Therapeutic Potential of Autophagy Modulation in Cholangiocarcinoma

Abstract
1. Introduction
2. Autophagy in Cancer
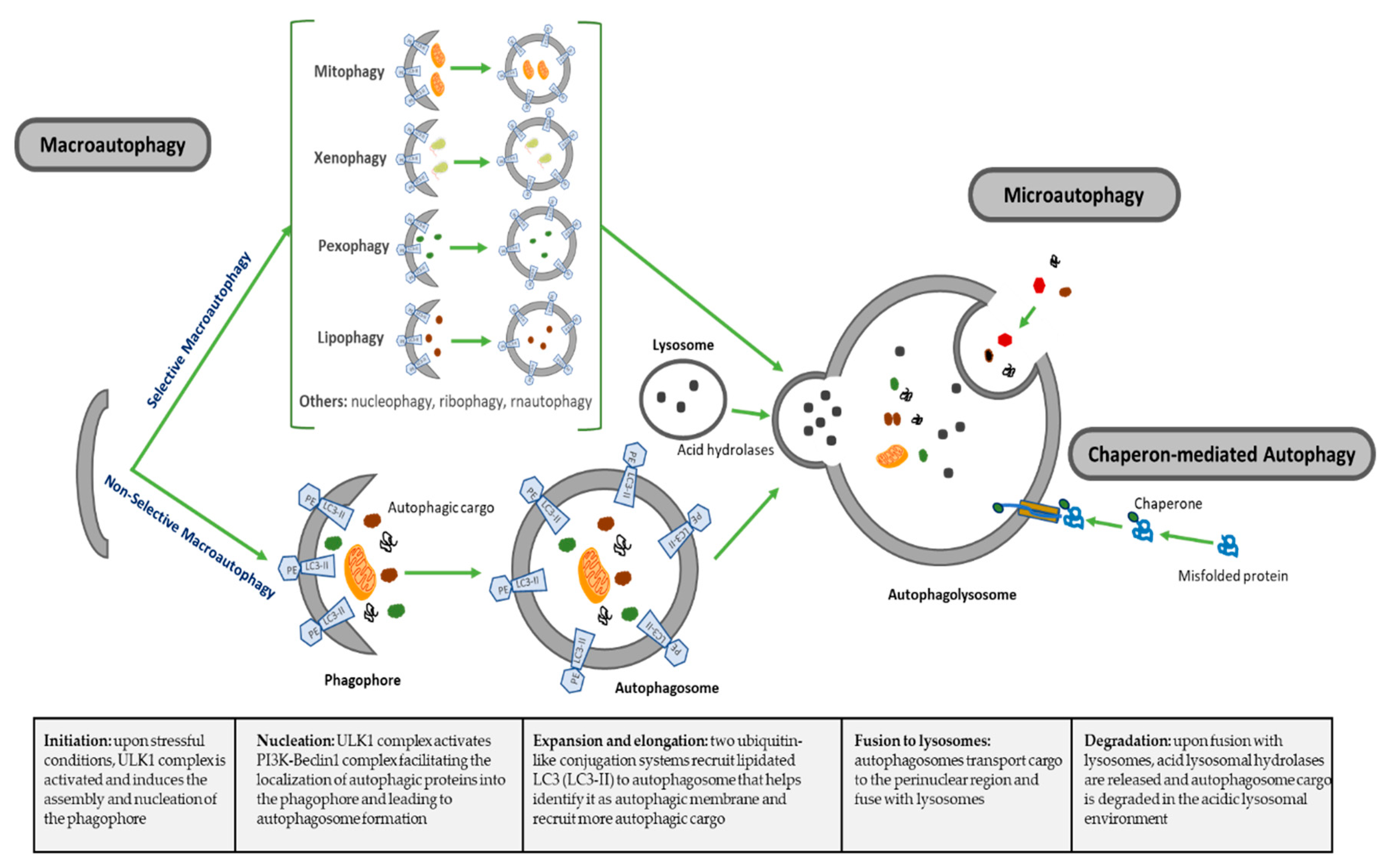
2.1. Autophagy Molecular Process
2.2. Autophagy as a Tumor Suppressor
2.3. Autophagy as a Tumor Promoter
3. Cholangiocarcinoma Genetic and Epigenetic Alterations and Autophagy
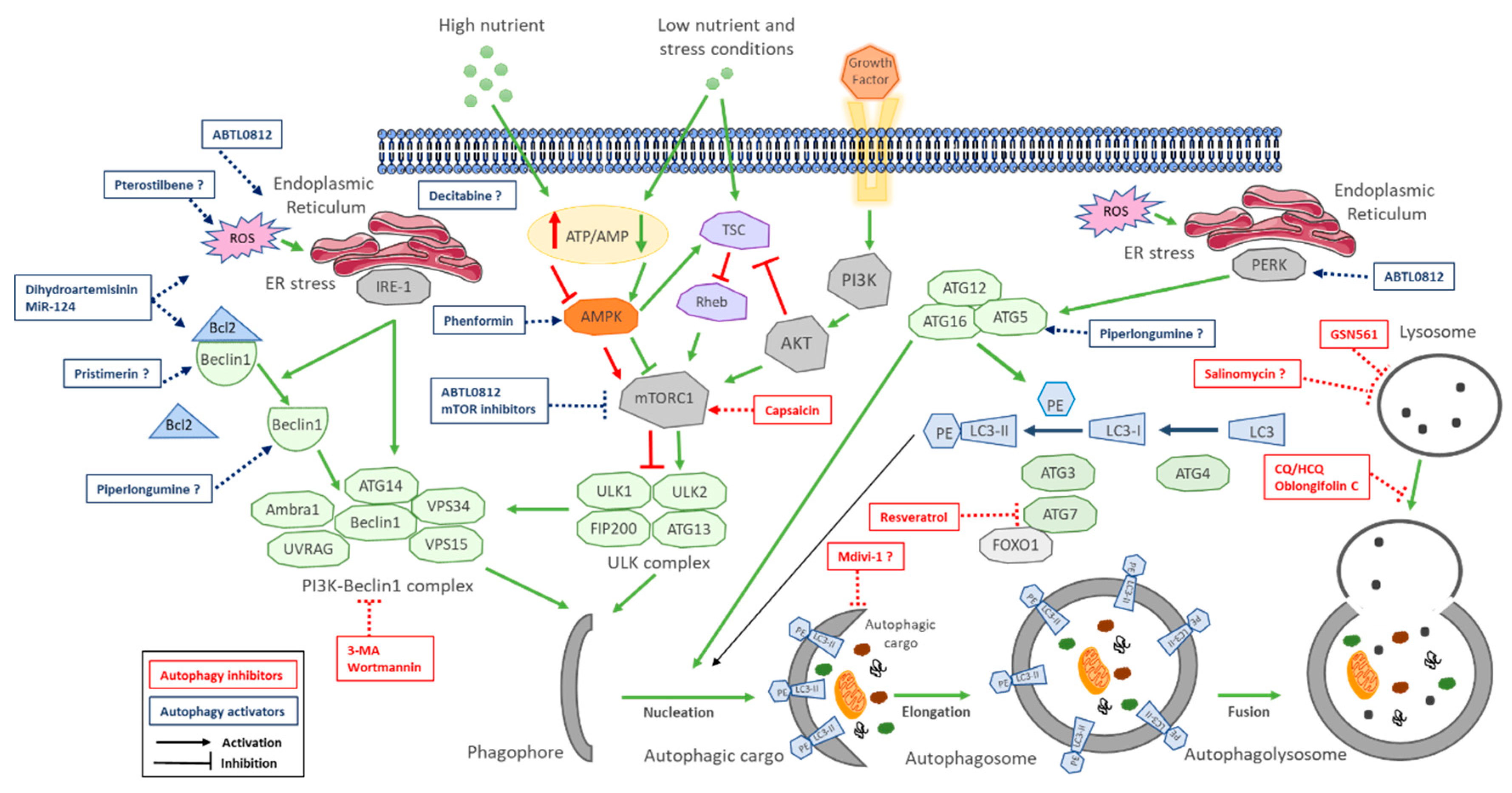
4. Autophagy Modulation in Cholangiocarcinoma
5. Clinical Development of Autophagy Modulators in Cholangiocarcinoma
6. Autophagy Modulators in Cholangiocarcinoma
6.1. Autophagy Inhibitors
6.2. Autophagy Activators
7. Discussion and Future Perspectives
Conflicts of Interest
References
- Strazzabosco, M.; Fabris, L. Functional anatomy of normal bile ducts. Anat. Rec. 2008, 291, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Razumilava, N.; Gores, G.J. Cholangiocarcinoma. Lancet 2014, 383, 2168–2179. [Google Scholar] [CrossRef]
- Nakanuma, Y.; Sato, Y.; Harada, K.; Sasaki, M.; Xu, J.; Ikeda, H. Pathological classification of intrahepatic cholangiocarcinoma based on a new concept. World J. Hepatol. 2010, 2, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Banales, J.M.; Cardinale, V.; Carpino, G.; Marzioni, M.; Andersen, J.B.; Invernizzi, P.; Lind, G.E.; Folseraas, T.; Forbes, S.J.; Fouassier, L.; et al. Expert consensus document: Cholangiocarcinoma: Current knowledge and future perspectives consensus statement from the European Network for the Study of Cholangiocarcinoma (ENS-CCA). Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 261–280. [Google Scholar] [CrossRef] [PubMed]
- Benavides, M.; Antón, A.; Gallego, J.; Gómez, M.A.; Jiménez-Gordo, A.; La Casta, A.; Laquente, B.; Macarulla, T.; Rodríguez-Mowbray, J.R.; Maure, J. Biliary tract cancers: SEOM clinical guidelines. Clin. Transl. Oncol. 2015, 17, 982–987. [Google Scholar] [CrossRef] [PubMed]
- Valle, J.W.; Borbath, I.; Khan, S.A.; Huguet, F.; Gruenberger, T.; Arnold, D. Biliary cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2016, 27, v28–v37. [Google Scholar] [CrossRef] [PubMed]
- Bragazzi, M.C.; Ridola, L.; Safarikia, S.; di Matteo, S.; Costantini, D.; Nevi, L.; Cardinale, V. New insights into cholangiocarcinoma: Multiple stems and related cell lineages of origin. Ann. Gastroenterol. Hell. Soc. Gastroenterol. 2018, 31, 42–55. [Google Scholar] [CrossRef] [PubMed]
- Patel, T. New insights into the molecular pathogenesis of intrahepatic cholangiocarcinoma. J. Gastroenterol. 2014, 49, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Cardinale, V.; Bragazzi, M.C.; Carpino, G.; Torrice, A.; Fraveto, A.; Gentile, R.; Pasqualino, V.; Melandro, F.; Aliberti, C.; Bastianelli, C.; et al. Cholangiocarcinoma: Increasing burden of classifications. Hepatobiliary Surg. Nutr. 2013, 2, 272–280. [Google Scholar] [PubMed]
- Cardinale, V.; Carpino, G.; Reid, L.; Gaudio, E.; Alvaro, D. Multiple cells of origin in cholangiocarcinoma underlie biological, epidemiological and clinical heterogeneity. World J. Gastrointest. Oncol. 2012, 4, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Bridgewater, J.; Galle, P.R.; Khan, S.A.; Llovet, J.M.; Park, J.W.; Patel, T.; Pawlik, T.M.; Gores, G.J. Guidelines for the diagnosis and management of intrahepatic cholangiocarcinoma. J. Hepatol. 2014, 60, 1268–1289. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, S.; Gores, G.J. Current diagnostic and management options in perihilar cholangiocarcinoma. Digestion 2014, 89, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Blechacz, B. Cholangiocarcinoma: Current knowledge and new developments. Gut Liver 2017, 11, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.D.; Pinson, C.W.; Berlin, J.; Chari, R.S. Diagnosis and treatment of cholangiocarcinoma. Oncologist 2004, 9, 43–57. [Google Scholar] [CrossRef]
- Valle, J.; Wasan, H.; Palmer, D.H.; Cunningham, D.; Anthoney, A.; Maraveyas, A.; Madhusudan, S.; Iveson, T.; Hughes, S.; Pereira, S.P.; et al. Cisplatin plus Gemcitabine versus Gemcitabine for biliary tract cancer. N. Engl. J. Med. 2010, 362, 1273–1281. [Google Scholar] [CrossRef]
- Valle, J.W.; Furuse, J.; Jitlal, M.; Beare, S.; Mizuno, N.; Wasan, H.; Bridgewater, J.; Okusaka, T. Cisplatin and gemcitabine for advanced biliary tract cancer: A meta-analysis of two randomised trials. Ann. Oncol. 2014, 25, 391–398. [Google Scholar] [CrossRef]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef]
- Yu, L.; Chen, Y.; Tooze, S.A. Autophagy pathway: Cellular and molecular mechanisms. Autophagy 2018, 14, 207–215. [Google Scholar] [CrossRef]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Levine, B.; Green, D.R.; Kroemer, G. Pharmacological modulation of autophagy: Therapeutic potential and persisting obstacles. Nat. Rev. Drug Discov. 2017, 16, 487–511. [Google Scholar] [CrossRef]
- Yun, C.W.; Lee, S.H. The roles of autophagy in cancer. Int. J. Mol. Sci. 2018, 19, 3466. [Google Scholar] [CrossRef]
- Ávalos, Y.; Canales, J.; Bravo-Sagua, R.; Criollo, A.; Lavandero, S.; Quest, A.F.G. Tumor suppression and promotion by autophagy. BioMed Res. Int. 2014, 2014, 603980. [Google Scholar] [CrossRef] [PubMed]
- Pietrocola, F.; Pol, J.; Vacchelli, E.; Baracco, E.E.; Levesque, S.; Castoldi, F.; Maiuri, M.C.; Madeo, F.; Kroemer, G. Autophagy induction for the treatment of cancer. Autophagy 2016, 12, 1962–1964. [Google Scholar] [CrossRef] [PubMed]
- Chude, C.I.; Amaravadi, R.K. Targeting autophagy in cancer: Update on clinical trials and novel inhibitors. Int. J. Mol. Sci. 2017, 18, 1279. [Google Scholar] [CrossRef] [PubMed]
- Marinković, M.; Šprung, M.; Buljubašić, M.; Novak, I. Autophagy modulation in cancer: Current knowledge on action and therapy. Oxidative Med. Cell. Longev. 2018, 2018, 8023821. [Google Scholar] [CrossRef] [PubMed]
- Levine, B. Cell biology: Autophagy and cancer. Nature 2007, 446, 745–747. [Google Scholar] [CrossRef] [PubMed]
- Cudjoe, E.K.; Kyte, S.L.; Saleh, T.; Landry, J.W.; Gewirtz, D.A. Autophagy inhibition and chemosensitization in cancer therapy. In Targeting Cell Survival Pathways to Enhance Response to Chemotherapy; Elsevier: Amsterdam, The Netherlands, 2019; pp. 259–273. [Google Scholar]
- Dong, L.W.; Hou, Y.J.; Tan, Y.X.; Tang, L.; Pan, Y.F.; Wang, M.; Wang, H.Y. Prognostic significance of Beclin 1 in intrahepatic cholangiocellular carcinoma. Autophagy 2011, 7, 1222–1229. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.T.; Cao, Q.H.; Chen, M.Y.; Xia, Q.; Fan, X.J.; Ma, X.K.; Lin, Q.; Jia, C.C.; Dong, M.; Ruan, D.Y.; et al. Beclin 1 deficiency correlated with lymph node metastasis, predicts a distinct outcome in intrahepatic and extrahepatic cholangiocarcinoma. PLoS ONE 2013, 8, e80317. [Google Scholar] [CrossRef]
- Ma, J.; Weng, L.; Wang, Z.; Jia, Y.; Liu, B.; Wu, S.; Cao, Y.; Sun, X.; Yin, X.; Shang, M.; et al. MiR-124 induces autophagy-related cell death in cholangiocarcinoma cells through direct targeting of the EZH2-STAT3 signaling axis. Exp. Cell Res. 2018, 366, 103–113. [Google Scholar] [CrossRef]
- He, W.; Zhang, A.; Qi, L.; Na, C.; Jiang, R.; Fan, Z.; Chen, J. FOXO1, a potential therapeutic target, regulates autophagic flux, oxidative stress, mitochondrial dysfunction, and apoptosis in human cholangiocarcinoma QBC939 cells. Cell. Physiol. Biochem. 2018, 45, 1506–1514. [Google Scholar] [CrossRef]
- Chen, L.; Fu, H.; Lu, T.; Cai, J.; Liu, W.; Yao, J.; Liang, J.; Zhao, H.; Zhang, J.; Zheng, J.; et al. An integrated nomogram combining clinical factors and microtubule-associated Protein 1 light chain 3B expression to predict postoperative prognosis in patients with intrahepatic cholangiocarcinomas. Cancer Res. Treat. 2019. [Google Scholar] [CrossRef]
- Nakanuma, Y.; Sasaki, M.; Harada, K. Autophagy and senescence in fibrosing cholangiopathies. J. Hepatol. 2015, 62, 934–945. [Google Scholar] [CrossRef] [PubMed]
- Greer, S.U.; Ogmundsdottir, M.H.; Chen, J.; Lau, B.T.; Delacruz, R.G.C.; Sandoval, I.T.; Kristjansdottir, S.; Jones, D.A.; Haslem, D.S.; Romero, R.; et al. Genetic risk of cholangiocarcinoma is linked to the autophagy gene ATG7. BioRxiv 2019, 836767. [Google Scholar] [CrossRef]
- Kaur, J.; Debnath, J. Autophagy at the crossroads of catabolism and anabolism. Nat. Rev. Mol. Cell Biol. 2015, 16, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132. [Google Scholar] [CrossRef]
- Itakura, E.; Kishi, C.; Inoue, K.; Mizushima, N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol. Biol. Cell 2008, 19, 5360–5372. [Google Scholar] [CrossRef]
- Ichimura, Y.; Kirisako, T.; Takao, T.; Satomi, Y.; Shimonishi, Y.; Ishihara, N.; Mizushima, N.; Tanida, I.; Kominami, E.; Ohsumi, M.; et al. A ubiquitin-like system mediates protein lipidation. Nature 2000, 408, 488–492. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef]
- Nakamura, S.; Yoshimori, T. New insights into autophagosome-lysosome fusion. J. Cell Sci. 2017, 130, 1209–1216. [Google Scholar] [CrossRef]
- Jiang, P.; Nishimura, T.; Sakamaki, Y.; Itakura, E.; Hatta, T.; Natsume, T.; Mizushima, N. The HOPS complex mediates autophagosome-lysosome fusion through interaction with syntaxin 17. Mol. Biol. Cell 2014, 25, 1327–1337. [Google Scholar] [CrossRef]
- Liang, C.; Lee, J.-S.; Inn, K.-S.; Jung, J.U. Beclin1-Binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat. Cell Biol. 2008, 10, 776–787. [Google Scholar] [CrossRef] [PubMed]
- Karantza-Wadsworth, V.; Patel, S.; Kravchuk, O.; Chen, G.; Mathew, R.; Jin, S.; White, E. Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev. 2007, 21, 1621–1635. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; White, E. Tumor suppression by autophagy through the management of metabolic stress. Autophagy 2008, 4, 563–566. [Google Scholar] [CrossRef]
- Manning, B.D.; Toker, A. AKT/PKB signaling: Navigating the network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [PubMed]
- Arico, S.; Petiot, A.; Bauvy, C.; Dubbelhuis, P.F.; Meijer, A.J.; Codogno, P.; Ogier-Denis, E. The tumor suppressor PTEN positively regulates macroautophagy by inhibiting the phosphatidylinositol 3-kinase/protein kinase B pathway. J. Biol. Chem. 2001, 276, 35243–35246. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Shao, S.H.; Xu, Z.-X.; Hennessy, B.; Ding, Z.; Larrea, M.; Kondo, S.; Dumont, D.J.; Gutterman, J.U.; Walker, C.L.; et al. The energy sensing LKB1-AMPK pathway regulates p27kip1 phosphorylation mediating the decision to enter autophagy or apoptosis. Nat. Cell Biol. 2007, 9, 218–224. [Google Scholar] [CrossRef]
- Parkhitko, A.; Myachina, F.; Morrison, T.A.; Hindi, K.M.; Auricchio, N.; Karbowniczek, M.; Wu, J.J.; Finkel, T.; Kwiatkowski, D.J.; Yu, J.J.U.; et al. Tumorigenesis in tuberous sclerosis complex is autophagy and p62/sequestosome 1 (SQSTM1)-dependent. Proc. Natl. Acad. Sci. USA 2011, 108, 12455–12460. [Google Scholar] [CrossRef]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402, 672–676. [Google Scholar] [CrossRef]
- Choi, A.M.K.; Ryter, S.W.; Levine, B. Mechanisms of disease: Autophagy in human health and disease. N. Engl. J. Med. 2013, 368, 651–662. [Google Scholar] [CrossRef]
- Mathew, R.; Kongara, S.; Beaudoin, B.; Cristina, M.; Karp, K.B.; Degenhardt, K.; Chen, G.; Jin, S.; White, E. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev. 2007, 21, 1367–1381. [Google Scholar] [CrossRef]
- Mathew, R.; Karp, C.M.; Beaudoin, B.; Vuong, N.; Chen, G.; Chen, H.Y.; Bray, K.; Reddy, A.; Bhanot, G.; Gelinas, C.; et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell 2009, 137, 1062–1075. [Google Scholar] [CrossRef] [PubMed]
- Rouschop, K.; Wouters, B. Regulation of autophagy through multiple independent hypoxic signaling pathways. Curr. Mol. Med. 2009, 9, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Onodera, J.; Ohsumi, Y. Autophagy is required for maintenance of amino acid levels and protein synthesis under nitrogen starvation. J. Biol. Chem. 2005, 280, 31582–31586. [Google Scholar] [CrossRef] [PubMed]
- Rabinowitz, J.D.; White, E. Autophagy and metabolism. Science 2010, 330, 1344–1348. [Google Scholar] [CrossRef]
- Guo, J.Y.; White, E. Autophagy is required for mitochondrial function, lipid metabolism, growth, and fate of KRASG12D-driven lung tumors. Autophagy 2013, 9, 1636–1638. [Google Scholar] [CrossRef]
- Yang, S.; Wang, X.; Contino, G.; Liesa, M.; Sahin, E.; Ying, H.; Bause, A.; Li, Y.; Stommel, J.M.; Dell’antonio, G.; et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011, 25, 717–729. [Google Scholar] [CrossRef]
- Lock, R.; Roy, S.; Kenific, C.M.; Su, J.S.; Salas, E.; Ronen, S.M.; Debnath, J. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol. Biol. Cell 2011, 22, 165–178. [Google Scholar] [CrossRef]
- Lock, R.; Kenific, C.M.; Leidal, A.M.; Salas, E.; Debnath, J. Autophagy-Dependent production of secreted factors facilitates oncogenic RAS-Driven invasion. Cancer Discov. 2014, 4, 466–479. [Google Scholar] [CrossRef]
- Pérez-Hernández, M.; Arias, A.; Martínez-García, D.; Pérez-Tomás, R.; Quesada, R.; Soto-Cerrato, V. Targeting autophagy for cancer treatment and tumor chemosensitization. Cancers 2019, 11, 1599. [Google Scholar]
- Chen, N.; Karantza-Wadsworth, V. Role and regulation of autophagy in cancer. Biochim. Biophys. Acta Mol. Cell Res. 2009, 1793, 1516–1523. [Google Scholar] [CrossRef]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhou, Y.; Li, Y.; Yang, L.; Ma, Y.; Peng, X.; Yang, S.; Liu, J.; Li, H. Autophagy: A novel mechanism of chemoresistance in cancers. BioMed. Pharmacother. 2019, 119, 109415. [Google Scholar] [CrossRef] [PubMed]
- Chang, M. Tamoxifen resistance in breast cancer. Biomol. Ther. 2012, 20, 256–267. [Google Scholar] [CrossRef] [PubMed]
- Kanzawa, T.; Germano, I.M.; Komata, T.; Ito, H.; Kondo, Y.; Kondo, S. Role of autophagy in temozolomide-induced cytotoxicity for malignant glioma cells. Cell Death Differ. 2004, 11, 448–457. [Google Scholar] [CrossRef]
- Opipari, A.W.; Tan, L.; Boitano, A.E.; Sorenson, D.R.; Aurora, A.; Liu, J.R. Resveratrol-Induced autophagocytosis in ovarian cancer cells. Cancer Res. 2004, 64, 696–703. [Google Scholar] [CrossRef]
- Kanzawa, T.; Zhang, L.; Xiao, L.; Germano, I.M.; Kondo, Y.; Kondo, S. Arsenic trioxide induces autophagic cell death in malignant glioma cells by upregulation of mitochondrial cell death protein BNIP3. Oncogene 2005, 24, 980–991. [Google Scholar] [CrossRef]
- Ito, H.; Daido, S.; Kanzawa, T.; Kondo, S.; Kondo, Y. Radiation-Induced autophagy is associated with LC3 and its inhibition sensitizes malignant glioma cells. Int. J. Oncol. 2005, 26, 1401–1410. [Google Scholar] [CrossRef]
- Labib, P.L.; Goodchild, G.; Pereira, S.P. Molecular pathogenesis of cholangiocarcinoma. BMC Cancer 2019, 19, 185. [Google Scholar] [CrossRef]
- Simile, M.M.; Bagella, P.; Vidili, G.; Spanu, A.; Manetti, R.; Seddaiu, M.A.; Babudieri, S.; Madeddu, G.; Serra, P.A.; Altana, M. Targeted therapies in cholangiocarcinoma: Emerging evidence from clinical trials. Medicina 2019, 55, 42. [Google Scholar] [CrossRef]
- Sia, D.; Hoshida, Y.; Villanueva, A.; Roayaie, S.; Ferrer, J.; Tabak, B.; Peix, J.; Sole, M.; Tovar, V.; Alsinet, C.; et al. Integrative molecular analysis of intrahepatic cholangiocarcinoma reveals 2 classes that have different outcomes. Gastroenterology 2013, 144, 829–840. [Google Scholar] [CrossRef]
- Simbolo, M.; Fassan, M.; Ruzzenente, A.; Mafficini, A.; Laura, D.; Wood, V.C.; Melisi, D.; Malleo, G.; Vicentini, C.; Malpeli, G.; et al. Multigene mutational profiling of cholangiocarcinomas identifies actionable molecular subgroups. Oncotarget 2014, 5, 2839–2852. [Google Scholar] [CrossRef] [PubMed]
- Arai, Y.; Totoki, Y.; Hosoda, F.; Shirota, T.; Hama, N.; Nakamura, H.; Ojima, H.; Furuta, K.; Shimada, K.; Okusaka, T.; et al. Fibroblast growth factor receptor 2 tyrosine kinase fusions define a unique molecular subtype of cholangiocarcinoma. Hepatology 2014, 59, 1427–1434. [Google Scholar] [CrossRef] [PubMed]
- Maroni, L.; Pierantonelli, I.; Banales, J.M.; Benedetti, A.; Marzioni, M. The significance of genetics for cholangiocarcinoma development. Ann. Transl. Med. 2013, 1, 28. [Google Scholar] [PubMed]
- Cardinale, V.; Semeraro, R.; Torrice, A.; Gatto, M.; Napoli, C.; Bragazzi, M.C.; Gentile, R.; Alvaro, D. Intra-Hepatic and extra-hepatic cholangiocarcinoma: New insight into epidemiology and risk factors. World J. Gastrointest. Oncol. 2010, 2, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.; Han, Y.; Hughart, N.; McCarra, J.; Alpini, G.; Meng, F. Interleukin-6 and its receptor, key players in hepatobiliary inflammation and cancer. Transl. Gastrointest. Cancer 2012, 1, 58–70. [Google Scholar]
- Jaiswal, M.; LaRusso, N.F.; Burgart, L.J.; Gores, G.J. Inflammatory cytokines induce DNA damage and inhibit DNA repair in cholangiocarcinoma cells by a nitric oxide-dependent mechanism. Cancer Res. 2000, 60, 184–190. [Google Scholar]
- Nzeako, U.C.; Guicciardi, M.E.; Yoon, J.H.; Bronk, S.F.; Gores, G.J. COX-2 inhibits Fas-mediated apoptosis in cholangiocarcinoma cells. Hepatology 2002, 35, 552–559. [Google Scholar] [CrossRef]
- Lin, Y.; Jiang, M.; Chen, W.; Zhao, T.; Wei, Y. Cancer and ER stress: Mutual crosstalk between autophagy, oxidative stress and inflammatory response. BioMed. Pharmacother. 2019, 118, 109249. [Google Scholar] [CrossRef]
- Qi, Y.; Zhang, M.; Li, H.; Frank, J.A.; Dai, L.; Liu, H.; Zhang, Z.; Wang, C.; Chen, G. Autophagy inhibition by sustained overproduction of IL6 contributes to arsenic carcinogenesis. Cancer Res. 2014, 74, 3740–3752. [Google Scholar] [CrossRef]
- Monkkonen, T.; Debnath, J. Inflammatory signaling cascades and autophagy in cancer. Autophagy 2018, 14, 190–198. [Google Scholar] [CrossRef]
- Ngabire, D.; Kim, G.D. Autophagy and inflammatory response in the tumor microenvironment. Int. J. Mol. Sci. 2017, 18, 2016. [Google Scholar] [CrossRef] [PubMed]
- Robertson, S.; Hyder, O.; Dodson, R.; Nayar, S.K.; Poling, J.; Beierl, K.; Eshleman, J.R.; Lin, M.; Pawlik, T.M.; Anders, R.A. The frequency of KRAS and BRAF mutations in intrahepatic cholangiocarcinomas and their correlation with clinical outcome. Hum. Pathol. 2013, 44, 2768–2773. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.A.; Thomas, H.C.; Toledano, M.B.; Cox, I.J.; Taylor-Robinson, S.D. p53 mutations in human cholangiocarcinoma: A review. Liver Int. 2005, 25, 704–716. [Google Scholar] [CrossRef] [PubMed]
- O’Dell, M.R.; Huang, J.L.; Christa, L.; Miller, W.; Deshpande, V.; Rothberg, P.; Grose, V.; Rossi, R.M.; Zhu, A.X.; Land, H.; et al. Kras G12D and p53 mutation cause primary intrahepatic cholangiocarcinoma. Cancer Res. 2012, 72, 1557–1567. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.L.; Hezel, A.F. Autophagy in intra-hepatic cholangiocarcinoma. Autophagy 2012, 8, 1148–1149. [Google Scholar] [CrossRef]
- Eng, C.H.; Wang, Z.; Tkach, D.; Toral-Barza, L.; Ugwonali, S.; Liu, S.; Stephanie, L.; Fitzgerald, E.G.; Frias, E.; Cochran, N.; et al. Macroautophagy is dispensable for growth of KRAS mutant tumors and chloroquine efficacy. Proc. Natl. Acad. Sci. USA 2016, 113, 182–187. [Google Scholar] [CrossRef]
- Guo, J.Y.; Karsli-Uzunbas, G.; Mathew, R.; Aisner, S.C.; Kamphorst, J.J.; Strohecker, A.M.; Chen, G.; Price, S.; Lu, W.; Teng, X.; et al. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev. 2013, 27, 1447–1461. [Google Scholar] [CrossRef]
- Belmont, P.J.; Jiang, P.; McKee, T.D.; Xie, T.; Isaacson, J.; Baryla, N.E.; Roper, J.; Sinnamon, M.J.; Lee, N.V.; Kan, J.L.; et al. Resistance to dual blockade of the kinases PI3K and mTOR in KRAS-mutant colorectal cancer models results in combined sensitivity to inhibition of the receptor tyrosine kinase EGFR. Sci. Signal. 2014, 7. [Google Scholar] [CrossRef]
- Strohecker, A.M.; White, E. Autophagy promotes BrafV600E-driven lung tumorigenesis by preserving mitochondrial metabolism. Autophagy 2014, 10, 384–385. [Google Scholar] [CrossRef]
- Seton-Rogers, S. Eliminating protective autophagy in KRAS-mutant cancers. Nat. Rev. Cancer. 2019, 19, 247. [Google Scholar] [CrossRef]
- Mertens, J.C.; Rizvi, S.; Gores, G.J. Targeting cholangiocarcinoma. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1454–1460. [Google Scholar] [CrossRef] [PubMed]
- Socoteanu, M.P.; Mott, F.; Alpini, G.; Frankel, A.E. c-Met targeted therapy of cholangiocarcinoma. World J. Gastroenterol. 2008, 14, 2990–2994. [Google Scholar] [CrossRef]
- Miyamoto, M.; Ojima, H.; Iwasaki, M.; Shimizu, H.; Kokubu, A.; Hiraoka, N.; Kosuge, T.; Yoshikawa, D.; Kono, T.; Furukawa, H.; et al. Prognostic significance of overexpression of c-Met oncoprotein in cholangiocarcinoma. Br. J. Cancer 2011, 105, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, J.-H.; Chai, K.; Tashiro, S.-I.; Onodera, S.; Ikejima, T. Inhibition of c-Met promoted apoptosis, autophagy and loss of the mitochondrial transmembrane potential in oridonin-induced A549 lung cancer cells. J. Pharm. Pharmacol. 2013, 65, 1622–1642. [Google Scholar] [CrossRef] [PubMed]
- Leone, F.; Cavalloni, G.; Pignochino, Y.; Sarotto, I.; Ferraris, R.; Piacibello, W.; Venesio, T.; Capussotti, L.; Risio, M.; Aglietta, M. Somatic mutations of epidermal growth factor receptor in bile duct and gallbladder carcinoma. Clin. Cancer Res. 2006, 12, 1680–1685. [Google Scholar] [CrossRef] [PubMed]
- Lai, G.H.; Zhang, Z.; Shen, X.N.; Ward, D.J.; Dewitt, J.L.; Holt, S.E.; Rozich, R.A.; Hixson, D.C.; Sirica, A.E. erbB-2/neu transformed rat cholangiocytes recapitulate key cellular and molecular features of human bile duct cancer. Gastroenterology 2005, 129, 2047–2057. [Google Scholar] [CrossRef] [PubMed]
- Schmukler, E.; Pinkas-Kramarski, R. Inhibition of ErbB receptors and autophagy in cancer therapy. In Autophagy: Cancer, Other Pathologies, Inflammation, Immunity, Infection, and Aging 5; Elsevier Inc.: Amsterdam, The Netherlands, 2015; pp. 65–80. [Google Scholar]
- Wu, Y.-M.; Su, F.; Kalyana-Sundaram, S.; Khazanov, N.; Ateeq, B.; Cao, X.; Robert, J.; Pankaj Vats, L.; Wang, R.; Li, S.-F.; et al. Identification of targetable FGFR gene fusions in diverse cancers. Cancer Discov. 2013, 3, 636–647. [Google Scholar] [CrossRef]
- Yuan, H.; Li, Z.-M.; Shao, J.; Ji, W.-X.; Xia, W.; Lu, S. FGF2/FGFR1 regulates autophagy in FGFR1-amplified non-small cell lung cancer cells. J. Exp. Clin. Cancer Res. 2017, 36, 72. [Google Scholar] [CrossRef]
- Chen, Y.; Xie, X.; Li, X.; Wang, P.; Jing, Q.; Yue, J.; Liu, Y.; Cheng, Z.; Li, J.; Song, H.; et al. FGFR antagonist induces protective autophagy in FGFR1-amplified breast cancer cell. Biochem. Biophys. Res. Commun. 2016, 474, 1–7. [Google Scholar] [CrossRef]
- Kang, Y.K.; Kim, W.H.; Jang, J.J. Expression of G1-S modulators (p53, p16, p27, cyclin D1, Rb) and Smad4/Dpc4 in intrahepatic cholangiocarcinoma. Hum. Pathol. 2002, 33, 877–883. [Google Scholar] [CrossRef]
- Wang, F.; Xia, X.; Yang, C.; Shen, J.; Mai, J.; Kim, H.C.; Kirui, D.; Kang, Y.; Fleming, J.B.; Koay, E.J.; et al. SMAD4 gene mutation renders pancreatic cancer resistance to radiotherapy through promotion of autophagy. Clin. Cancer Res. 2018, 24, 3176–3185. [Google Scholar] [CrossRef] [PubMed]
- Cong, W.M.; Bakker, A.; Swalsky, P.A.; Raja, S.; Woods, J.; Thomas, S.; Demetris, A.J.; Finkelstein, S.D. Multiple genetic alterations involved in the tumorigenesis of human cholangiocarcinoma: A molecular genetic and clinicopathological study. J. Cancer Res. Clin. Oncol. 2001, 127, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Nitta, T.; Sato, Y.; Nakanuma, Y. Autophagy may occur at an early stage of cholangiocarcinogenesis via biliary intraepithelial neoplasia. Hum. Pathol. 2015, 46, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.J.; Dong, L.W.; Tan, Y.W.; Yang, G.Z.; Pan, Y.F.; Li, Z.; Tang, L.; Wang, M.; Wang, Q.; Wang, H.Y.; et al. Inhibition of active autophagy induces apoptosis and increases chemosensitivity in cholangiocarcinoma. Lab. Investig. 2011, 91, 1146–1157. [Google Scholar] [CrossRef]
- Isomoto, H. Epigenetic alterations associated with cholangiocarcinoma. Oncol. Rep. 2009, 22, 227–232. [Google Scholar] [CrossRef]
- Baek, S.H.; Kim, K.I. Epigenetic control of autophagy: Nuclear events gain more attention. Mol. Cell 2017, 65, 781–785. [Google Scholar] [CrossRef]
- Gradilone, S.A.; Radtke, B.N.; Bogert, P.S.; Huang, B.Q.; Gajdos, G.B.; LaRusso, N.F. HDAC6 inhibition restores ciliary expression and decreases tumor growth. Cancer Res. 2013, 73, 2259–2270. [Google Scholar] [CrossRef]
- Gradilone, S.A.; Habringer, S.; Masyuk, T.V.; Howard, B.N.; Masyuk, A.I.; Larusso, N.F. HDAC6 is overexpressed in cystic cholangiocytes and its inhibition reduces cystogenesis. Am. J. Pathol. 2014, 184, 600–608. [Google Scholar] [CrossRef]
- Kaliszczak, M.; van Hechanova, E.; Li, Y.; Alsadah, H.; Parzych, K.; Auner, H.W.; Aboagye, E.O. The HDAC6 inhibitor C1A modulates autophagy substrates in diverse cancer cells and induces cell death. Br. J. Cancer 2018, 119, 1278–1287. [Google Scholar] [CrossRef]
- Morine, Y.; Shimada, M.; Iwahashi, S.; Utsunomiya, T.; Imura, S.; Ikemoto, T.; Mori, H.; Hanaoka, J.; Miyake, H. Role of histone deacetylase expression in intrahepatic cholangiocarcinoma. Surgery 2012, 151, 412–419. [Google Scholar] [CrossRef]
- Collins, P.L.; Oltz, E.M. Histone methylation keeps the brakes on autophagy. Mol. Cell. Biol. 2013, 33, 3974–3975. [Google Scholar] [CrossRef] [PubMed]
- Borger, D.R.; Tanabe, K.K.; Kenneth, C.F.; Lopez, H.U.; Fantin, V.R.; Straley, K.S.; Schenkein, D.P.; Hezel, A.F.; Ancukiewicz, M.; Liebman, H.M.; et al. Frequent mutation of isocitrate dehydrogenase (IDH)1 and IDH2 in cholangiocarcinoma identified through broad-based tumor genotyping. Oncologist 2012, 17, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Dong, Q.; Zhang, C.; Kuan, P.F.; Liu, Y.; Jeck, W.R.; Andersen, J.B.; Jiang, W.; Savich, G.L.; Tan, T.X.; et al. Mutations in isocitrate dehydrogenase 1 and 2 occur frequently in intrahepatic cholangiocarcinomas and share hypermethylation targets with glioblastomas. Oncogene 2013, 32, 3091–3100. [Google Scholar] [CrossRef] [PubMed]
- Rahim, S.A.A.; Dirkse, A.; Oudin, A.; Schuster, A.; Bohler, J.; Barthelemy, V.; Muller, A.; Vallar, L.; Janji, B.; Golebiewska, A.; et al. Regulation of hypoxia-induced autophagy in glioblastoma involves ATG9A. Br. J. Cancer 2017, 117, 813–825. [Google Scholar] [CrossRef] [PubMed]
- Howell, J.A.; Khan, S.A. The role of miRNAs in cholangiocarcinoma. Hepatic Oncol. 2016, 3, 167–180. [Google Scholar] [CrossRef]
- Meng, F.; Henson, R.; Lang, M.; Wehbe, H.; Maheshwari, S.; MEndell, J.T.; Jiang, J.; Schmittgen, T.D.; Patel, T. Involvement of human Micro-RNA in growth and response to chemotherapy in human cholangiocarcinoma cell lines. Gastroenterology 2006, 130, 2113–2129. [Google Scholar] [CrossRef]
- Gozuacik, D.; Akkoc, Y.; Ozturk, D.G.; Kocak, M. Autophagy-Regulating microRNAs and Cancer. Front. Oncol. 2017, 7, 65. [Google Scholar] [CrossRef]
- Frankel, L.B.; Lund, A.H. MicroRNA regulation of autophagy. Carcinogenesis 2012, 33, 2018–2025. [Google Scholar] [CrossRef]
- Saitoh, M. Involvement of partial EMT in cancer progression. J. Biochem. 2018, 164, 257–264. [Google Scholar] [CrossRef]
- Krebs, A.M.; Mitschke, J.; Lasierra Losada, M.; Schmalhofer, O.; Boerries, M.; Busch, H.; Boettcher, M.; Mougiakakos, D.; Reichardt, W.; Bronsert, P.; et al. The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat. Cell Biol. 2017, 19, 518–529. [Google Scholar] [CrossRef]
- Chen, H.T.; Liu, H.; Mao, M.J.; Tan, Y.; Mo, X.Q.; Meng, X.J.; Cao, M.T.; Zhong, C.Y.; Liu, Y.; Shan, H.; et al. Crosstalk between autophagy and epithelial-mesenchymal transition and its application in cancer therapy. Mol. Cancer 2019, 18, 101. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Yao, W.; Yang, T.; Yang, Y.; Liu, Y.; Shen, Q.; Zhang, J.; Qi, W.; Wang, J. aPKC-ι/P-Sp1/Snail signaling induces epithelial-mesenchymal transition and immunosuppression in cholangiocarcinoma. Hepatology 2017, 66, 1165–1182. [Google Scholar] [CrossRef] [PubMed]
- Kimura-Tsuchiya, R.; Ishikawa, T.; Kokura, S.; Mizushima, K.; Adachi, S.; Okajima, M.; Matsuyama, T.; Okayama, T.; Sakamoto, N.; Katada, K.; et al. The inhibitory effect of heat treatment against epithelial-mesenchymal transition (EMT) in human pancreatic adenocarcinoma cell lines. J. Clin. Biochem. Nutr. 2014, 55, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Nitta, T.; Sato, Y.; Ren, X.S.; Harada, K.; Sasaki, M.; Hirano, S.; Nakanuma, Y. Autophagy may promote carcinoma cell invasion and correlate with poor prognosis in cholangiocarcinoma. Int. J. Clin. Exp. Pathol. 2014, 7, 4913–4921. [Google Scholar] [PubMed]
- Kong, X.-X.; Zhang, H.-Y.; Chen, Z.-Q.; Fan, X.-F.; Gon, Y.-S. Inhibition of Beclin 1 enhances apoptosis by H2O2 in glioma U251 cells. Sheng Li Xue Bao 2011, 63, 238–244. [Google Scholar] [PubMed]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.-L.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Invest. 2003, 112, 1809–1820. [Google Scholar] [CrossRef] [PubMed]
- Frampton, G.; Ueno, Y.; Quinn, M.; McMillin, M.; Pae, H.Y.; Galindo, C.; Leyva-Illades, D.; DeMorrow, S. The novel growth factor, progranulin, stimulates mouse cholangiocyte proliferation via sirtuin-1-mediated inactivation of FOXO1. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G1202–G1211. [Google Scholar] [CrossRef]
- Molenaar, R.J.; Coelen, R.J.S.; Khurshed, M.; Roos, E.; Caan, M.W.A.; van Linde, M.E.; Kouwenhoven, M.; Bramer, J.A.M.; Bovée, J.V.M.G.; Mathôt, R.A.; et al. Study protocol of a phase IB/II clinical trial of metformin and chloroquine in patients with IDH1-mutated or IDH2-mutated solid tumours. BMJ Open 2017, 7, e014961. [Google Scholar] [CrossRef]
- Beljanski, V.; Knaak, C.; Smith, C.D. A novel sphingosine kinase inhibitor induces autophagy in tumor cells. J. Pharmacol. Exp. Ther. 2010, 333, 454–464. [Google Scholar] [CrossRef]
- Moolthiya, P.; Tohtong, R.; Keeratichamroen, S.; Leelawat, K. Role of mTOR inhibitor in cholangiocarcinoma cell progression. Oncol. Lett. 2014, 7, 854–860. [Google Scholar] [CrossRef]
- Yothaisong, S.; Dokduang, H.; Techasen, A.; Namwat, N.; Yongvanit, P.; Bhudhisawasdi, V.; Puapairoj, A.; Gregory, J. Riggins & watcharin loilome. Increased activation of PI3K/AKT signaling pathway is associated with cholangiocarcinoma metastasis and PI3K/mTOR inhibition presents a possible therapeutic strategy. Tumor Biol. 2013, 34, 3637–3648. [Google Scholar]
- Nitta, T.; Mitsuhashi, T.; Hatanaka, Y.; Miyamoto, M.; Oba, K.; Tsuchikawa, T.; Suzuki, Y.; Hatanaka, K.C.; Hirano, S.; Matsuno, Y. Prognostic significance of epithelial-mesenchymal transition-related markers in extrahepatic cholangiocarcinoma: Comprehensive immunohistochemical study using a tissue microarray. Br. J. Cancer 2014, 111, 1363–1372. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Sheng, J.; Shen, L.; Su, J.; Xu, Y.; Xie, Q.; Wu, Y.; Zhang, X.; Sun, L. Autophagy inhibitor chloroquine increases sensitivity to cisplatin in QBC939 cholangiocarcinoma cells by mitochondrial ROS. PLoS ONE 2017, 12, e0173712. [Google Scholar] [CrossRef] [PubMed]
- Jia, B.; Xue, Y.; Yan, X.; Li, J.; Wu, Y.; Guo, R.; Zhang, J.; Zhang, L.; Li, Y.; Liu, Y.; et al. Autophagy inhibitor chloroquine induces apoptosis of cholangiocarcinoma cells via endoplasmic reticulum stress. Oncol. Lett. 2018, 16, 3509–3516. [Google Scholar] [CrossRef]
- Brun, S.; Bassissi, F.; Serdjebi, C.; Novello, M.; Tracz, J.; Autelitano, F.; Guillemot, M.; Fabre, P.; Courcambeck, J.; Ansaldi, C.; et al. GNS561, a new lysosomotropic small molecule, for the treatment of intrahepatic cholangiocarcinoma. Investig. New Drugs 2019, 37, 1135–1145. [Google Scholar] [CrossRef]
- Klose, J.; Guerlevik, E.; Trostel, T.; Kühnel, F.; Schmidt, T.; Schneider, M.; Ulrich, A. Salinomycin inhibits cholangiocarcinoma growth by inhibition of autophagic flux. Oncotarget 2018, 9, 3619–3630. [Google Scholar] [CrossRef]
- Hong, Z.-F.; Zhao, W.-X.; Yin, Z.-Y.; Xie, C.-R.; Xu, Y.-P.; Chi, X.-Q.; Zhang, S.; Wang, X.-M. Capsaicin enhances the drug sensitivity of cholangiocarcinoma through the inhibition of chemotherapeutic-induced autophagy. PLoS ONE 2015, 10, e0121538. [Google Scholar] [CrossRef]
- Zhang, A.; He, W.; Shi, H.; Huang, X.; Ji, G. Natural compound oblongifolin C inhibits autophagic flux, and induces apoptosis and mitochondrial dysfunction in human cholangiocarcinoma QBC939 cells. Mol. Med. Rep. 2016, 14, 3179–3183. [Google Scholar] [CrossRef]
- Tusskorn, O.; Khunluck, T.; Prawan, A.; Senggunprai, L.; Kukongviriyapan, V. Mitochondrial division inhibitor-1 potentiates cisplatin-induced apoptosis via the mitochondrial death pathway in cholangiocarcinoma cells. BioMed. Pharmacother. 2019, 111, 109–118. [Google Scholar] [CrossRef]
- Chu, H.; Li, M.; Wang, X. Capsaicin induces apoptosis and autophagy in human melanoma cells. Oncol. Lett. 2019, 17, 4827–4834. [Google Scholar] [CrossRef]
- Puissant, A.; Robert, G.; Fenouille, N.; Luciano, F.; Cassuto, J.-P.; Raynaud, S.; Auberger, P. Resveratrol promotes autophagic cell death in chronic myelogenous leukemia cells via JNK-mediated p62/SQSTM1 expression and AMPK activation. Cancer Res. 2010, 70, 1042–1052. [Google Scholar] [CrossRef] [PubMed]
- Signorelli, P.; Munoz-Olaya, J.M.; Gagliostro, V.; Casas, J.; Ghidoni, R.; Fabriàs, G. Dihydroceramide intracellular increase in response to resveratrol treatment mediates autophagy in gastric cancer cells. Cancer Lett. 2009, 282, 238–243. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jiang, W.; Li, C.; Xiong, X.; Guo, H.; Tian, Q.; Li, X. Autophagy suppression accelerates apoptosis induced by norcantharidin in cholangiocarcinoma. Pathol. Oncol. Res. 2019. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Luo, G.; Cheng, Y.; Yu, W.; Chen, R.; Xiao, B.; Xiang, Y.; Feng, C.; Fu, W.; Duan, C.; et al. Compound C induces protective autophagy in human cholangiocarcinoma cells via Akt/mTOR-independent pathway. J. Cell. Biochem. 2018, 119, 5538–5550. [Google Scholar] [CrossRef]
- Kwak, T.W.; Kim, D.H.; Jeong, Y.I.; Kang, D.H. Antitumor activity of vorinostat-incorporated nanoparticles against human cholangiocarcinoma cells. J. Nanobiotechnol. 2015, 13, 60. [Google Scholar] [CrossRef]
- Opydo-Chanek, M.; Gonzalo, O.; Marzo, I. Multifaceted anticancer activity of BH3 mimetics: Current evidence and future prospects. Biochem. Pharmacol. 2017, 136, 12–23. [Google Scholar] [CrossRef]
- Carracedo, A.; Gironella, M.; Lorente, M.; Garcia, S.; Guzmán, M.; Velasco, G.; Iovanna, J.L. Cannabinoids induce apoptosis of pancreatic tumor cells via endoplasmic reticulum stress-related genes. Cancer Res. 2006, 66, 6748–6755. [Google Scholar] [CrossRef]
- Russo, M.; Russo, G.L. Autophagy inducers in cancer. Biochem. Pharmacol. 2018, 153, 51–61. [Google Scholar] [CrossRef]
- Chen, S.; Huang, H.; Lin, H.; Fang, C. Piperlongumine induces autophagy in biliary cancer cells via reactive oxygen species-activated Erk signaling pathway. Int. J. Mol. Med. 2019, 44, 1687–1696. [Google Scholar] [CrossRef]
- Wang, D.; Guo, H.; Yang, H.; Wang, D.; Gao, P.; Wei, W. Pterostilbene, an active constituent of blueberries, suppresses proliferation potential of human cholangiocarcinoma via enhancing the autophagic flux. Front. Pharmacol. 2019, 10, 1238. [Google Scholar] [CrossRef]
- Sun, J.M.; Xu, H.T.; Zhao, L.; Zhang, Y.B.; Kang, P.C.; Song, Z.F.; Liu, H.S.; Cui, Y.F. Induction of cell-cycle arrest and apoptosis in human cholangiocarcinoma cells by pristimerin. J. Cell. Biochem. 2019, 120, 12002–12009. [Google Scholar] [CrossRef] [PubMed]
- Thongchot, S.; Vidoni, C.; Ferraresi, A.; Loilome, W.; Yongvanit, P.; Namwat, N.; Isidoro, C. Dihydroartemisinin induces apoptosis and autophagy-dependent cell death in cholangiocarcinoma through a DAPK1-BECLIN1 pathway. Mol. Carcinog. 2018, 57, 1735–1750. [Google Scholar] [CrossRef] [PubMed]
- Thongsom, S.; Suginta, W.; Lee, K.J.; Choe, H.; Talabnin, C. Piperlongumine induces G2/M phase arrest and apoptosis in cholangiocarcinoma cells through the ROS-JNK-ERK signaling pathway. Apoptosis 2017, 22, 1473–1484. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Li, H.; Yang, R.; Zhou, S.; Zou, S. Decitabine inhibits the cell growth of cholangiocarcinoma in cultured cell lines and mouse xenografts. Oncol. Lett. 2014, 8, 1919–1924. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Ouyang, Q.; Cheng, Q.; Wang, J.; Feng, F.; Qiao, L.; Gan, W.; Shi, Y.; Wu, D.; Jiang, X. Phenformin inhibits cell proliferation and induces cell apoptosis and autophagy in cholangiocarcinoma. Mol. Med. Rep. 2018, 17, 6028–6032. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Guardiola, P.; Casas, J.; Megías-Roda, E.; Solé, S.; Perez-Montoyo, H.; Yeste-Velasco, M.; Erazo, T.; Diéguez-Martínez, N.; Espinosa-Gil, S.; Muñoz-Pinedoet, C.; et al. The anticancer drug ABTL0812 induces ER stress-mediated cytotoxic autophagy by increasing dihydroceramide levels in cancer cells. Autophagy 2020, in press. [Google Scholar]
- Gravina, G.L.; Festuccia, C.; Marampon, F.; Popov, V.M.; Pestell, R.G.; Zani, B.M.; Tombolini, V. Biological rationale for the use of DNA methyltransferase inhibitors as new strategy for modulation of tumor response to chemotherapy and radiation. Mol. Cancer 2010, 9, 305. [Google Scholar] [CrossRef]
- Perez-Montoyo, H.; Olaizola, P.; Muñoz-Guardiola, P.; Megias-Roda, E.; Sole, S.; Gil, M.; Vidal, L.; Gascon, P.; Jose, M.; Carles Domenech, L.; et al. ABTL0812, a novel phase-2 clinical stage pro-autophagic anti-cancer compound with potential clinical activity in cholangiocarcinoma. In Proceedings of the Cholangiocarcinoma Foundation Annual Conference, Salt Lake City, UT, USA, 30 January–1 February 2019. [Google Scholar]
- Erazo, T.; Lorente, M.; López-Plana, A.; Muñoz-Guardiola, P.; Fernández-Nogueira, P.; García-Martínez, J.A.; Bragado, P.; Fuster, G.; Salazar, M.; Espadaler, J.; et al. The new antitumor drug ABTL0812 inhibits the Akt/mTORC1 axis by upregulating Tribbles-3 pseudokinase. Clin. Cancer Res. 2016, 22, 2508–2519. [Google Scholar] [CrossRef]
- Sofer, A.; Lei, K.; Johannessen, C.M.; Ellisen, L.W. Regulation of mTOR and cell growth in response to energy stress by REDD1. Mol. Cell. Biol. 2005, 25, 5834–5845. [Google Scholar] [CrossRef]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta Mol. Cell Res. 2013, 1833, 3460–3470. [Google Scholar] [CrossRef]
- Ord, T.; Ord, T. Mammalian pseudokinase TRIB3 in normal physiology and disease: Charting the progress in old and new avenues. Curr. Protein Pept. Sci. 2017, 18, 819–842. [Google Scholar] [CrossRef] [PubMed]
- Salazar, M.; Carracedo, A.; Salanueva, I.J.; Hernández-Tiedra, S.; Egia, A.; Lorente, M.; Vázquez, P.; Torres, S.; Iovanna, J.L.; Guzmán, M.; et al. TRB3 links ER stress to autophagy in cannabinoid anti-tumoral action. Autophagy 2009, 5, 1048–1049. [Google Scholar] [CrossRef] [PubMed]
- Salazar, M.; Lorente, M.; García-Taboada, E.; Pérez Gómez, E.; Dávila, D.; Zúñiga-García, P.; María Flores, J.; Rodríguez, A.; Hegedus, Z.; Mosén-Ansorena, D.; et al. Loss of tribbles pseudokinase-3 promotes Akt-driven tumorigenesis via FOXO inactivation. Cell Death Differ. 2015, 22, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Leelawat, S.; Leelawat, K.; Narong, S.; Matangkasombut, O. The dual effects of Δ9-tetrahydrocannabinol on cholangiocarcinoma cells: Anti-invasion activity at low concentration and apoptosis induction at high concentration. Cancer Investig. 2010, 28, 357–363. [Google Scholar] [CrossRef]
- Alfon, J.; Vidal, L.; Gaba, L.; Victoria, I.; Gil, M.; Laquente, B.; Brunet, M.; Colom, H.; Ramis, J.; Perez-Montoyo, H.; et al. Determination of recommended phase II dose of ABTL0812, a novel regulator of Akt/mTOR axis, by pharmacokinetic-pharmacodynamic modelling. Ann. Oncol. 2016, 27, 114–135. [Google Scholar] [CrossRef]
- Felip, I.; Moiola, C.P.; Megino-Luque, C.; Lopez-Gil, C.; Cabrera, S.; Solé-Sánchez, S.; Muñoz-Guardiola, P.; Megias-Roda, E.; Pérez-Montoyo, H.; Alfon, J.; et al. Therapeutic potential of the new TRIB3-mediated cell autophagy anticancer drug ABTL0812 in endometrial cancer. Gynecol. Oncol. 2019, 153, 425–435. [Google Scholar] [CrossRef]
- López-Plana, A.; Fernández-Nogueira, P.; Muñoz-Guardiola, P.; Solé-Sánchez, S.; Megías-Roda, E.; Pérez-Montoyo, H.; Jauregui, P.; Yeste-Velasco, M.; Erazo, M.G.T.; Ametller, E.; et al. The novel pro-autophagy anticancer drug ABTL0812 potentiates chemotherapy in adenocarcinoma and squamous non-small cell lung cancer. Int. J. Cancer 2020. [Google Scholar] [CrossRef]
- Shuda, M.; Kondoh, N.; Imazeki, N.; Tanaka, K.; Okada, T.; Mori, K.; Hada, A.; Arai, M.; Wakatsuki, T.; Matsubara, O.; et al. Activation of the ATF6, XBP1 and grp78 genes in human hepatocellular carcinoma: A possible involvement of the ER stress pathway in hepatocarcinogenesis. J. Hepatol. 2003, 38, 605–614. [Google Scholar] [CrossRef]
- Schönthal, A.H. Endoplasmic reticulum stress and autophagy as targets for cancer therapy. Cancer Lett. 2009, 275, 163–169. [Google Scholar] [CrossRef]
- Thongchot, S.; Yongvanit, P.; Loilome, W.; Seubwai, W.; Phunicom, K.; Tassaneeyakul, W.; Pairojkul, C.; Promkotra, W.; Techasen, A.; Namwat, N. High expression of HIF-1α, BNIP3 and PI3KC3: Hypoxia-Induced autophagy predicts cholangiocarcinoma survival and metastasis. Asian Pac. J. Cancer Prev. 2014, 15, 5873–5878. [Google Scholar] [CrossRef]
- Kang, R.; Zeh, H.J.; Lotze, M.T.; Tang, D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011, 18, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.X.; Qiu, H.J.; Zeng, F.; Rao, H.L.; Yang, G.F.; Kung, H.F.; Zhu, X.F.; Zeng, Y.X.; Cai, M.Y.; Xie, D. Decreased expression of Beclin 1 correlates closely with Bcl-xL expression and poor prognosis of ovarian carcinoma. PLoS ONE 2013, 8, e60516. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.B.; Fan, X.J.; Chen, M.Y.; Xiang, J.; Huang, P.Y.; Guo, L.; Wu, X.Y.; Xu, J.; Long, Z.J.; Zhao, Y.; et al. Elevated Beclin 1 expression is correlated with HIF-1α in predicting poor prognosis of nasopharyngeal carcinoma. Autophagy 2010, 6, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Wang, X.; Zhao, X.; Kong, C.; Li, Z.; Liu, Y.; Jiang, X.; Zhang, X. The autophagy-related genes Beclin1 and LC3 in the prognosis of pancreatic cancer. Int. J. Clin. Exp. Pathol. 2019, 12, 2989. [Google Scholar]
- Han, Y.; Xue, X.F.; Shen, H.G.; Guo, X.B.; Wang, X.; Yuan, B.; Guo, X.P.; Kuang, Y.T.; Zhi, Q.M.; Zhao, H. Prognostic significance of Beclin-1 expression in colorectal cancer: A meta-analysis. Asian Pac. J. Cancer Prev. 2014, 15, 4583–4587. [Google Scholar] [CrossRef]
- Ding, Z.B.; Shi, Y.H.; Zhou, J.; Qiu, S.J.; Xu, Y.; Dai, Z.; Shi, G.M.; Wang, X.Y.; Ke, A.W.; Wu, B.; et al. Association of autophagy defect with a malignant phenotype and poor prognosis of hepatocellular carcinoma. Cancer Res. 2008, 68, 9167–9175. [Google Scholar] [CrossRef]
- Al-Shenawy, H.A.S. Expression of Beclin-1, an autophagy-related marker, in chronic hepatitis and hepatocellular carcinoma and its relation with apoptotic markers. APMIS 2016, 124, 229–237. [Google Scholar] [CrossRef]
- Osman, N.A.A.; El-Rehim, D.M.A.; Kamal, I.M. Defective Beclin-1 and elevated hypoxia-inducible factor (HIF)-1α expression are closely linked to tumorigenesis, differentiation, and progression of hepatocellular carcinoma. Tumor Biol. 2015, 36, 4293–4299. [Google Scholar] [CrossRef]
- Bi, C.; Liu, M.; Rong, W.; Wu, F.; Zhang, Y.; Lin, S.; Liu, Y.; Wu, J.; Wang, L. High Beclin-1 and ARID1A expression corelates with poor survival and high recurrence in intrahepatic cholangiocarcinoma: A histopathological retrospective study. BMC Cancer 2019, 19, 213. [Google Scholar] [CrossRef]
- Hernández-Tiedra, S.; Fabriàs, G.; Dávila, D.; Salanueva, Í.J.; Casas, J.; Montes, L.R.; Antón, Z.; García-Taboada, E.; Salazar-Roa, M.; Lorente, M.; et al. Dihydroceramide accumulation mediates cytotoxic autophagy of cancer cells via autolysosome destabilization. Autophagy 2016, 12, 2213–2229. [Google Scholar] [CrossRef]
- Hirose, Y.; Nagahashi, M.; Katsuta, E.; Yuza, K.; Miura, K.; Sakata, J.; Kobayashi, T.; Ichikawa, H.; Shimada, Y.; Kameyama, H.; et al. Generation of sphingosine-1-phosphate is enhanced in biliary tract cancer patients and is associated with lymphatic metastasis. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Xia, P.; Wang, J.J.; Zhao, B.B.; Song, C.L. The role of beclin-1 expression in patients with gastric cancer: A meta-analysis. Tumor Biol. 2013, 34, 3303–3307. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lv, X.-W.; Shi, J.-P.; Hu, X.-S. Mechanisms involved in ceramide-induced cell cycle arrest in human hepatocarcinoma cells. World J. Gastroenterol. 2007, 13, 1129–1134. [Google Scholar] [CrossRef] [PubMed]
- Seubwai, W.; Kraiklang, R.; Wongkham, C.; Wongkham, S. Hypoxia enhances aggressiveness of cholangiocarcinoma cells. Asian Pac. J. Cancer Prev. 2012, 13, 53–58. [Google Scholar] [PubMed]
- Follo, C.; Cheng, Y.; Richards, W.G.; Bueno, R.; Broaddus, V.C. Autophagy facilitates the release of immunogenic signals following chemotherapy in 3D models of mesothelioma. Mol. Carcinog. 2019, 58, 1754–1769. [Google Scholar] [CrossRef] [PubMed]
- Valle, A.S.; Anel, A.; Naval, J.; Marzo, I. Immunogenic cell death and immunotherapy of multiple myeloma. Front. Cell Dev. Biol. 2019, 7, 1–22. [Google Scholar]
- Zhou, J.; Wang, G.; Chen, Y.; Wang, H.; Hua, Y.; Cai, Z. Immunogenic cell death in cancer therapy: Present and emerging inducers. J. Cell. Mol. Med. 2019, 23, 4854–4865. [Google Scholar] [CrossRef]
- Kang, J.; Jeong, J.H.; Hwang, H.S.; Lee, S.S.; Park, D.H.; Oh, D.W.; Song, T.J.; Kim, K.H.; Hwang, S.; Hwang, D.W.; et al. Efficacy and safety of pembrolizumab in patients with PD-L1 positive advanced biliary tract cancer (BTC): A prospective cohort study. J. Clin. Oncol. 2019, 37, 4082. [Google Scholar] [CrossRef]
- Hu-Lieskovan, S.; Mok, S.; Homet Moreno, B.; Tsoi, J.; Robert, L.; Goedert, L.; Pinheiro, E.M.; Koya, R.C.; Graeber, T.G.; Comin-Anduix, B.; et al. Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in BRAFV600E melanoma. Sci. Transl. Med. 2015, 7, 279ra41. [Google Scholar] [CrossRef]
- Jahan, N.; Talat, H.; Alonso, A.; Saha, D.; Curry, W.T. Triple combination immunotherapy with GVAX, anti-PD-1 monoclonal antibody, and agonist anti-OX40 monoclonal antibody is highly effective against murine intracranial glioma. Oncoimmunology 2019, 8, e1577108. [Google Scholar] [CrossRef]
- Minor, D.R.; Puzanov, I.; Callahan, M.K.; Hug, B.A.; Hoos, A. Severe gastrointestinal toxicity with administration of trametinib in combination with dabrafenib and ipilimumab. Pigment Cell Melanoma Res. 2015, 28, 611–612. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yan, J.; Wang, L.; Xiao, F.; Yang, Y.; Guo, X.; Wang, H. Beclin1 inhibition promotes autophagy and decreases gemcitabine-induced apoptosis in Miapaca2 pancreatic cancer cells. Cancer Cell Int. 2013, 13, 26. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Types of Autophagy | Features | Mechanism | Selectivity of Cargo |
|---|---|---|---|
| Macroautophagy | Nonselective macroautophagy: multistep process of nonselective degradation and recycling of cellular misfolded, aggregated or mutated proteins and damaged organelles. Mediated by the formation of autophagosomes and their fusion to lysosomes | Cytoplasm degraded in a bulk manner, including proteins, organelles and cytoplasmic components. Most-described autophagic process | Nonselective |
| Selective macroautophagy: multistep process of selective degradation and recycling of specific targets organelles, proteins and cellular components. Mediated by the formation of autophagosomes and their fusion to lysosomes | Lipophagy: lipids droplets autophagic degradation | Selective | |
| Pexophagy: peroxisomes autophagic degradation | Selective | ||
| Mitophagy: mitochondria autophagic degradation | Selective | ||
| Xenophagy: microbes autophagic degradation | Selective | ||
| Others: autophagic degradation of nucleus (nucleophagy), ribosomes (ribophagy), RNA (rnautophagy), etc. | Selective | ||
| Microautophagy | Direct uptake of cytoplasmic substances into the lysosomes for degradation. No autophagosome formation needed | Cytoplasmic substrates are engulfed via direct invagination, protrusion or septation of the lysosomal limiting membrane | Nonselective |
| Chaperon-mediated autophagy (CMA) | Uptake of soluble cytosolic proteins that are directly translocated across the lysosome membrane for degradation. No autophagosome formation needed | Chaperone-dependent recognition of specific sites in proteins to form the CMA substrate-chaperone complex, which is recognized by lysosomal membrane-bound receptors to unfold proteins and translocate them across lysosomal membranes | Highly selective for proteins |
| Autophagy Inhibitors | |||||
| Compound | Mechanism of Action | Preclinical Models | Effects on CCA | Level of Inhibition | Reference |
| Wortmannin (cell permeable fungal metabolite) and 3-MA (synthetic 3 methyl adenine) | Specific class III PI3K (VPS34) inhibitors. VPS34 is needed to recruit Atg12-Atg5 conjugates to preautophagosomal structure | In vitro: QBC939, RBE and HCCC9810. In vivo: QBC939 xenografts | Apoptosis induction in vitro and inhibition of tumor growth, decreasing mRNA levels of ATG5 and Beclin1 in tumors | Initiation: inhibits Vps34 (class III PI3K) complex | Hou et al. 2011 [106] |
| Chloroquine (antimalaria agent) | Alters acidic environment of lysosomes, induces sustained ER stress and CHOP-mediated apoptosis | In vitro: CCKS1 and HuCCT1 cells | Attenuate invasive activity of CCA cells under starvation conditions and in TGF-β1-induced EMT | Fusion: Inhibits autophagosome fusion with lysosomes | Nitta et al. 2014 [126] |
| Capsaicin (major pungent component of chili peppers) | Interferes with NF-kB and AP-1 signaling | In vitro: QBC939, SK-ChA-1 and MZ-ChA-1. In vivo: QBC939 xenograft | Inhibition of 5-FU induced autophagy in vitro and in vivo via activation of PI3K/Akt/mTOR pathway, increasing sensitivity to 5-FU | Initiation: activates mTOR | Hong et al. 2015 [139] |
| Oblongifolin C (natural small molecule extracted from herbs) | Induces mitochondrial apoptotic pathway | In vitro: QBC939 | Induces apoptosis and mitochondrial dysfunction | Fusion: Inhibits autophagosome fusion with lysosomes | Zang et al. 2016 [140] |
| Chloroquine (antimalaria agent) | Alters acidic environment of lysosomes, induces sustained ER stress and CHOP-mediated apoptosis | In vitro: QBC939 cells | Reduces antioxidant capacity of cells increases ROS and sensitizes cells to cisplatin | Fusion: Inhibits autophagosome fusion with lysosomes | Qu et al. 2017 [135] |
| Salinomycin (polyether antibiotic) | Interferes with WNT signaling and acts as potassium ionophore | In vitro: TFK-1 and EGI-1 cells. In vivo: s.c. and intrahepatic murine models KRAs and p53 mutated | Inhibits proliferation and transmembrane migration mediated by dysfunctional mitochondria in vitro and inhibits tumor growth in vivo | * Fusion: Inhibits autophagosome fusion with lysosomes | Klose et al. 2018 [138] |
| Chloroquine (antimalaria agent) | Alters acidic environment of lysosomes, induces sustained ER stress and CHOP-mediated apoptosis | In vitro: QBC939 cells | Induces apoptosis through activation of multiple death pathways and increases sensitivity to cisplatin | Fusion: Inhibits autophagosome fusion with lysosomes | Jia et al. 2018 [136] |
| Resveratrol (natural phenol, phytoalexin, produced by plants against infections) | Sirt1 agonist. Promotes deacetylation of FOXO1, blocking FOXO1 binding to Atg7 | In vitro: QBC939 cells | Induces apoptosis by increasing oxidative stress and mitochondrial dysfunction. | Initiation: inhibits Foxo1-Atg7 activation | He et al. 2018 [30] |
| Mdivi1-selective Drp-1 inhibitor | Impedes mitochondrial dynamics | In vitro: KKU-156 and KKU-214 | Potentiates cisplatin-induced apoptosis inducing mitochondrial dysfunction and ROS | * Elongation inhibits mitophagy | Tusskorn et al. 2019 [141] |
| GNS561 (lysosomotropic small molecule) | Lysosomal dysregulation through lysosome permeabilizes and releases hydrolytic enzymes to the cytosol | In vitro: HuCCT1 and RBE iCCAs. In vivo: chicken chorioallantoic membrane xenograft model | In vitro: reduces cell proliferation and induces apoptosis. In vivo: reduced tumor growth | Fusion: Inhibits lysosomal proteases | Brun et al. 2019 [137] |
| Autophagy Activators | |||||
| Compound | Mechanism of Action | Preclinical Models | Effects on CCA | Level of Activation | Reference |
| Decitabine (cytosine analog) DNA demethylating agent | DNA methyl transferase inhibitor | In vitro: TFK-1 and QBC939. In vivo: TFK-1 xenograft | Induces apoptosis and autophagy-dependent caspase-independent cell death in vitro and reduces tumor growth in vivo | * Initiation: epigenetic control of autophagy | Wang et al. 2014 [156] |
| Phenformin (biguanide compound paralog of metformin) | In vitro: RBE and Huh28. In vivo: RBE xenograft | Induces apoptosis and autophagy in vitro (Atg7, Atg5 and Beclin1 upregulation) and reduces tumor growth in vivo | Initiation: AMPK-mediated mTOR inhibition | Hu et al. 2017 [157] | |
| Dihydroartemisinin (active compound from Artemisia annua) | ROS-mediated ER stress through DAPK activation promoting the disruption Beclin11-Bcl2 | In vitro: KKU-452, KKU-023 and KKU-100, KKU-223 and MMNK-1 | Induces apoptosis-dependent and autophagy-mediated apoptosis-independent cell death | Initiation: disruption of Beclin1-Bcl2 | Thongchot et al. 2018 [154] |
| MiR-124 (associated with STAT3 regulation) | Targets EZH2 and STAT3 signaling pathway inducing ER stress | In vitro: HuCCT1, KMBC and MZChA1. In vivo MZChA1 transfected to stably express low levels of miR-124 or shEZH2 | Induces autophagy-related cell death via EZH2-STAT3 signaling axis in vitro and tumor-suppressive function in vivo | Initiation: disruption of Beclin1-Bcl2 | Ma et al. 2018 [29] |
| Piperlongumine (small molecule extracted from plants) | Inhibits the antioxidant enzyme glutathione S-transferase P, leading to elevated ROS via multiple pathways (p38/JNK, MAPK-C/EBO and NN-KB) | In vitro: HuCCT-1 | Induces apoptosis and autophagy through ROS-activated Erk signaling | * Initiation: disruption of Beclin1-Bcl2 | Chen et al. 2019 [123] |
| Pterostilbene (active constituent of blueberries; natural demethylated analogue of resveratrol | Involves overlap among intrinsic and extrinsic apoptotic pathway, cell cycle arrest, DNA damage, mitochondrial depolarization and autophagy | In vitro: RBE and HCCC-9810. In vivo: HCCC-9810 | Induces dose-dependent and time-dependent cytotoxic effects and inhibits colony formation upregulating Beclin1, ATG5 and ATG7 and inhibits tumor growth in vivo | * Initiation: disruption of Beclin1-Bcl2 | Wang et al. 2019 [152] |
| Pristimerin (triterpenoid isolated from herbs) | Has multiple targets (Li et al. 2018 | In vitro: QBC and RBE. In vivo: QBC939 xenografts | Induces apoptosis and autophagy in dose-dependent manner, decreasing apoptosis-related proteins Bcl-2, Bcl-xL and porcaspase-3 in vitro and inhibits tumor growth in vivo | * Initiation: disruption of Beclin1-Bcl2 | Sun et al. 2019 [153] |
| ABTL0812 (hydroxylated variant of linoleic acid) | Induces robust and sustained ER stress, and TRIB3-mediated Akt/mTOR axis inhibition, leading to cytotoxic autophagy | In vitro: EGI-1 and TFK-1 | Induces ER stress-mediated cytotoxic autophagy (elevated ATF4, CHOP and TRIB3) | Initiation: mTOR inhibition and ER stress mediated autophagy initiation | Muñoz-Guardiola et al. 2020 [158] |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perez-Montoyo, H. Therapeutic Potential of Autophagy Modulation in Cholangiocarcinoma. Cells 2020, 9, 614. https://doi.org/10.3390/cells9030614
Perez-Montoyo H. Therapeutic Potential of Autophagy Modulation in Cholangiocarcinoma. Cells. 2020; 9(3):614. https://doi.org/10.3390/cells9030614
Chicago/Turabian StylePerez-Montoyo, Hector. 2020. "Therapeutic Potential of Autophagy Modulation in Cholangiocarcinoma" Cells 9, no. 3: 614. https://doi.org/10.3390/cells9030614
APA StylePerez-Montoyo, H. (2020). Therapeutic Potential of Autophagy Modulation in Cholangiocarcinoma. Cells, 9(3), 614. https://doi.org/10.3390/cells9030614