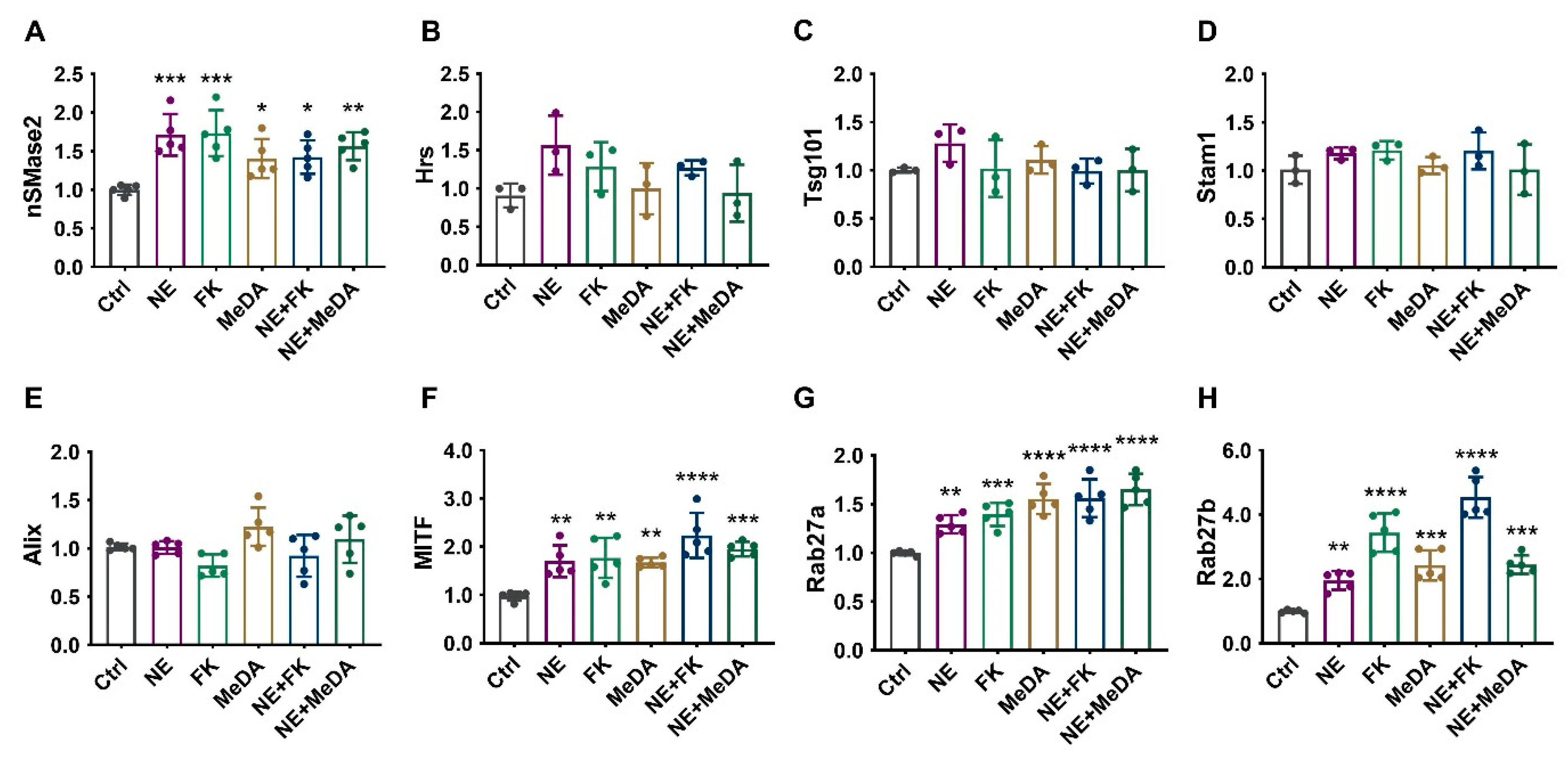
3.5. Effects of Small Molecule Modulators on Genes Affecting Exosome Biogenesis and Secretion
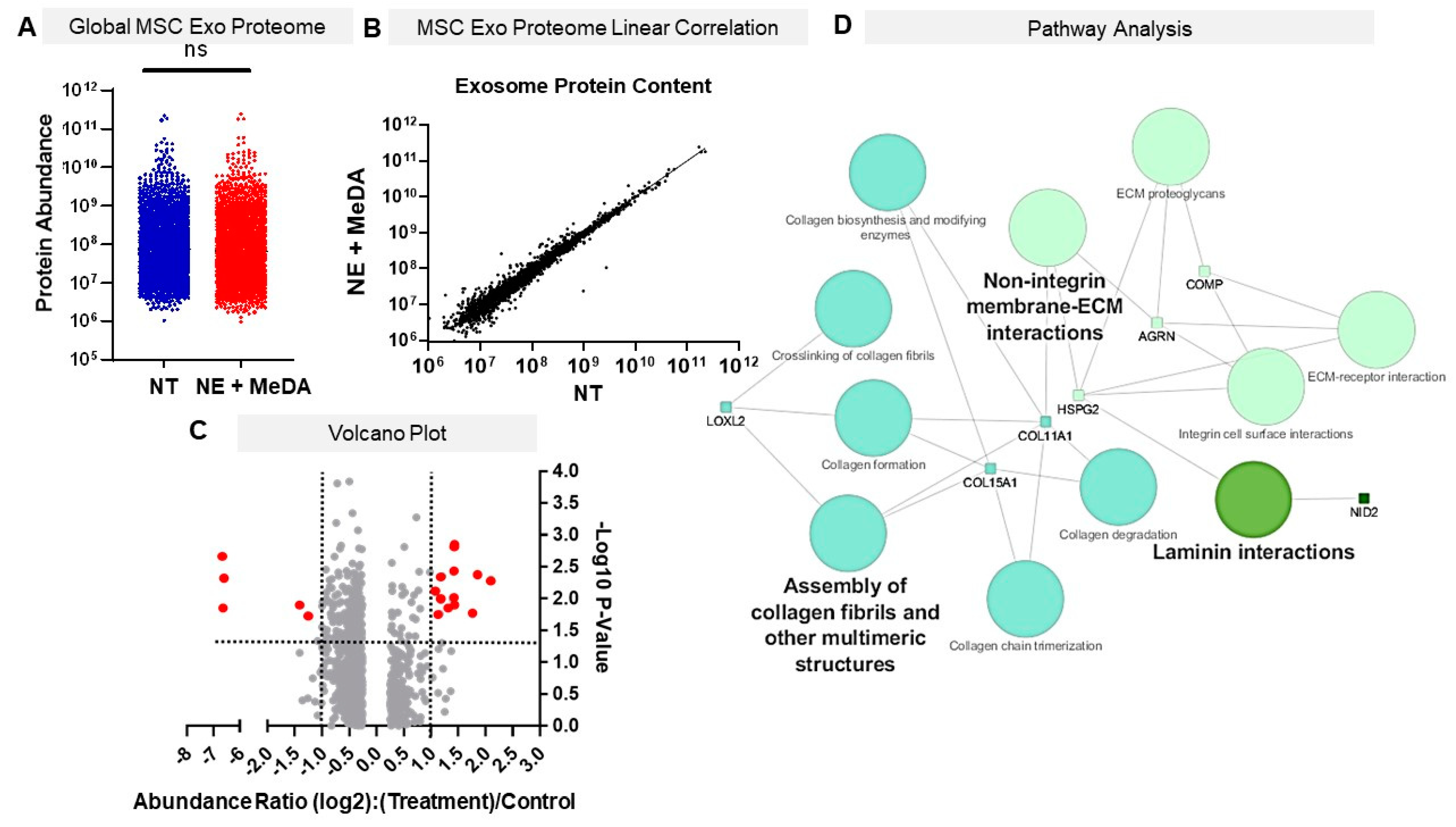
3.6. Effects of Small Molecule Modulators on Exosomal Protein Composition
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MSC | mesenchymal stem cell |
| FT | fenoterol |
| NE | norepinephrine |
| FK | foskolin |
| MeDA | N-methyldopamine |
| Mepn | mephenesin |
| HCF | human cardiac fibroblast |
| HUVEC | human umbilical vein endothelial cell |
| BMDM | bone-marrow derived macrophage |
| MVB | multi-vesicular body |
| ESCRT | endosomal sorting complex required for transport |
References
- Kourembanas, S. Exosomes: Vehicles of intercellular signaling, biomarkers, and vectors of cell therapy. Annu. Rev. Physiol. 2015, 77, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Doyle, L.M.; Wang, M.Z. Overview of extracellular vesicles, their origin, composition, purpose, and methods for exosome isolation and analysis. Cells 2019, 8, 727. [Google Scholar] [CrossRef] [PubMed]
- Kishore, R.; Khan, M. More Than Tiny Sacks: Stem Cell Exosomes as Cell-Free Modality for Cardiac Repair. Circ. Res. 2016, 118, 330–343. [Google Scholar] [CrossRef] [PubMed]
- Bebelman, M.; Smit, M.J.; Pegtel, D.M.; Baglio, S.R. Biogenesis and function of extracellular vesicles in cancer. Pharmacol. Ther. 2018, 188, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ståhl, A.-L.; Johansson, K.; Mossberg, M.; Kahn, R.; Karpman, D. Exosomes and microvesicles in normal physiology, pathophysiology, and renal diseases. Pediatr. Nephrol. 2019, 34, 11–30. [Google Scholar] [CrossRef]
- Ferguson, S.W.; Nguyen, J. Exosomes as therapeutics: The implications of molecular composition and exosomal heterogeneity. J. Control. Release 2016, 228, 179–190. [Google Scholar] [CrossRef]
- Katsuda, T.; Kosaka, N.; Takeshita, F.; Ochiya, T. The therapeutic potential of mesenchymal stem cell-derived extracellular vesicles. Proteomics 2013, 13. [Google Scholar] [CrossRef]
- Nakamura, Y.; Miyaki, S.; Ishitobi, H.; Matsuyama, S.; Nakasa, T.; Kamei, N.; Akimoto, T.; Higashi, Y.; Ochi, M. Mesenchymal-stem-cell-derived exosomes accelerate skeletal muscle regeneration. FEBS Lett. 2015, 589, 1257–1265. [Google Scholar] [CrossRef]
- Ferguson, S.W.; Wang, J.; Lee, C.J.; Liu, M.; Neelamegham, S.; Canty, J.M.; Nguyen, J. The microRNA regulatory landscape of MSC-derived exosomes: A systems view. Sci. Rep. 2018, 8, 1419. [Google Scholar] [CrossRef]
- Chen, T.S.; Arslan, F.; Yin, Y.; Tan, S.S.; Lai, R.C.; Choo, A.B.H.; Padmanabhan, J.; Lee, C.N.; de Kleijn, D.; Lim, S.K. Enabling a robust scalable manufacturing process for therapeutic exosomes through oncogenic immortalization of human ESC-derived MSCs. J. Transl. Med. 2011, 9, 47. [Google Scholar] [CrossRef]
- Severino, V.; Alessio, N.; Farina, A.; Sandomenico, A.; Cipollaro, M.; Peluso, G.; Galderisi, U.; Chambery, A. Insulin-like growth factor binding proteins 4 and 7 released by senescent cells promote premature senescence in mesenchymal stem cells. Cell Death Dis. 2013, 4, e911. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Lu, K.; Zhang, N.; Zhao, Y.; Ma, Q.; Shen, J.; Lin, Y.; Xiang, P.; Tang, Y.; Hu, X. Myocardial reparative functions of exosomes from mesenchymal stem cells are enhanced by hypoxia treatment of the cells via transferring microRNA-210 in an nSMase2-dependent way. Artif. Cells Nanomed. Biotechnol. 2018, 46, 1659–1670. [Google Scholar] [CrossRef] [PubMed]
- Böker, K.O.; Lemus-Diaz, N.; Ferreira, R.R.; Schiller, L.; Schneider, S.; Gruber, J. The impact of the CD9 tetraspanin on lentivirus infectivity and exosome secretion. Mol. Ther. 2018, 26, 634–647. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-King, H.; García, N.A.; Ontoria-Oviedo, I.; Ciria, M.; Montero, J.A.; Sepúlveda, P. Hypoxia inducible factor-1α potentiates jagged 1-mediated angiogenesis by mesenchymal stem cell-derived exosomes. Stem Cells 2017, 35, 1747–1759. [Google Scholar] [CrossRef]
- Patel, D.B.; Luthers, C.R.; Lerman, M.J.; Fisher, J.P.; Jay, S.M. Enhanced extracellular vesicle production and ethanol-mediated vascularization bioactivity via a 3D-printed scaffold-perfusion bioreactor system. Acta Biomater. 2019, 95, 236–244. [Google Scholar] [CrossRef]
- Yan, I.K.; Shukla, N.; Borrelli, D.A.; Patel, T. Use of a Hollow Fiber Bioreactor to Collect Extracellular Vesicles from Cells in Culture. In Extracellular RNA: Methods and Protocols; Patel, T., Ed.; Springer: New York, NY, USA, 2018; pp. 35–41. [Google Scholar]
- Ferguson, S.; Kim, S.; Lee, C.; Deci, M.; Nguyen, J. The Phenotypic Effects of Exosomes Secreted from Distinct Cellular Sources: A Comparative Study Based on miRNA Composition. AAPS J. 2018, 20, 67. [Google Scholar] [CrossRef]
- Wang, J.; Lee, C.; Deci, M.B.; Jasiewicz, N.; Canty, J.; Nguyen, J. MiR-101a loaded Extracellular Nanovesicles as Bioactive Carriers for Cardiac Repair. bioRxiv 2020. [Google Scholar] [CrossRef]
- Deci, M.B.; Ferguson, S.W.; Scatigno, S.L.; Nguyen, J. Modulating Macrophage Polarization through CCR2 Inhibition and Multivalent Engagement. Mol. Pharm. 2018, 15, 2721–2731. [Google Scholar] [CrossRef]
- Deci, M.B.; Liu, M.; Gonya, J.; Lee, C.J.; Li, T.; Ferguson, S.W.; Bonacquisti, E.E.; Wang, J.; Nguyen, J. Carrier-Free CXCR4-Targeted Nanoplexes Designed for Polarizing Macrophages to Suppress Tumor Growth. Cell. Mol. Bioeng. 2019, 12, 375–388. [Google Scholar] [CrossRef]
- Amend, S.R.; Valkenburg, K.C.; Pienta, K.J. Murine hind limb long bone dissection and bone marrow isolation. J. Vis. Exp. 2016, 110, e53936. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.-H.; Pagès, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [PubMed]
- Datta, A.; Kim, H.; McGee, L.; Johnson, A.E.; Talwar, S.; Marugan, J.; Southall, N.; Hu, X.; Lal, M.; Mondal, D.; et al. High-throughput screening identified selective inhibitors of exosome biogenesis and secretion: A drug repurposing strategy for advanced cancer. Sci. Rep. 2018, 8, 8161. [Google Scholar] [CrossRef]
- Rong, X.; Liu, J.; Yao, X.; Jiang, T.; Wang, Y.; Xie, F. Human bone marrow mesenchymal stem cells-derived exosomes alleviate liver fibrosis through the Wnt/β-catenin pathway. Stem Cell Res. Ther. 2019, 10, 98. [Google Scholar] [CrossRef]
- Antimisiaris, S.G.; Mourtas, S.; Marazioti, A. Exosomes and exosome-inspired vesicles for targeted drug delivery. Pharmaceutics 2018, 10, 218. [Google Scholar] [CrossRef]
- Colombo, M.; Raposo, G.; Théry, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef]
- Trajkovic, K.; Hsu, C.; Chiantia, S.; Rajendran, L.; Wenzel, D.; Wieland, F.; Schwille, P.; Brügger, B.; Simons, M. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 2008, 319, 1244–1247. [Google Scholar] [CrossRef]
- Colombo, M.; Moita, C.; van Niel, G.; Kowal, J.; Vigneron, J.; Benaroch, P.; Manel, N.; Moita, L.F.; Théry, C.; Raposo, G. Analysis of ESCRT functions in exosome biogenesis, composition and secretion highlights the heterogeneity of extracellular vesicles. J. Cell Sci. 2013, 126, 5553–5565. [Google Scholar] [CrossRef]
- Im, E.-J.; Lee, C.-H.; Moon, P.-G.; Rangaswamy, G.G.; Lee, B.; Lee, J.M.; Lee, J.-C.; Jee, J.-G.; Bae, J.-S.; Kwon, T.-K. Sulfisoxazole inhibits the secretion of small extracellular vesicles by targeting the endothelin receptor A. Nat. Commun. 2019, 10, 1387. [Google Scholar] [CrossRef]
- Chiaverini, C.; Beuret, L.; Flori, E.; Abbe, P.; Bille, K.; Bahadoran, P.; Ortonne, J.-P.; Bertolotto, C.; Ballotti, R. Microphthalmia-associated transcription factor regulates RAB27A gene expression and controls melanosome transport. J. Biol. Chem. 2008, 283, 12635–12642. [Google Scholar] [CrossRef] [PubMed]
- Ostrowski, M.; Carmo, N.B.; Krumeich, S.; Fanget, I.; Raposo, G.; Savina, A.; Moita, C.F.; Schauer, K.; Hume, A.N.; Freitas, R.P.; et al. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat. Cell Biol. 2010, 12, 19. [Google Scholar] [CrossRef]
- Essandoh, K.; Yang, L.; Wang, X.; Huang, W.; Qin, D.; Hao, J.; Wang, Y.; Zingarelli, B.; Peng, T.; Fan, G.C. Blockade of exosome generation with GW4869 dampens the sepsis-induced inflammation and cardiac dysfunction. Biochim. Biophys. Acta 2015, 1852, 2362–2371. [Google Scholar] [CrossRef] [PubMed]
- Datta, A.; Kim, H.; Lal, M.; McGee, L.; Johnson, A.; Moustafa, A.A.; Jones, J.C.; Mondal, D.; Ferrer, M.; Abdel-Mageed, A.B. Manumycin A suppresses exosome biogenesis and secretion via targeted inhibition of Ras/Raf/ERK1/2 signaling and hnRNP H1 in castration-resistant prostate cancer cells. Cancer Lett. 2017, 408, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Kupcsik, L. Estimation of Cell Number Based on Metabolic Activity: The MTT Reduction Assay. In Mammalian Cell Viability: Methods and Protocols; Stoddart, M.J., Ed.; Humana Press: Totowa, NJ, USA, 2011; pp. 13–19. [Google Scholar]
- Cypess, A.M.; Weiner, L.S.; Roberts-Toler, C.; Elía, E.F.; Kessler, S.H.; Kahn, P.A.; English, J.; Chatman, K.; Trauger, S.A.; Doria, A. Activation of human brown adipose tissue by a β3-adrenergic receptor agonist. Cell Metab. 2015, 21, 33–38. [Google Scholar] [CrossRef]
- Pascucci, L.; Coccè, V.; Bonomi, A.; Ami, D.; Ceccarelli, P.; Ciusani, E.; Viganò, L.; Locatelli, A.; Sisto, F.; Doglia, S.M. Paclitaxel is incorporated by mesenchymal stromal cells and released in exosomes that inhibit in vitro tumor growth: A new approach for drug delivery. J. Control. Release 2014, 192, 262–270. [Google Scholar] [CrossRef]
- Ferguson, S.W.; Megna, J.S.; Nguyen, J. Composition, Physicochemical and Biological Properties of Exosomes Secreted From Cancer Cells. In Diagnostic and Therapeutic Applications of Exosomes in Cancer; Academic Press: Cambridge, MA, USA, 2018; pp. 27–57. [Google Scholar]
- Kosaka, N.; Iguchi, H.; Hagiwara, K.; Yoshioka, Y.; Takeshita, F.; Ochiya, T. Neutral sphingomyelinase 2 (nSMase2)-dependent exosomal transfer of angiogenic microRNAs regulate cancer cell metastasis. J. Biol. Chem. 2013, 288, 10849–10859. [Google Scholar] [CrossRef]
- McAndrews, K.M.; Kalluri, R. Mechanisms associated with biogenesis of exosomes in cancer. Mol. Cancer 2019, 18, 52. [Google Scholar] [CrossRef]
- Koumangoye, R.B.; Sakwe, A.M.; Goodwin, J.S.; Patel, T.; Ochieng, J. Detachment of Breast Tumor Cells Induces Rapid Secretion of Exosomes Which Subsequently Mediate Cellular Adhesion and Spreading. PLoS ONE 2011, 6, e24234. [Google Scholar] [CrossRef]
- Mu, W.; Rana, S.; Zoller, M. Host matrix modulation by tumor exosomes promotes motility and invasiveness. Neoplasia 2013, 15, 875–887. [Google Scholar] [CrossRef]
- Sung, B.H.; Ketova, T.; Hoshino, D.; Zijlstra, A.; Weaver, A.M. Directional cell movement through tissues is controlled by exosome secretion. Nat. Commun. 2015, 6, 7164. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, J.; Ishihara, A.; Fukunaga, K.; Sasaki, K.; White, M.J.V.; Briquez, P.S.; Hubbell, J.A. Laminin heparin-binding peptides bind to several growth factors and enhance diabetic wound healing. Nat. Commun. 2018, 9, 2163. [Google Scholar] [CrossRef] [PubMed]
- Alaseirlis, D.A.; Li, Y.; Cilli, F.; Fu, F.H.; Wang, J.H. Decreasing inflammatory response of injured patellar tendons results in increased collagen fibril diameters. Connect. Tissue Res. 2005, 46, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Rosso, F.; Giordano, A.; Barbarisi, M.; Barbarisi, A. From cell-ECM interactions to tissue engineering. J. Cell. Physiol. 2004, 199, 174–180. [Google Scholar] [CrossRef]
- White, J.F.; Werkmeister, J.A.; Darby, I.A.; Bisucci, T.; Birk, D.E.; Ramshaw, J.A. Collagen fibril formation in a wound healing model. J. Struct. Biol. 2002, 137, 23–30. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Bonacquisti, E.E.; Brown, A.D.; Nguyen, J. Boosting the Biogenesis and Secretion of Mesenchymal Stem Cell-Derived Exosomes. Cells 2020, 9, 660. https://doi.org/10.3390/cells9030660
Wang J, Bonacquisti EE, Brown AD, Nguyen J. Boosting the Biogenesis and Secretion of Mesenchymal Stem Cell-Derived Exosomes. Cells. 2020; 9(3):660. https://doi.org/10.3390/cells9030660
Chicago/Turabian StyleWang, Jinli, Emily E. Bonacquisti, Adam D. Brown, and Juliane Nguyen. 2020. "Boosting the Biogenesis and Secretion of Mesenchymal Stem Cell-Derived Exosomes" Cells 9, no. 3: 660. https://doi.org/10.3390/cells9030660
APA StyleWang, J., Bonacquisti, E. E., Brown, A. D., & Nguyen, J. (2020). Boosting the Biogenesis and Secretion of Mesenchymal Stem Cell-Derived Exosomes. Cells, 9(3), 660. https://doi.org/10.3390/cells9030660