The Quest for Cellular Prion Protein Functions in the Aged and Neurodegenerating Brain

 ,
, 

Abstract
1. Introduction
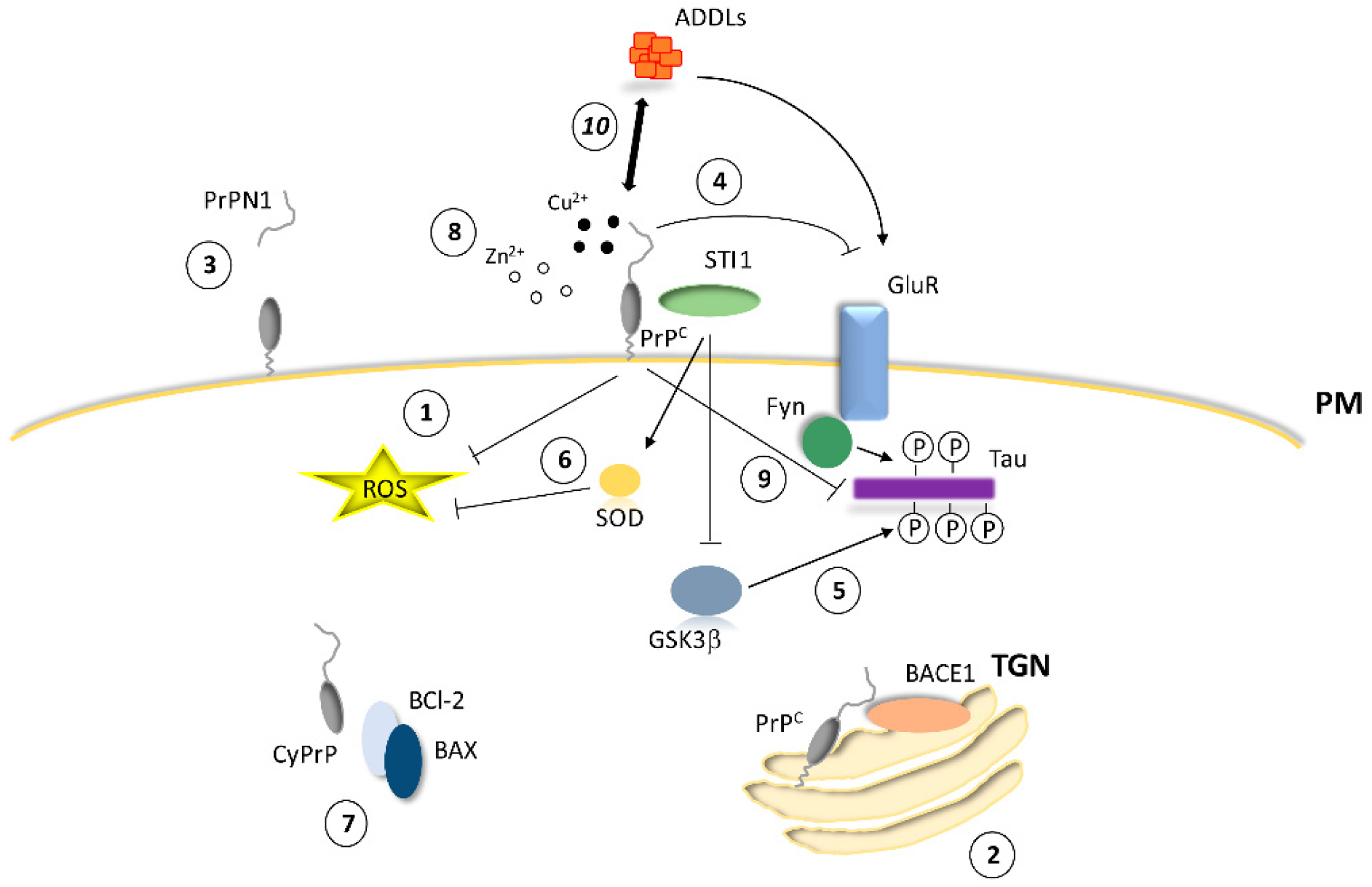
2. PrPC and Neuroprotection
2.1. Antioxidant Activity
2.2. Antiapoptotic Activity
2.3. Regulation of Calcium Homeostasis and Ionotropic Glutamate Receptors by PrPC
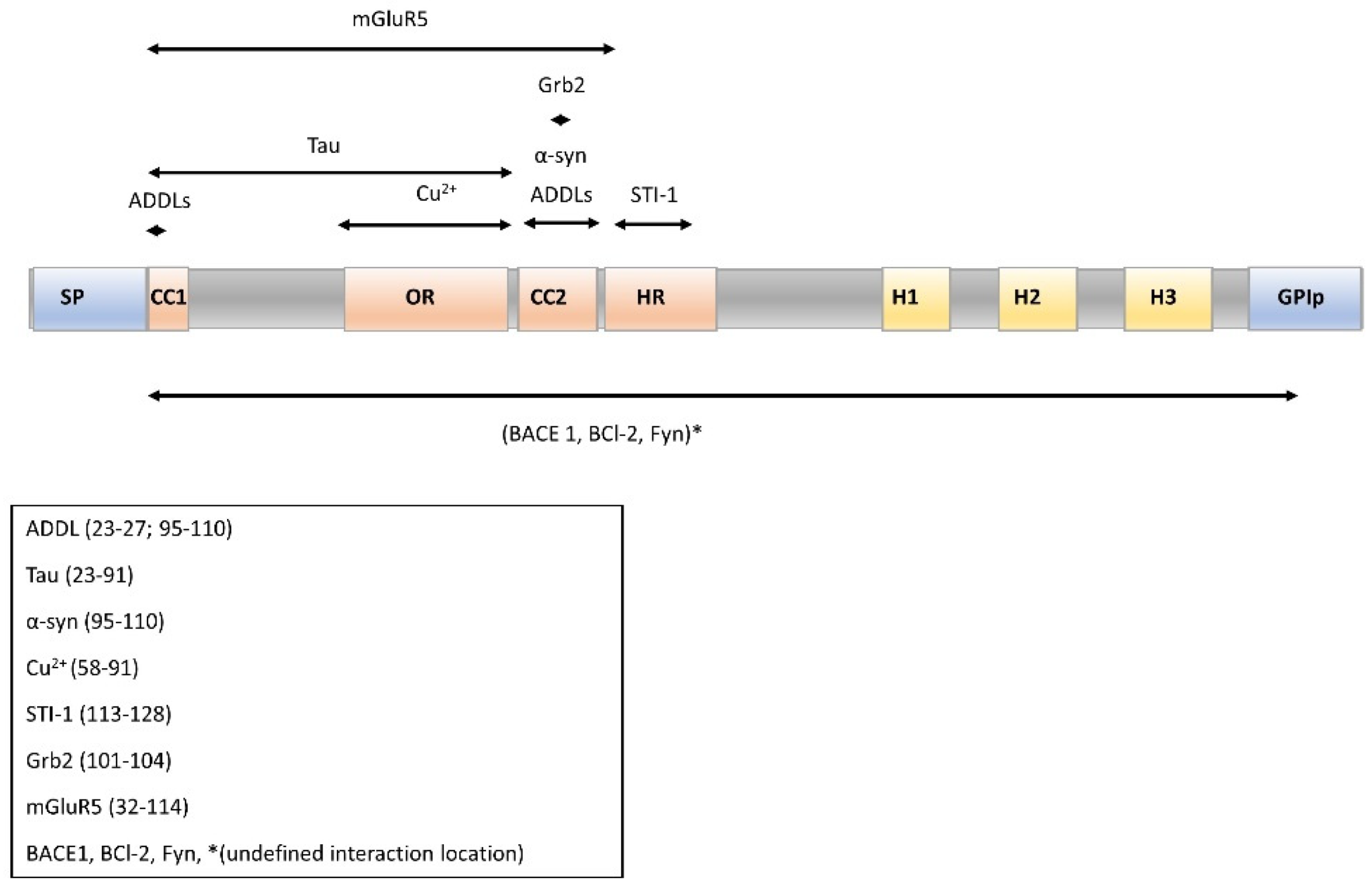
2.4. Molecular Partners of PrPC for Interaction and Cell Signaling
2.5. Physiological Processing of PrPC and Neuroprotective Metabolites
3. Functions of PrPC during Aging and Neurodegeneration
3.1. PrPC in Alzheimer’s Disease (AD) and Other Tauopathies
3.2. Neuroprotective Role of PrPC in Huntington’s and Parkinson’s Diseases
4. Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
References
- Prusiner, S.B. Prions. Proc. Natl. Acad. Sci. USA 1998, 95, 13363–13383. [Google Scholar] [CrossRef]
- Prusiner, S.B. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144. [Google Scholar] [CrossRef]
- Bolton, D.C.; McKinley, M.P.; Prusiner, S.B. Identification of a protein that purifies with the scrapie prion. Science 1982, 218, 1309–1311. [Google Scholar] [CrossRef]
- Ford, M.J.; Burton, L.J.; Morris, R.J.; Hall, S.M. Selective expression of prion protein in peripheral tissues of the adult mouse. Neuroscience 2002, 113, 177–192. [Google Scholar] [CrossRef]
- Miele, G.; Alejo Blanco, A.R.; Baybutt, H.; Horvat, S.; Manson, J.; Clinton, M. Embryonic activation and developmental expression of the murine prion protein gene. Gene Expr. 2003, 11, 1–12. [Google Scholar] [CrossRef]
- Nicolas, O.; Gavin, R.; del Rio, J.A. New insights into cellular prion protein (PrPc) functions: The “ying and yang” of a relevant protein. Brain Res. Rev. 2009, 61, 170–184. [Google Scholar] [CrossRef]
- Aguzzi, A.; Sigurdson, C.; Heikenwaelder, M. Molecular mechanisms of prion pathogenesis. Annu. Rev. Pathol. 2008, 3, 11–40. [Google Scholar] [CrossRef]
- Collinge, J. Prion diseases of humans and animals: Their causes and molecular basis. Annu. Rev. Neurosci. 2001, 24, 519–550. [Google Scholar] [CrossRef]
- Aguzzi, A.; Baumann, F.; Bremer, J. The prion’s elusive reason for being. Annu. Rev. Neurosci. 2008, 31, 439–477. [Google Scholar] [CrossRef]
- Aguzzi, A.; Rajendran, L. The transcellular spread of cytosolic amyloids, prions, and prionoids. Neuron 2009, 64, 783–790. [Google Scholar] [CrossRef]
- Fernandez-Borges, N.; Erana, H.; Elezgarai, S.R.; Harrathi, C.; Gayosso, M.; Castilla, J. Infectivity versus Seeding in Neurodegenerative Diseases Sharing a Prion-Like Mechanism. Int J. Cell Biol. 2013, 2013, 583498. [Google Scholar] [CrossRef]
- Cushman, M.; Johnson, B.S.; King, O.D.; Gitler, A.D.; Shorter, J. Prion-like disorders: Blurring the divide between transmissibility and infectivity. J. Cell Sci. 2010, 123, 1191–1201. [Google Scholar] [CrossRef]
- Ayers, J.I.; Cashman, N.R. Prion-like mechanisms in amyotrophic lateral sclerosis. Handb. Clin. Neurol. 2018, 153, 337–354. [Google Scholar] [CrossRef]
- Cascarina, S.M.; Ross, E.D. Natural and pathogenic protein sequence variation affecting prion-like domains within and across human proteomes. BMC Genom. 2020, 21, 23. [Google Scholar] [CrossRef]
- Ren, P.H.; Lauckner, J.E.; Kachirskaia, I.; Heuser, J.E.; Melki, R.; Kopito, R.R. Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates. Nat. Cell Biol. 2009, 11, 219–225. [Google Scholar] [CrossRef]
- Luk, K.C.; Kehm, V.M.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M. Intracerebral inoculation of pathological alpha-synuclein initiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. J. Exp. Med. 2012, 209, 975–986. [Google Scholar] [CrossRef]
- Masuda-Suzukake, M.; Nonaka, T.; Hosokawa, M.; Oikawa, T.; Arai, T.; Akiyama, H.; Mann, D.M.; Hasegawa, M. Prion-like spreading of pathological alpha-synuclein in brain. Brain 2013, 136, 1128–1138. [Google Scholar] [CrossRef]
- Aulic, S.; Le, T.T.; Moda, F.; Abounit, S.; Corvaglia, S.; Casalis, L.; Gustincich, S.; Zurzolo, C.; Tagliavini, F.; Legname, G. Defined alpha-synuclein prion-like molecular assemblies spreading in cell culture. BMC Neurosci. 2014, 15, 69. [Google Scholar] [CrossRef]
- Lee, H.J.; Patel, S.; Lee, S.J. Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. J. Neurosci. 2005, 25, 6016–6024. [Google Scholar] [CrossRef]
- Desplats, P.; Lee, H.J.; Bae, E.J.; Patrick, C.; Rockenstein, E.; Crews, L.; Spencer, B.; Masliah, E.; Lee, S.J. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc. Natl. Acad. Sci. USA 2009, 106, 13010–13015. [Google Scholar] [CrossRef]
- Del Rio, J.A.; Ferrer, I.; Gavin, R. Role of cellular prion protein in interneuronal amyloid transmission. Prog. Neurobiol. 2018, 165–167, 87–102. [Google Scholar] [CrossRef]
- Urrea, L.; Segura-Feliu, M.; Masuda-Suzukake, M.; Hervera, A.; Pedraz, L.; Garcia-Aznar, J.M.; Vila, M.; Samitier, J.; Torrents, E.; Ferrer, I.; et al. Involvement of Cellular Prion Protein in alpha-Synuclein Transport in Neurons. Mol. Neurobiol. 2018, 55, 1847–1860. [Google Scholar] [CrossRef]
- Ferreira, D.G.; Temido-Ferreira, M.; Vicente Miranda, H.; Batalha, V.L.; Coelho, J.E.; Szego, E.M.; Marques-Morgado, I.; Vaz, S.H.; Rhee, J.S.; Schmitz, M.; et al. alpha-synuclein interacts with PrP(C) to induce cognitive impairment through mGluR5 and NMDAR2B. Nat. Neurosci. 2017, 20, 1569–1579. [Google Scholar] [CrossRef]
- Nath, S.; Agholme, L.; Kurudenkandy, F.R.; Granseth, B.; Marcusson, J.; Hallbeck, M. Spreading of neurodegenerative pathology via neuron-to-neuron transmission of beta-amyloid. J. Neurosci. 2012, 32, 8767–8777. [Google Scholar] [CrossRef]
- Domert, J.; Rao, S.B.; Agholme, L.; Brorsson, A.C.; Marcusson, J.; Hallbeck, M.; Nath, S. Spreading of amyloid-beta peptides via neuritic cell-to-cell transfer is dependent on insufficient cellular clearance. Neurobiol. Dis. 2014, 65, 82–92. [Google Scholar] [CrossRef]
- Eisele, Y.S.; Obermuller, U.; Heilbronner, G.; Baumann, F.; Kaeser, S.A.; Wolburg, H.; Walker, L.C.; Staufenbiel, M.; Heikenwalder, M.; Jucker, M. Peripherally applied Abeta-containing inoculates induce cerebral beta-amyloidosis. Science 2010, 330, 980–982. [Google Scholar] [CrossRef]
- Clavaguera, F.; Bolmont, T.; Crowther, R.A.; Abramowski, D.; Frank, S.; Probst, A.; Fraser, G.; Stalder, A.K.; Beibel, M.; Staufenbiel, M.; et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell Biol. 2009, 11, 909–913. [Google Scholar] [CrossRef]
- Frost, B.; Ollesch, J.; Wille, H.; Diamond, M.I. Conformational diversity of wild-type Tau fibrils specified by templated conformation change. J. Biol. Chem. 2009, 284, 3546–3551. [Google Scholar] [CrossRef]
- Sydow, A.; Mandelkow, E.M. ‘Prion-like’ propagation of mouse and human tau aggregates in an inducible mouse model of tauopathy. Neurodegener. Dis. 2010, 7, 28–31. [Google Scholar] [CrossRef]
- Guo, J.L.; Lee, V.M. Seeding of normal Tau by pathological Tau conformers drives pathogenesis of Alzheimer-like tangles. J. Biol. Chem. 2011, 286, 15317–15331. [Google Scholar] [CrossRef]
- Clavaguera, F.; Akatsu, H.; Fraser, G.; Crowther, R.A.; Frank, S.; Hench, J.; Probst, A.; Winkler, D.T.; Reichwald, J.; Staufenbiel, M.; et al. Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc. Natl. Acad. Sci. USA 2013, 110, 9535–9540. [Google Scholar] [CrossRef]
- Michelitsch, M.D.; Weissman, J.S. A census of glutamine/asparagine-rich regions: Implications for their conserved function and the prediction of novel prions. Proc. Natl. Acad. Sci. USA 2000, 97, 11910–11915. [Google Scholar] [CrossRef]
- Sabate, R.; Rousseau, F.; Schymkowitz, J.; Ventura, S. What makes a protein sequence a prion? PLoS Comput. Biol. 2015, 11, e1004013. [Google Scholar] [CrossRef]
- Jaunmuktane, Z.; Mead, S.; Ellis, M.; Wadsworth, J.D.; Nicoll, A.J.; Kenny, J.; Launchbury, F.; Linehan, J.; Richard-Loendt, A.; Walker, A.S.; et al. Evidence for human transmission of amyloid-beta pathology and cerebral amyloid angiopathy. Nature 2015, 525, 247–250. [Google Scholar] [CrossRef]
- Frontzek, K.; Lutz, M.I.; Aguzzi, A.; Kovacs, G.G.; Budka, H. Amyloid-beta pathology and cerebral amyloid angiopathy are frequent in iatrogenic Creutzfeldt-Jakob disease after dural grafting. Swiss Med. Wkly 2016, 146, w14287. [Google Scholar] [CrossRef]
- Kovacs, G.G.; Lutz, M.I.; Ricken, G.; Strobel, T.; Hoftberger, R.; Preusser, M.; Regelsberger, G.; Honigschnabl, S.; Reiner, A.; Fischer, P.; et al. Dura mater is a potential source of Abeta seeds. Acta Neuropathol. 2016, 131, 911–923. [Google Scholar] [CrossRef]
- Hansen, L.A.; Masliah, E.; Terry, R.D.; Mirra, S.S. A neuropathological subset of Alzheimer’s disease with concomitant Lewy body disease and spongiform change. Acta Neuropathol. 1989, 78, 194–201. [Google Scholar] [CrossRef]
- Race, B.; Phillips, K.; Kraus, A.; Chesebro, B. Phosphorylated human tau associates with mouse prion protein amyloid in scrapie-infected mice but does not increase progression of clinical disease. Prion 2016, 10, 319–330. [Google Scholar] [CrossRef][Green Version]
- Debatin, L.; Streffer, J.; Geissen, M.; Matschke, J.; Aguzzi, A.; Glatzel, M. Association between deposition of beta-amyloid and pathological prion protein in sporadic Creutzfeldt-Jakob disease. Neurodegener. Dis. 2008, 5, 347–354. [Google Scholar] [CrossRef]
- Hainfellner, J.A.; Wanschitz, J.; Jellinger, K.; Liberski, P.P.; Gullotta, F.; Budka, H. Coexistence of Alzheimer-type neuropathology in Creutzfeldt-Jakob disease. Acta Neuropathol. 1998, 96, 116–122. [Google Scholar] [CrossRef]
- Barcikowska, M.; Kwiecinski, H.; Liberski, P.P.; Kowalski, J.; Brown, P.; Gajdusek, D.C. Creutzfeldt-Jakob disease with Alzheimer-type A beta-reactive amyloid plaques. Histopathology 1995, 26, 445–450. [Google Scholar] [CrossRef]
- Leuba, G.; Saini, K.; Savioz, A.; Charnay, Y. Early-onset familial Alzheimer disease with coexisting beta-amyloid and prion pathology. JAMA 2000, 283, 1689–1691. [Google Scholar] [CrossRef]
- Alzualde, A.; Indakoetxea, B.; Ferrer, I.; Moreno, F.; Barandiaran, M.; Gorostidi, A.; Estanga, A.; Ruiz, I.; Calero, M.; van Leeuwen, F.W.; et al. A novel PRNP Y218N mutation in Gerstmann-Straussler-Scheinker disease with neurofibrillary degeneration. J. Neuropathol. Exp. Neurol. 2010, 69, 789–800. [Google Scholar] [CrossRef]
- Irwin, D.J.; Lee, V.M.; Trojanowski, J.Q. Parkinson’s disease dementia: Convergence of alpha-synuclein, tau and amyloid-beta pathologies. Nat. Rev. Neurosci. 2013, 14, 626–636. [Google Scholar] [CrossRef]
- Cali, I.; Cohen, M.L.; Haik, S.; Parchi, P.; Giaccone, G.; Collins, S.J.; Kofskey, D.; Wang, H.; McLean, C.A.; Brandel, J.P.; et al. Iatrogenic Creutzfeldt-Jakob disease with Amyloid-beta pathology: An international study. Acta Neuropathol. Commun. 2018, 6, 5. [Google Scholar] [CrossRef]
- Dasari, A.K.R.; Kayed, R.; Wi, S.; Lim, K.H. Tau Interacts with the C-Terminal Region of alpha-Synuclein, Promoting Formation of Toxic Aggregates with Distinct Molecular Conformations. Biochemistry 2019, 58, 2814–2821. [Google Scholar] [CrossRef]
- Han, J.; Zhang, J.; Yao, H.; Wang, X.; Li, F.; Chen, L.; Gao, C.; Gao, J.; Nie, K.; Zhou, W.; et al. Study on interaction between microtubule associated protein tau and prion protein. Sci. China C Life Sci. 2006, 49, 473–479. [Google Scholar] [CrossRef]
- Wang, X.F.; Dong, C.F.; Zhang, J.; Wan, Y.Z.; Li, F.; Huang, Y.X.; Han, L.; Shan, B.; Gao, C.; Han, J.; et al. Human tau protein forms complex with PrP and some GSS- and fCJD-related PrP mutants possess stronger binding activities with tau in vitro. Mol. Cell Biochem. 2008, 310, 49–55. [Google Scholar] [CrossRef]
- Ferrer, I.; Blanco, R.; Carmona, M.; Puig, B.; Ribera, R.; Rey, M.J.; Ribalta, T. Prion protein expression in senile plaques in Alzheimer’s disease. Acta Neuropathol. 2001, 101, 49–56. [Google Scholar] [CrossRef]
- Lauren, J.; Gimbel, D.A.; Nygaard, H.B.; Gilbert, J.W.; Strittmatter, S.M. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 2009, 457, 1128–1132. [Google Scholar] [CrossRef]
- Meyne, F.; Gloeckner, S.F.; Ciesielczyk, B.; Heinemann, U.; Krasnianski, A.; Meissner, B.; Zerr, I. Total prion protein levels in the cerebrospinal fluid are reduced in patients with various neurological disorders. J. Alzheimers Dis. 2009, 17, 863–873. [Google Scholar] [CrossRef] [PubMed]
- Llorens, F.; Villar-Pique, A.; Schmitz, M.; Diaz-Lucena, D.; Wohlhage, M.; Hermann, P.; Goebel, S.; Schmidt, I.; Glatzel, M.; Hauw, J.J.; et al. Plasma total prion protein as a potential biomarker for neurodegenerative dementia: Diagnostic accuracy in the spectrum of prion diseases. Neuropathol. Appl. Neurobiol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Aguzzi, A.; Lakkaraju, A.K. Cell Biology of Prions and Prionoids: A Status Report. Trends Cell Biol. 2016, 26, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Houston, F.; Andreoletti, O. Animal prion diseases: The risks to human health. Brain Pathol. 2019, 29, 248–262. [Google Scholar] [CrossRef]
- Asher, D.M.; Gregori, L. Human transmissible spongiform encephalopathies: Historic view. Handb. Clin. Neurol. 2018, 153, 1–17. [Google Scholar] [CrossRef]
- O’Carroll, A.; Coyle, J.; Gambin, Y. Prions and Prion-like assemblies in neurodegeneration and immunity: The emergence of universal mechanisms across health and disease. Semin. Cell Dev. Biol. 2019. [Google Scholar] [CrossRef]
- Bueler, H.; Fischer, M.; Lang, Y.; Bluethmann, H.; Lipp, H.P.; DeArmond, S.J.; Prusiner, S.B.; Aguet, M.; Weissmann, C. Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature 1992, 356, 577–582. [Google Scholar] [CrossRef]
- Rossi, D.; Cozzio, A.; Flechsig, E.; Klein, M.A.; Rulicke, T.; Aguzzi, A.; Weissmann, C. Onset of ataxia and Purkinje cell loss in PrP null mice inversely correlated with Dpl level in brain. EMBO J. 2001, 20, 694–702. [Google Scholar] [CrossRef]
- Manson, J.C.; Clarke, A.R.; Hooper, M.L.; Aitchison, L.; McConnell, I.; Hope, J. 129/Ola mice carrying a null mutation in PrP that abolishes mRNA production are developmentally normal. Mol. Neurobiol. 1994, 8, 121–127. [Google Scholar] [CrossRef]
- Nuvolone, M.; Hermann, M.; Sorce, S.; Russo, G.; Tiberi, C.; Schwarz, P.; Minikel, E.; Sanoudou, D.; Pelczar, P.; Aguzzi, A. Strictly co-isogenic C57BL/6J-Prnp-/- mice: A rigorous resource for prion science. J. Exp. Med. 2016, 213, 313–327. [Google Scholar] [CrossRef]
- Steele, A.D.; Lindquist, S.; Aguzzi, A. The prion protein knockout mouse: A phenotype under challenge. Prion 2007, 1, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Del Rio, J.A.; Gavin, R. Functions of the cellular prion protein, the end of Moore’s law, and Ockham’s razor theory. Prion 2016, 10, 25–40. [Google Scholar] [CrossRef] [PubMed]
- Spielhaupter, C.; Schatzl, H.M. PrPC directly interacts with proteins involved in signaling pathways. J. Biol. Chem. 2001, 276, 44604–44612. [Google Scholar] [CrossRef] [PubMed]
- Schmitt-Ulms, G.; Legname, G.; Baldwin, M.A.; Ball, H.L.; Bradon, N.; Bosque, P.J.; Crossin, K.L.; Edelman, G.M.; DeArmond, S.J.; Cohen, F.E.; et al. Binding of neural cell adhesion molecules (N-CAMs) to the cellular prion protein. J. Mol. Biol. 2001, 314, 1209–1225. [Google Scholar] [CrossRef]
- Zafar, S.; von Ahsen, N.; Oellerich, M.; Zerr, I.; Schulz-Schaeffer, W.J.; Armstrong, V.W.; Asif, A.R. Proteomics approach to identify the interacting partners of cellular prion protein and characterization of Rab7a interaction in neuronal cells. J. Proteome Res. 2011, 10, 3123–3135. [Google Scholar] [CrossRef]
- Kuffer, A.; Lakkaraju, A.K.; Mogha, A.; Petersen, S.C.; Airich, K.; Doucerain, C.; Marpakwar, R.; Bakirci, P.; Senatore, A.; Monnard, A.; et al. The prion protein is an agonistic ligand of the G protein-coupled receptor Adgrg6. Nature 2016, 536, 464–468. [Google Scholar] [CrossRef]
- La Vitola, P.; Beeg, M.; Balducci, C.; Santamaria, G.; Restelli, E.; Colombo, L.; Caldinelli, L.; Pollegioni, L.; Gobbi, M.; Chiesa, R.; et al. Cellular prion protein neither binds to alpha-synuclein oligomers nor mediates their detrimental effects. Brain 2019, 142, 249–254. [Google Scholar] [CrossRef]
- Azzalin, A.; Del Vecchio, I.; Chiarelli, L.R.; Valentini, G.; Comincini, S.; Ferretti, L. Absence of interaction between doppel and GFAP, Grb2, PrPc proteins in human tumor astrocytic cells. Anticancer Res. 2005, 25, 4369–4374. [Google Scholar]
- Nuvolone, M.; Kana, V.; Hutter, G.; Sakata, D.; Mortin-Toth, S.M.; Russo, G.; Danska, J.S.; Aguzzi, A. SIRPalpha polymorphisms, but not the prion protein, control phagocytosis of apoptotic cells. J. Exp. Med. 2013, 210, 2539–2552. [Google Scholar] [CrossRef]
- Linden, R. The Biological Function of the Prion Protein: A Cell Surface Scaffold of Signaling Modules. Front. Mol. Neurosci. 2017, 10, 77. [Google Scholar] [CrossRef]
- Brown, D.R.; Qin, K.; Herms, J.W.; Madlung, A.; Manson, J.; Strome, R.; Fraser, P.E.; Kruck, T.; von Bohlen, A.; Schulz-Schaeffer, W.; et al. The cellular prion protein binds copper in vivo. Nature 1997, 390, 684–687. [Google Scholar] [CrossRef] [PubMed]
- Viles, J.H.; Cohen, F.E.; Prusiner, S.B.; Goodin, D.B.; Wright, P.E.; Dyson, H.J. Copper binding to the prion protein: Structural implications of four identical cooperative binding sites. Proc. Natl. Acad. Sci. USA 1999, 96, 2042–2047. [Google Scholar] [CrossRef] [PubMed]
- Evans, E.G.; Pushie, M.J.; Markham, K.A.; Lee, H.W.; Millhauser, G.L. Interaction between Prion Protein’s Copper-Bound Octarepeat Domain and a Charged C-Terminal Pocket Suggests a Mechanism for N-Terminal Regulation. Structure 2016, 24, 1057–1067. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, X.T.A.; Tran, T.H.; Cojoc, D.; Legname, G. Copper Binding Regulates Cellular Prion Protein Function. Mol. Neurobiol. 2019, 56, 6121–6133. [Google Scholar] [CrossRef]
- Vassallo, N.; Herms, J. Cellular prion protein function in copper homeostasis and redox signalling at the synapse. J. Neurochem. 2003, 86, 538–544. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.R.; Nicholas, R.S.; Canevari, L. Lack of prion protein expression results in a neuronal phenotype sensitive to stress. J. Neurosci. Res. 2002, 67, 211–224. [Google Scholar] [CrossRef] [PubMed]
- Wong, B.S.; Liu, T.; Li, R.; Pan, T.; Petersen, R.B.; Smith, M.A.; Gambetti, P.; Perry, G.; Manson, J.C.; Brown, D.R.; et al. Increased levels of oxidative stress markers detected in the brains of mice devoid of prion protein. J. Neurochem. 2001, 76, 565–572. [Google Scholar] [CrossRef]
- Klamt, F.; Dal-Pizzol, F.; Conte da Frota, M.L., Jr.; Walz, R.; Andrades, M.E.; da Silva, E.G.; Brentani, R.R.; Izquierdo, I.; Fonseca Moreira, J.C. Imbalance of antioxidant defense in mice lacking cellular prion protein. Free Radic Biol. Med. 2001, 30, 1137–1144. [Google Scholar] [CrossRef]
- Brown, D.R.; Schulz-Schaeffer, W.J.; Schmidt, B.; Kretzschmar, H.A. Prion protein-deficient cells show altered response to oxidative stress due to decreased SOD-1 activity. Exp. Neurol. 1997, 146, 104–112. [Google Scholar] [CrossRef]
- Brown, D.R.; Besinger, A. Prion protein expression and superoxide dismutase activity. Biochem. J. 1998, 334 (Pt. 2), 423–429. [Google Scholar] [CrossRef]
- Kuwahara, C.; Takeuchi, A.M.; Nishimura, T.; Haraguchi, K.; Kubosaki, A.; Matsumoto, Y.; Saeki, K.; Matsumoto, Y.; Yokoyama, T.; Itohara, S.; et al. Prions prevent neuronal cell-line death. Nature 1999, 400, 225–226. [Google Scholar] [CrossRef] [PubMed]
- Withee, J.L.; Sen, R.; Cyert, M.S. Ion tolerance of Saccharomyces cerevisiae lacking the Ca2+/CaM-dependent phosphatase (calcineurin) is improved by mutations in URE2 or PMA1. Genetics 1998, 149, 865–878. [Google Scholar] [PubMed]
- White, A.R.; Collins, S.J.; Maher, F.; Jobling, M.F.; Stewart, L.R.; Thyer, J.M.; Beyreuther, K.; Masters, C.L.; Cappai, R. Prion protein-deficient neurons reveal lower glutathione reductase activity and increased susceptibility to hydrogen peroxide toxicity. Am. J. Pathol. 1999, 155, 1723–1730. [Google Scholar] [CrossRef]
- Nishimura, T.; Sakudo, A.; Nakamura, I.; Lee, D.C.; Taniuchi, Y.; Saeki, K.; Matsumoto, Y.; Ogawa, M.; Sakaguchi, S.; Itohara, S.; et al. Cellular prion protein regulates intracellular hydrogen peroxide level and prevents copper-induced apoptosis. Biochem. Biophys. Res. Commun. 2004, 323, 218–222. [Google Scholar] [CrossRef]
- Qin, K.; Zhao, L.; Ash, R.D.; McDonough, W.F.; Zhao, R.Y. ATM-mediated transcriptional elevation of prion in response to copper-induced oxidative stress. J. Biol. Chem. 2009, 284, 4582–4593. [Google Scholar] [CrossRef]
- Sakudo, A.; Lee, D.C.; Saeki, K.; Nakamura, Y.; Inoue, K.; Matsumoto, Y.; Itohara, S.; Onodera, T. Impairment of superoxide dismutase activation by N-terminally truncated prion protein (PrP) in PrP-deficient neuronal cell line. Biochem. Biophys. Res. Commun. 2003, 308, 660–667. [Google Scholar] [CrossRef]
- McLennan, N.F.; Brennan, P.M.; McNeill, A.; Davies, I.; Fotheringham, A.; Rennison, K.A.; Ritchie, D.; Brannan, F.; Head, M.W.; Ironside, J.W.; et al. Prion protein accumulation and neuroprotection in hypoxic brain damage. Am. J. Pathol. 2004, 165, 227–235. [Google Scholar] [CrossRef]
- Hoshino, S.; Inoue, K.; Yokoyama, T.; Kobayashi, S.; Asakura, T.; Teramoto, A.; Itohara, S. Prions prevent brain damage after experimental brain injury: A preliminary report. Acta Neurochir. Suppl. 2003, 86, 297–299. [Google Scholar]
- Shyu, W.C.; Lin, S.Z.; Chiang, M.F.; Ding, D.C.; Li, K.W.; Chen, S.F.; Yang, H.I.; Li, H. Overexpression of PrPC by adenovirus-mediated gene targeting reduces ischemic injury in a stroke rat model. J. Neurosci. 2005, 25, 8967–8977. [Google Scholar] [CrossRef]
- Spudich, A.; Frigg, R.; Kilic, E.; Kilic, U.; Oesch, B.; Raeber, A.; Bassetti, C.L.; Hermann, D.M. Aggravation of ischemic brain injury by prion protein deficiency: Role of ERK-1/-2 and STAT-1. Neurobiol. Dis. 2005, 20, 442–449. [Google Scholar] [CrossRef]
- Weise, J.; Crome, O.; Sandau, R.; Schulz-Schaeffer, W.; Bahr, M.; Zerr, I. Upregulation of cellular prion protein (PrPc) after focal cerebral ischemia and influence of lesion severity. Neurosci. Lett. 2004, 372, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Williams, W.M.; Stadtman, E.R.; Moskovitz, J. Ageing and exposure to oxidative stress in vivo differentially affect cellular levels of PrP in mouse cerebral microvessels and brain parenchyma. Neuropathol. Appl. Neurobiol. 2004, 30, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Esiri, M.M.; Carter, J.; Ironside, J.W. Prion protein immunoreactivity in brain samples from an unselected autopsy population: Findings in 200 consecutive cases. Neuropathol. Appl. Neurobiol. 2000, 26, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Doeppner, T.R.; Kaltwasser, B.; Schlechter, J.; Jaschke, J.; Kilic, E.; Bahr, M.; Hermann, D.M.; Weise, J. Cellular prion protein promotes post-ischemic neuronal survival, angioneurogenesis and enhances neural progenitor cell homing via proteasome inhibition. Cell Death Dis. 2015, 6, e2024. [Google Scholar] [CrossRef]
- Bounhar, Y.; Zhang, Y.; Goodyer, C.G.; LeBlanc, A. Prion protein protects human neurons against Bax-mediated apoptosis. J. Biol. Chem. 2001, 276, 39145–39149. [Google Scholar] [CrossRef]
- Yin, X.M.; Oltvai, Z.N.; Korsmeyer, S.J. BH1 and BH2 domains of Bcl-2 are required for inhibition of apoptosis and heterodimerization with Bax. Nature 1994, 369, 321–323. [Google Scholar] [CrossRef]
- Kurschner, C.; Morgan, J.I. Analysis of interaction sites in homo- and heteromeric complexes containing Bcl-2 family members and the cellular prion protein. Brain Res. 1996, 37, 249–258. [Google Scholar] [CrossRef]
- Kurschner, C.; Morgan, J.I. The cellular prion protein (PrP) selectively binds to Bcl-2 in the yeast two-hybrid system. Brain Res. 1995, 30, 165–168. [Google Scholar] [CrossRef]
- Kim, B.H.; Lee, H.G.; Choi, J.K.; Kim, J.I.; Choi, E.K.; Carp, R.I.; Kim, Y.S. The cellular prion protein (PrPC) prevents apoptotic neuronal cell death and mitochondrial dysfunction induced by serum deprivation. Brain Res. 2004, 124, 40–50. [Google Scholar] [CrossRef]
- Herms, J.W.; Korte, S.; Gall, S.; Schneider, I.; Dunker, S.; Kretzschmar, H.A. Altered intracellular calcium homeostasis in cerebellar granule cells of prion protein-deficient mice. J. Neurochem. 2000, 75, 1487–1492. [Google Scholar] [CrossRef]
- Korte, S.; Vassallo, N.; Kramer, M.L.; Kretzschmar, H.A.; Herms, J. Modulation of L-type voltage-gated calcium channels by recombinant prion protein. J. Neurochem. 2003, 87, 1037–1042. [Google Scholar] [CrossRef][Green Version]
- Fuhrmann, M.; Bittner, T.; Mitteregger, G.; Haider, N.; Moosmang, S.; Kretzschmar, H.; Herms, J. Loss of the cellular prion protein affects the Ca2+ homeostasis in hippocampal CA1 neurons. J. Neurochem. 2006, 98, 1876–1885. [Google Scholar] [CrossRef] [PubMed]
- Krebs, B.; Wiebelitz, A.; Balitzki-Korte, B.; Vassallo, N.; Paluch, S.; Mitteregger, G.; Onodera, T.; Kretzschmar, H.A.; Herms, J. Cellular prion protein modulates the intracellular calcium response to hydrogen peroxide. J. Neurochem. 2007, 100, 358–367. [Google Scholar] [CrossRef]
- Jayaraman, T.; Ondrias, K.; Ondriasova, E.; Marks, A.R. Regulation of the inositol 1,4,5-trisphosphate receptor by tyrosine phosphorylation. Science 1996, 272, 1492–1494. [Google Scholar] [CrossRef] [PubMed]
- Rangel, A.; Burgaya, F.; Gavin, R.; Soriano, E.; Aguzzi, A.; Del Rio, J.A. Enhanced susceptibility of Prnp-deficient mice to kainate-induced seizures, neuronal apoptosis, and death: Role of AMPA/kainate receptors. J. Neurosci. Res. 2007, 85, 2741–2755. [Google Scholar] [CrossRef] [PubMed]
- Carulla, P.; Bribian, A.; Rangel, A.; Gavin, R.; Ferrer, I.; Caelles, C.; Del Rio, J.A.; Llorens, F. Neuroprotective role of PrPC against kainate-induced epileptic seizures and cell death depends on the modulation of JNK3 activation by GluR6/7-PSD-95 binding. Mol. Biol. Cell 2011, 22, 3041–3054. [Google Scholar] [CrossRef] [PubMed]
- Carulla, P.; Llorens, F.; Matamoros-Angles, A.; Aguilar-Calvo, P.; Espinosa, J.C.; Gavin, R.; Ferrer, I.; Legname, G.; Torres, J.M.; del Rio, J.A. Involvement of PrP(C) in kainate-induced excitotoxicity in several mouse strains. Sci. Rep. 2015, 5, 11971. [Google Scholar] [CrossRef] [PubMed]
- Gasperini, L.; Meneghetti, E.; Pastore, B.; Benetti, F.; Legname, G. Prion protein and copper cooperatively protect neurons by modulating NMDA receptor through S-nitrosylation. Antioxid Redox Signal. 2015, 22, 772–784. [Google Scholar] [CrossRef]
- Khosravani, H.; Zhang, Y.; Tsutsui, S.; Hameed, S.; Altier, C.; Hamid, J.; Chen, L.; Villemaire, M.; Ali, Z.; Jirik, F.R.; et al. Prion protein attenuates excitotoxicity by inhibiting NMDA receptors. J. Cell Biol. 2008, 181, 551–565. [Google Scholar] [CrossRef]
- Stys, P.K.; You, H.; Zamponi, G.W. Copper-dependent regulation of NMDA receptors by cellular prion protein: Implications for neurodegenerative disorders. J. Physiol. 2012, 590, 1357–1368. [Google Scholar] [CrossRef]
- Pham, N.; Dhar, A.; Khalaj, S.; Desai, K.; Taghibiglou, C. Down regulation of brain cellular prion protein in an animal model of insulin resistance: Possible implication in increased prevalence of stroke in pre-diabetics/diabetics. Biochem. Biophys. Res. Commun. 2014, 448, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Llorens, F.; Del Rio, J.A. Unraveling the neuroprotective mechanisms of PrP (C) in excitotoxicity. Prion 2012, 6, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Black, S.A.; Stys, P.K.; Zamponi, G.W.; Tsutsui, S. Cellular prion protein and NMDA receptor modulation: Protecting against excitotoxicity. Front. Cell Dev. Biol. 2014, 2, 45. [Google Scholar] [CrossRef] [PubMed]
- Graner, E.; Mercadante, A.F.; Zanata, S.M.; Forlenza, O.V.; Cabral, A.L.; Veiga, S.S.; Juliano, M.A.; Roesler, R.; Walz, R.; Minetti, A.; et al. Cellular prion protein binds laminin and mediates neuritogenesis. Brain Res. 2000, 76, 85–92. [Google Scholar] [CrossRef]
- Chen, S.; Mange, A.; Dong, L.; Lehmann, S.; Schachner, M. Prion protein as trans-interacting partner for neurons is involved in neurite outgrowth and neuronal survival. Mol. Cell. Neurosci. 2003, 22, 227–233. [Google Scholar] [CrossRef]
- Chiarini, L.B.; Freitas, A.R.; Zanata, S.M.; Brentani, R.R.; Martins, V.R.; Linden, R. Cellular prion protein transduces neuroprotective signals. EMBO J. 2002, 21, 3317–3326. [Google Scholar] [CrossRef]
- Mouillet-Richard, S.; Ermonval, M.; Chebassier, C.; Laplanche, J.L.; Lehmann, S.; Launay, J.M.; Kellermann, O. Signal transduction through prion protein. Science 2000, 289, 1925–1928. [Google Scholar] [CrossRef]
- Schneider, B.; Mutel, V.; Pietri, M.; Ermonval, M.; Mouillet-Richard, S.; Kellermann, O. NADPH oxidase and extracellular regulated kinases 1/2 are targets of prion protein signaling in neuronal and nonneuronal cells. Proc. Natl. Acad. Sci. USA 2003, 100, 13326–13331. [Google Scholar] [CrossRef]
- Pradines, E.; Loubet, D.; Schneider, B.; Launay, J.M.; Kellermann, O.; Mouillet-Richard, S. CREB-dependent gene regulation by prion protein: Impact on MMP-9 and beta-dystroglycan. Cell Signal. 2008, 20, 2050–2058. [Google Scholar] [CrossRef]
- Hernandez-Rapp, J.; Martin-Lanneree, S.; Hirsch, T.Z.; Pradines, E.; Alleaume-Butaux, A.; Schneider, B.; Baudry, A.; Launay, J.M.; Mouillet-Richard, S. A PrP(C)-caveolin-Lyn complex negatively controls neuronal GSK3beta and serotonin 1B receptor. Sci. Rep. 2014, 4, 4881. [Google Scholar] [CrossRef]
- Rambold, A.S.; Muller, V.; Ron, U.; Ben-Tal, N.; Winklhofer, K.F.; Tatzelt, J. Stress-protective signalling of prion protein is corrupted by scrapie prions. EMBO J. 2008, 27, 1974–1984. [Google Scholar] [CrossRef] [PubMed]
- Zanata, S.M.; Lopes, M.H.; Mercadante, A.F.; Hajj, G.N.; Chiarini, L.B.; Nomizo, R.; Freitas, A.R.; Cabral, A.L.; Lee, K.S.; Juliano, M.A.; et al. Stress-inducible protein 1 is a cell surface ligand for cellular prion that triggers neuroprotection. EMBO J. 2002, 21, 3307–3316. [Google Scholar] [CrossRef] [PubMed]
- Lopes, M.H.; Hajj, G.N.; Muras, A.G.; Mancini, G.L.; Castro, R.M.; Ribeiro, K.C.; Brentani, R.R.; Linden, R.; Martins, V.R. Interaction of cellular prion and stress-inducible protein 1 promotes neuritogenesis and neuroprotection by distinct signaling pathways. J. Neurosci. 2005, 25, 11330–11339. [Google Scholar] [CrossRef] [PubMed]
- Sakudo, A.; Lee, D.C.; Li, S.; Nakamura, T.; Matsumoto, Y.; Saeki, K.; Itohara, S.; Ikuta, K.; Onodera, T. PrP cooperates with STI1 to regulate SOD activity in PrP-deficient neuronal cell line. Biochem. Biophys. Res. Commun. 2005, 328, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Amin, L.; Nguyen, X.T.; Rolle, I.G.; D’Este, E.; Giachin, G.; Tran, T.H.; Serbec, V.C.; Cojoc, D.; Legname, G. Characterization of prion protein function by focal neurite stimulation. J. Cell Sci. 2016, 129, 3878–3891. [Google Scholar] [CrossRef]
- Lysek, D.A.; Wuthrich, K. Prion protein interaction with the C-terminal SH3 domain of Grb2 studied using NMR and optical spectroscopy. Biochemistry 2004, 43, 10393–10399. [Google Scholar] [CrossRef]
- Donne, D.G.; Viles, J.H.; Groth, D.; Mehlhorn, I.; James, T.L.; Cohen, F.E.; Prusiner, S.B.; Wright, P.E.; Dyson, H.J. Structure of the recombinant full-length hamster prion protein PrP(29-231): The N terminus is highly flexible. Proc. Natl. Acad. Sci. USA 1997, 94, 13452–13457. [Google Scholar] [CrossRef]
- Riek, R.; Hornemann, S.; Wider, G.; Glockshuber, R.; Wuthrich, K. NMR characterization of the full-length recombinant murine prion protein, mPrP(23-231). Febs Lett. 1997, 413, 282–288. [Google Scholar] [CrossRef]
- Riek, R.; Hornemann, S.; Wider, G.; Billeter, M.; Glockshuber, R.; Wuthrich, K. NMR structure of the mouse prion protein domain PrP(121-321). Nature 1996, 382, 180–182. [Google Scholar] [CrossRef]
- Chen, S.G.; Teplow, D.B.; Parchi, P.; Teller, J.K.; Gambetti, P.; Autilio-Gambetti, L. Truncated forms of the human prion protein in normal brain and in prion diseases. J. Biol. Chem. 1995, 270, 19173–19180. [Google Scholar] [CrossRef]
- Mange, A.; Beranger, F.; Peoc’h, K.; Onodera, T.; Frobert, Y.; Lehmann, S. Alpha- and beta- cleavages of the amino-terminus of the cellular prion protein. Biol. Cell 2004, 96, 125–132. [Google Scholar] [CrossRef] [PubMed]
- McMahon, H.E.; Mange, A.; Nishida, N.; Creminon, C.; Casanova, D.; Lehmann, S. Cleavage of the amino terminus of the prion protein by reactive oxygen species. J. Biol. Chem. 2001, 276, 2286–2291. [Google Scholar] [CrossRef] [PubMed]
- Resenberger, U.K.; Harmeier, A.; Woerner, A.C.; Goodman, J.L.; Muller, V.; Krishnan, R.; Vabulas, R.M.; Kretzschmar, H.A.; Lindquist, S.; Hartl, F.U.; et al. The cellular prion protein mediates neurotoxic signalling of beta-sheet-rich conformers independent of prion replication. EMBO J. 2011, 30, 2057–2070. [Google Scholar] [CrossRef]
- Guillot-Sestier, M.V.; Sunyach, C.; Druon, C.; Scarzello, S.; Checler, F. The alpha-secretase-derived N-terminal product of cellular prion, N1, displays neuroprotective function in vitro and in vivo. J. Biol. Chem. 2009, 284, 35973–35986. [Google Scholar] [CrossRef] [PubMed]
- Resenberger, U.K.; Winklhofer, K.F.; Tatzelt, J. Neuroprotective and neurotoxic signaling by the prion protein. Top. Curr. Chem. 2011, 305, 101–119. [Google Scholar] [CrossRef]
- Beland, M.; Motard, J.; Barbarin, A.; Roucou, X. PrP(C) homodimerization stimulates the production of PrPC cleaved fragments PrPN1 and PrPC1. J. Neurosci. 2012, 32, 13255–13263. [Google Scholar] [CrossRef]
- McDonald, A.J.; Millhauser, G.L. PrP overdrive: Does inhibition of alpha-cleavage contribute to PrP(C) toxicity and prion disease? Prion 2014, 8. [Google Scholar] [CrossRef]
- Mitteregger, G.; Vosko, M.; Krebs, B.; Xiang, W.; Kohlmannsperger, V.; Nolting, S.; Hamann, G.F.; Kretzschmar, H.A. The role of the octarepeat region in neuroprotective function of the cellular prion protein. Brain Pathol. 2007, 17, 174–183. [Google Scholar] [CrossRef]
- Lee, K.J.; Panzera, A.; Rogawski, D.; Greene, L.E.; Eisenberg, E. Celular prion protein protects (PrPC) protects neuronal cells from the effect of huntingtin aggregation. J. Cell Sci 2007, 120, 2663–2671. [Google Scholar] [CrossRef][Green Version]
- Kiachopoulos, S.; Heske, J.; Tatzelt, J.; Winklhofer, K.F. Misfolding of the prion protein at the plasma membrane induces endocytosis, intracellular retention and degradation. Traffic 2004, 5, 426–436. [Google Scholar] [CrossRef]
- Prado, M.A.; Alves-Silva, J.; Magalhaes, A.C.; Prado, V.F.; Linden, R.; Martins, V.R.; Brentani, R.R. PrPc on the road: Trafficking of the cellular prion protein. J. Neurochem. 2004, 88, 769–781. [Google Scholar] [CrossRef] [PubMed]
- Americo, T.A.; Chiarini, L.B.; Linden, R. Signaling induced by hop/STI-1 depends on endocytosis. Biochem. Biophys. Res. Commun. 2007, 358, 620–625. [Google Scholar] [CrossRef] [PubMed]
- Caetano, F.A.; Lopes, M.H.; Hajj, G.N.; Machado, C.F.; Pinto Arantes, C.; Magalhaes, A.C.; Vieira Mde, P.; Americo, T.A.; Massensini, A.R.; Priola, S.A.; et al. Endocytosis of prion protein is required for ERK1/2 signaling induced by stress-inducible protein 1. J. Neurosci. 2008, 28, 6691–6702. [Google Scholar] [CrossRef] [PubMed]
- Mironov, A., Jr.; Latawiec, D.; Wille, H.; Bouzamondo-Bernstein, E.; Legname, G.; Williamson, R.A.; Burton, D.; DeArmond, S.J.; Prusiner, S.B.; Peters, P.J. Cytosolic prion protein in neurons. J. Neurosci. 2003, 23, 7183–7193. [Google Scholar] [CrossRef] [PubMed]
- Roucou, X.; Guo, Q.; Zhang, Y.; Goodyer, C.G.; LeBlanc, A.C. Cytosolic prion protein is not toxic and protects against Bax-mediated cell death in human primary neurons. J. Biol. Chem. 2003, 278, 40877–40881. [Google Scholar] [CrossRef]
- Lin, D.T.; Jodoin, J.; Baril, M.; Goodyer, C.G.; Leblanc, A.C. Cytosolic prion protein is the predominant anti-Bax prion protein form: Exclusion of transmembrane and secreted prion protein forms in the anti-Bax function. Biochim. Et Biophys. Acta 2008, 1783, 2001–2012. [Google Scholar] [CrossRef][Green Version]
- Hegde, R.S.; Mastrianni, J.A.; Scott, M.R.; DeFea, K.A.; Tremblay, P.; Torchia, M.; DeArmond, S.J.; Prusiner, S.B.; Lingappa, V.R. A transmembrane form of the prion protein in neurodegenerative disease. Science 1998, 279, 827–834. [Google Scholar] [CrossRef]
- Rane, N.S.; Chakrabarti, O.; Feigenbaum, L.; Hegde, R.S. Signal sequence insufficiency contributes to neurodegeneration caused by transmembrane prion protein. J. Cell Biol. 2010, 188, 515–526. [Google Scholar] [CrossRef]
- Stewart, R.S.; Piccardo, P.; Ghetti, B.; Harris, D.A. Neurodegenerative illness in transgenic mice expressing a transmembrane form of the prion protein. J. Neurosci. 2005, 25, 3469–3477. [Google Scholar] [CrossRef]
- Chakrabarti, O.; Ashok, A.; Hegde, R.S. Prion protein biosynthesis and its emerging role in neurodegeneration. Trends Biochem. Sci. 2009, 34, 287–295. [Google Scholar] [CrossRef] [PubMed]
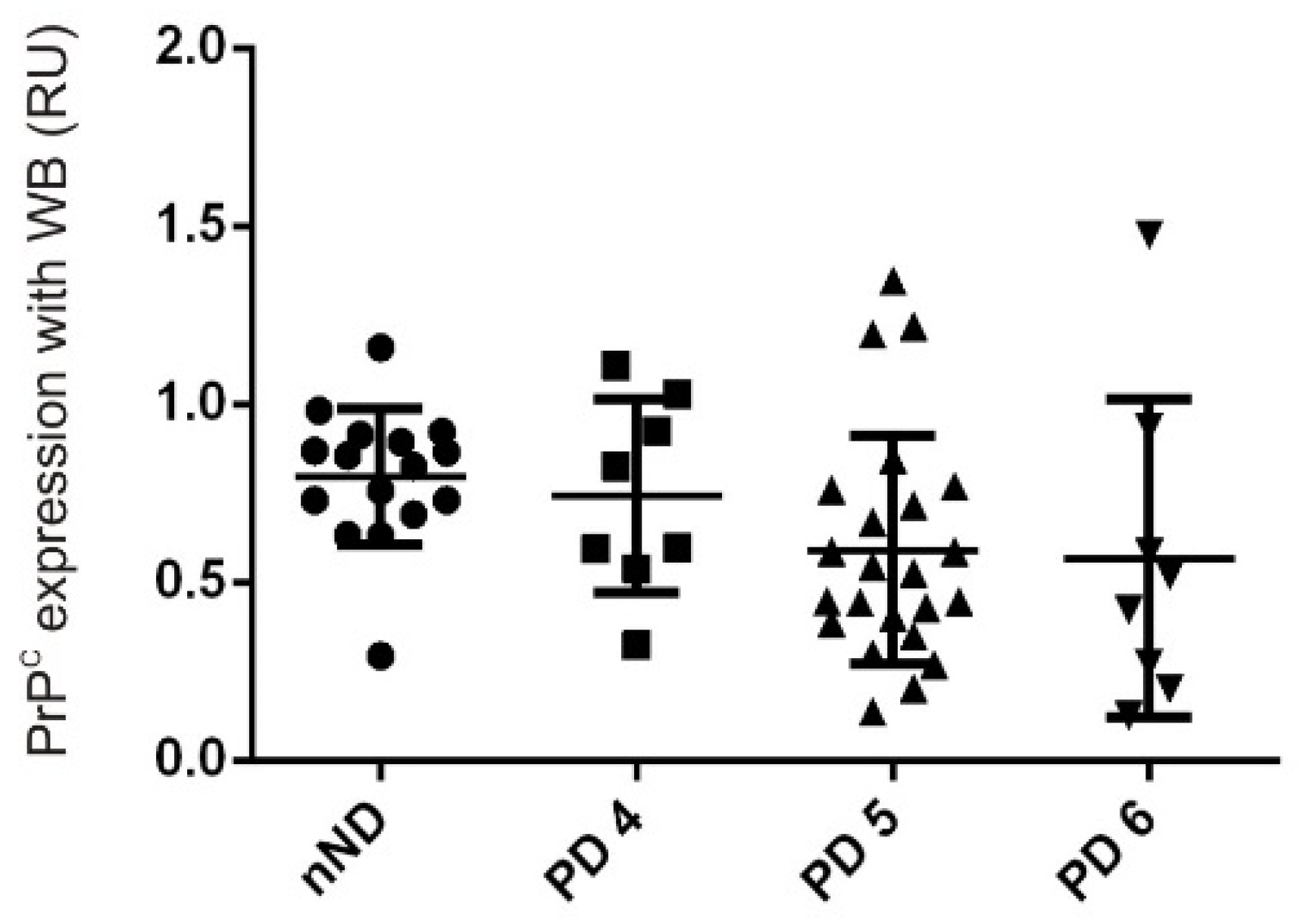
- Whitehouse, I.J.; Jackson, C.; Turner, A.J.; Hooper, N.M. Prion protein is reduced in aging and in sporadic but not in familial Alzheimer’s disease. J. Alzheimers Dis. 2010, 22, 1023–1031. [Google Scholar] [CrossRef] [PubMed]
- Beal, M.F. Aging, energy, and oxidative stress in neurodegenerative diseases. Ann. Neurol. 1995, 38, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.A.; Rottkamp, C.A.; Nunomura, A.; Raina, A.K.; Perry, G. Oxidative stress in Alzheimer’s disease. Biochim. Et Biophys. Acta 2000, 1502, 139–144. [Google Scholar] [CrossRef]
- Wong, B.S.; Pan, T.; Liu, T.; Li, R.; Petersen, R.B.; Jones, I.M.; Gambetti, P.; Brown, D.R.; Sy, M.S. Prion disease: A loss of antioxidant function? Biochem. Biophys. Res. Commun. 2000, 275, 249–252. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Yadav, S.P.; Surewicz, W.K. Interaction between human prion protein and amyloid-beta (Abeta) oligomers: Role OF N-terminal residues. J. Biol. Chem. 2010, 285, 26377–26383. [Google Scholar] [CrossRef] [PubMed]
- Nieznanski, K.; Surewicz, K.; Chen, S.; Nieznanska, H.; Surewicz, W.K. Interaction between prion protein and Abeta amyloid fibrils revisited. Acs Chem. Neurosci. 2014, 5, 340–345. [Google Scholar] [CrossRef]
- Aulic, S.; Masperone, L.; Narkiewicz, J.; Isopi, E.; Bistaffa, E.; Ambrosetti, E.; Pastore, B.; De Cecco, E.; Scaini, D.; Zago, P.; et al. alpha-Synuclein Amyloids Hijack Prion Protein to Gain Cell Entry, Facilitate Cell-to-Cell Spreading and Block Prion Replication. Sci. Rep. 2017, 7, 10050. [Google Scholar] [CrossRef]
- Parkin, E.T.; Watt, N.T.; Hussain, I.; Eckman, E.A.; Eckman, C.B.; Manson, J.C.; Baybutt, H.N.; Turner, A.J.; Hooper, N.M. Cellular prion protein regulates beta-secretase cleavage of the Alzheimer’s amyloid precursor protein. Proc. Natl. Acad. Sci. USA 2007, 104, 11062–11067. [Google Scholar] [CrossRef]
- Griffiths, H.H.; Whitehouse, I.J.; Baybutt, H.; Brown, D.; Kellett, K.A.; Jackson, C.D.; Turner, A.J.; Piccardo, P.; Manson, J.C.; Hooper, N.M. Prion protein interacts with BACE1 protein and differentially regulates its activity toward wild type and Swedish mutant amyloid precursor protein. J. Biol. Chem. 2011, 286, 33489–33500. [Google Scholar] [CrossRef]
- Beland, M.; Bedard, M.; Tremblay, G.; Lavigne, P.; Roucou, X. Abeta induces its own prion protein N-terminal fragment (PrPN1)-mediated neutralization in amorphous aggregates. Neurobiol. Aging 2014, 35, 1537–1548. [Google Scholar] [CrossRef]
- Nieznanski, K.; Choi, J.K.; Chen, S.; Surewicz, K.; Surewicz, W.K. Soluble prion protein inhibits amyloid-beta (Abeta) fibrillization and toxicity. J. Biol. Chem. 2012, 287, 33104–33108. [Google Scholar] [CrossRef] [PubMed]
- Ostapchenko, V.G.; Beraldo, F.H.; Mohammad, A.H.; Xie, Y.F.; Hirata, P.H.; Magalhaes, A.C.; Lamour, G.; Li, H.; Maciejewski, A.; Belrose, J.C.; et al. The prion protein ligand, stress-inducible phosphoprotein 1, regulates amyloid-beta oligomer toxicity. J. Neurosci. 2013, 33, 16552–16564. [Google Scholar] [CrossRef] [PubMed]
- Watt, N.T.; Griffiths, H.H.; Hooper, N.M. Lipid rafts: Linking prion protein to zinc transport and amyloid-beta toxicity in Alzheimer’s disease. Front. Cell Dev. Biol. 2014, 2, 41. [Google Scholar] [CrossRef] [PubMed]
- Fluharty, B.R.; Biasini, E.; Stravalaci, M.; Sclip, A.; Diomede, L.; Balducci, C.; La Vitola, P.; Messa, M.; Colombo, L.; Forloni, G.; et al. An N-terminal fragment of the prion protein binds to amyloid-beta oligomers and inhibits their neurotoxicity in vivo. J. Biol. Chem. 2013, 288, 7857–7866. [Google Scholar] [CrossRef] [PubMed]
- Rial, D.; Piermartiri, T.C.; Duarte, F.S.; Tasca, C.I.; Walz, R.; Prediger, R.D. Overexpression of cellular prion protein (PrP(C)) prevents cognitive dysfunction and apoptotic neuronal cell death induced by amyloid-beta (Abeta(1)(-)(4)(0)) administration in mice. Neuroscience 2012, 215, 79–89. [Google Scholar] [CrossRef]
- Voigtlander, T.; Kloppel, S.; Birner, P.; Jarius, C.; Flicker, H.; Verghese-Nikolakaki, S.; Sklaviadis, T.; Guentchev, M.; Budka, H. Marked increase of neuronal prion protein immunoreactivity in Alzheimer’s disease and human prion diseases. Acta Neuropathol. 2001, 101, 417–423. [Google Scholar] [CrossRef]
- Vergara, C.; Ordonez-Gutierrez, L.; Wandosell, F.; Ferrer, I.; del Rio, J.A.; Gavin, R. Role of PrP(C) Expression in Tau Protein Levels and Phosphorylation in Alzheimer’s Disease Evolution. Mol. Neurobiol. 2015, 51, 1206–1220. [Google Scholar] [CrossRef]
- Steinacker, P.; Hawlik, A.; Lehnert, S.; Jahn, O.; Meier, S.; Gorz, E.; Braunstein, K.E.; Krzovska, M.; Schwalenstocker, B.; Jesse, S.; et al. Neuroprotective function of cellular prion protein in a mouse model of amyotrophic lateral sclerosis. Am. J. Pathol. 2010, 176, 1409–1420. [Google Scholar] [CrossRef]
- Huang, S.; Chen, L.; Bladen, C.; Stys, P.K.; Zamponi, G.W. Differential modulation of NMDA and AMPA receptors by cellular prion protein and copper ions. Mol. Brain 2018, 11, 62. [Google Scholar] [CrossRef]
- Beraldo, F.H.; Arantes, C.P.; Santos, T.G.; Queiroz, N.G.; Young, K.; Rylett, R.J.; Markus, R.P.; Prado, M.A.; Martins, V.R. Role of alpha7 nicotinic acetylcholine receptor in calcium signaling induced by prion protein interaction with stress-inducible protein 1. J. Biol. Chem. 2010, 285, 36542–36550. [Google Scholar] [CrossRef]
- Plattner, F.; Angelo, M.; Giese, K.P. The roles of cyclin-dependent kinase 5 and glycogen synthase kinase 3 in tau hyperphosphorylation. J. Biol. Chem. 2006, 281, 25457–25465. [Google Scholar] [CrossRef] [PubMed]
- Vassar, R.; Bennett, B.D.; Babu-Khan, S.; Kahn, S.; Mendiaz, E.A.; Denis, P.; Teplow, D.B.; Ross, S.; Amarante, P.; Loeloff, R.; et al. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 1999, 286, 735–741. [Google Scholar] [CrossRef] [PubMed]
- Avila, J. Tau aggregation into fibrillar polymers: Taupathies. Febs Lett. 2000, 476, 89–92. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Evolution of the neuropathology of Alzheimer’s disease. Acta Neurol. Scand. Suppl. 1996, 165, 3–12. [Google Scholar] [CrossRef]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef]
- Hardy, J. The relationship between amyloid and tau. J. Mol. Neurosci. 2003, 20, 203–206. [Google Scholar] [CrossRef]
- Lambert, M.P.; Barlow, A.K.; Chromy, B.A.; Edwards, C.; Freed, R.; Liosatos, M.; Morgan, T.E.; Rozovsky, I.; Trommer, B.; Viola, K.L.; et al. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. USA 1998, 95, 6448–6453. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect Med. 2011, 1, a006189. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological staging of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Buee, L.; Delacourte, A. Comparative biochemistry of tau in progressive supranuclear palsy, corticobasal degeneration, FTDP-17 and Pick’s disease. Brain Pathol. 1999, 9, 681–693. [Google Scholar] [CrossRef]
- Umeda, Y.; Taniguchi, S.; Arima, K.; Piao, Y.S.; Takahashi, H.; Iwatsubo, T.; Mann, D.; Hasegawa, M. Alterations in human tau transcripts correlate with those of neurofilament in sporadic tauopathies. Neurosci. Lett. 2004, 359, 151–154. [Google Scholar] [CrossRef] [PubMed]
- Hoglinger, G.U.; Respondek, G.; Kovacs, G.G. New classification of tauopathies. Rev. Neurol. (Paris) 2018, 174, 664–668. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.G. Tauopathies. Handb Clin. Neurol. 2017, 145, 355–368. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Spillantini, M.G.; Jakes, R.; Rutherford, D.; Crowther, R.A. Multiple isoforms of human microtubule-associated protein tau: Sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron 1989, 3, 519–526. [Google Scholar] [CrossRef]
- Burack, M.A.; Halpain, S. Site-specific regulation of Alzheimer-like tau phosphorylation in living neurons. Neuroscience 1996, 72, 167–184. [Google Scholar] [CrossRef]
- Cruz, J.C.; Tsai, L.H. Cdk5 deregulation in the pathogenesis of Alzheimer’s disease. Trends Mol. Med. 2004, 10, 452–458. [Google Scholar] [CrossRef]
- Hooper, C.; Killick, R.; Lovestone, S. The GSK3 hypothesis of Alzheimer’s disease. J. Neurochem. 2008, 104, 1433–1439. [Google Scholar] [CrossRef]
- Ferrer, I.; Gomez-Isla, T.; Puig, B.; Freixes, M.; Ribe, E.; Dalfo, E.; Avila, J. Current advances on different kinases involved in tau phosphorylation, and implications in Alzheimer’s disease and tauopathies. Curr. Alzheimer Res. 2005, 2, 3–18. [Google Scholar] [CrossRef]
- Bulbarelli, A.; Lonati, E.; Cazzaniga, E.; Gregori, M.; Masserini, M. Pin1 affects Tau phosphorylation in response to Abeta oligomers. Mol. Cell. Neurosci. 2009, 42, 75–80. [Google Scholar] [CrossRef]
- De Felice, F.G.; Wu, D.; Lambert, M.P.; Fernandez, S.J.; Velasco, P.T.; Lacor, P.N.; Bigio, E.H.; Jerecic, J.; Acton, P.J.; Shughrue, P.J.; et al. Alzheimer’s disease-type neuronal tau hyperphosphorylation induced by A beta oligomers. Neurobiol. Aging 2008, 29, 1334–1347. [Google Scholar] [CrossRef]
- Dohler, F.; Sepulveda-Falla, D.; Krasemann, S.; Altmeppen, H.; Schluter, H.; Hildebrand, D.; Zerr, I.; Matschke, J.; Glatzel, M. High molecular mass assemblies of amyloid-beta oligomers bind prion protein in patients with Alzheimer’s disease. Brain 2014, 137, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Gunther, E.C.; Strittmatter, S.M. Beta-amyloid oligomers and cellular prion protein in Alzheimer’s disease. J. Mol. Med. (Berl) 2010, 88, 331–338. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Caetano, F.A.; Beraldo, F.H.; Hajj, G.N.; Guimaraes, A.L.; Jurgensen, S.; Wasilewska-Sampaio, A.P.; Hirata, P.H.; Souza, I.; Machado, C.F.; Wong, D.Y.; et al. Amyloid-beta oligomers increase the localization of prion protein at the cell surface. J. Neurochem. 2011, 117, 538–553. [Google Scholar] [CrossRef] [PubMed]
- Kellett, K.A.; Hooper, N.M. Prion protein and Alzheimer disease. Prion 2009, 3, 190–194. [Google Scholar] [CrossRef] [PubMed]
- Rushworth, J.V.; Griffiths, H.H.; Watt, N.T.; Hooper, N.M. Prion protein-mediated toxicity of amyloid-beta oligomers requires lipid rafts and the transmembrane LRP1. J. Biol. Chem. 2013, 288, 8935–8951. [Google Scholar] [CrossRef] [PubMed]
- Gimbel, D.A.; Nygaard, H.B.; Coffey, E.E.; Gunther, E.C.; Lauren, J.; Gimbel, Z.A.; Strittmatter, S.M. Memory impairment in transgenic Alzheimer mice requires cellular prion protein. J. Neurosci. 2010, 30, 6367–6374. [Google Scholar] [CrossRef]
- You, H.; Tsutsui, S.; Hameed, S.; Kannanaya kal, T.J.; Chen, L.; Xia, P.; Engbers, J.D.; Lipton, S.A.; Stys, P.K.; Zamponi, G.W. Abeta neurotoxicity depends on interactions between copper ions, prion protein, and N-methyl-D-aspartate receptors. Proc. Natl. Acad. Sci. USA 2012, 109, 1737–1742. [Google Scholar] [CrossRef]
- Kudo, W.; Lee, H.P.; Zou, W.Q.; Wang, X.; Perry, G.; Zhu, X.; Smith, M.A.; Petersen, R.B.; Lee, H.G. Cellular prion protein is essential for oligomeric amyloid-beta-induced neuronal cell death. Hum. Mol. Genet. 2012, 21, 1138–1144. [Google Scholar] [CrossRef]
- Barry, A.E.; Klyubin, I.; Mc Donald, J.M.; Mably, A.J.; Farrell, M.A.; Scott, M.; Walsh, D.M.; Rowan, M.J. Alzheimer’s disease brain-derived amyloid-beta-mediated inhibition of LTP in vivo is prevented by immunotargeting cellular prion protein. J. Neurosci. 2011, 31, 7259–7263. [Google Scholar] [CrossRef]
- Um, J.W.; Nygaard, H.B.; Heiss, J.K.; Kostylev, M.A.; Stagi, M.; Vortmeyer, A.; Wisniewski, T.; Gunther, E.C.; Strittmatter, S.M. Alzheimer amyloid-beta oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat. Neurosci. 2012, 15, 1227–1235. [Google Scholar] [CrossRef]
- Um, J.W.; Strittmatter, S.M. Amyloid-beta induced signaling by cellular prion protein and Fyn kinase in Alzheimer disease. Prion 2013, 7, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, A.; Zamponi, G.W.; Ferguson, S.S. Glutamate receptors function as scaffolds for the regulation of beta-amyloid and cellular prion protein signaling complexes. Mol. Brain 2015, 8, 18. [Google Scholar] [CrossRef] [PubMed]
- Mattei, V.; Garofalo, T.; Misasi, R.; Circella, A.; Manganelli, V.; Lucania, G.; Pavan, A.; Sorice, M. Prion protein is a component of the multimolecular signaling complex involved in T cell activation. Febs Lett. 2004, 560, 14–18. [Google Scholar] [CrossRef]
- Canu, N.; Filesi, I.; Pristera, A.; Ciotti, M.T.; Biocca, S. Altered intracellular distribution of PrPC and impairment of proteasome activity in tau overexpressing cortical neurons. J. Alzheimers Dis. 2011, 27, 603–613. [Google Scholar] [CrossRef] [PubMed]
- Haas, L.T.; Kostylev, M.A.; Strittmatter, S.M. Therapeutic molecules and endogenous ligands regulate the interaction between brain cellular prion protein (PrPC) and metabotropic glutamate receptor 5 (mGluR5). J. Biol. Chem. 2014, 289, 28460–28477. [Google Scholar] [CrossRef] [PubMed]
- Vilches, S.; Vergara, C.; Nicolas, O.; Sanclimens, G.; Merino, S.; Varon, S.; Acosta, G.A.; Albericio, F.; Royo, M.; Del Rio, J.A.; et al. Neurotoxicity of prion peptides mimicking the central domain of the cellular prion protein. PLoS ONE 2013, 8, e70881. [Google Scholar] [CrossRef]
- Goniotaki, D.; Lakkaraju, A.K.K.; Shrivastava, A.N.; Bakirci, P.; Sorce, S.; Senatore, A.; Marpakwar, R.; Hornemann, S.; Gasparini, F.; Triller, A.; et al. Inhibition of group-I metabotropic glutamate receptors protects against prion toxicity. PLoS Pathog. 2017, 13, e1006733. [Google Scholar] [CrossRef]
- Hernandez-Rapp, J.; Martin-Lanneree, S.; Hirsch, T.Z.; Launay, J.M.; Mouillet-Richard, S. Hijacking PrP(c)-dependent signal transduction: When prions impair Abeta clearance. Front. Aging Neurosci. 2014, 6, 25. [Google Scholar] [CrossRef]
- Larson, M.; Sherman, M.A.; Amar, F.; Nuvolone, M.; Schneider, J.A.; Bennett, D.A.; Aguzzi, A.; Lesne, S.E. The complex PrP(c)-Fyn couples human oligomeric Abeta with pathological tau changes in Alzheimer’s disease. J. Neurosci. 2012, 32, 16857–16871. [Google Scholar] [CrossRef]
- Kessels, H.W.; Nguyen, L.N.; Nabavi, S.; Malinow, R. The prion protein as a receptor for amyloid-beta. Nature 2010, 466, E3–E4, discussion E4-5. [Google Scholar] [CrossRef]
- Balducci, C.; Beeg, M.; Stravalaci, M.; Bastone, A.; Sclip, A.; Biasini, E.; Tapella, L.; Colombo, L.; Manzoni, C.; Borsello, T.; et al. Synthetic amyloid-beta oligomers impair long-term memory independently of cellular prion protein. Proc. Natl. Acad. Sci. USA 2010, 107, 2295–2300. [Google Scholar] [CrossRef] [PubMed]
- Calella, A.M.; Farinelli, M.; Nuvolone, M.; Mirante, O.; Moos, R.; Falsig, J.; Mansuy, I.M.; Aguzzi, A. Prion protein and Abeta-related synaptic toxicity impairment. EMBO Mol. Med. 2010, 2, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Younan, N.D.; Sarell, C.J.; Davies, P.; Brown, D.R.; Viles, J.H. The cellular prion protein traps Alzheimer’s Abeta in an oligomeric form and disassembles amyloid fibers. FASEB J. 2013, 27, 1847–1858. [Google Scholar] [CrossRef] [PubMed]
- Pietri, M.; Dakowski, C.; Hannaoui, S.; Alleaume-Butaux, A.; Hernandez-Rapp, J.; Ragagnin, A.; Mouillet-Richard, S.; Haik, S.; Bailly, Y.; Peyrin, J.M.; et al. PDK1 decreases TACE-mediated alpha-secretase activity and promotes disease progression in prion and Alzheimer’s diseases. Nat. Med. 2013, 19, 1124–1131. [Google Scholar] [CrossRef]
- Resenberger, U.K.; Winklhofer, K.F.; Tatzelt, J. Cellular prion protein mediates toxic signaling of amyloid beta. Neurodegener. Dis. 2012, 10, 298–300. [Google Scholar] [CrossRef]
- Chen, R.J.; Chang, W.W.; Lin, Y.C.; Cheng, P.L.; Chen, Y.R. Alzheimer’s Amyloid-beta Oligomers Rescue Cellular Prion Protein Induced Tau Reduction via Fyn Pathways. ACS Chem. Neurosci. 2013. [Google Scholar] [CrossRef]
- Schmitz, M.; Wulf, K.; Signore, S.C.; Schulz-Schaeffer, W.J.; Kermer, P.; Bahr, M.; Wouters, F.S.; Zafar, S.; Zerr, I. Impact of the cellular prion protein on amyloid-beta and 3PO-tau processing. J. Alzheimers Dis. 2014, 38, 551–565. [Google Scholar] [CrossRef]
- Ishizawa, K.; Mitsufuji, T.; Shioda, K.; Kobayashi, A.; Komori, T.; Nakazato, Y.; Kitamoto, T.; Araki, N.; Yamamoto, T.; Sasaki, A. An autopsy report of three kindred in a Gerstmann-Straussler-Scheinker disease P105L family with a special reference to prion protein, tau, and beta-amyloid. Brain Behav. 2018, 8, e01117. [Google Scholar] [CrossRef]
- Matamoros-Angles, A.; Gayosso, L.M.; Richaud-Patin, Y.; di Domenico, A.; Vergara, C.; Hervera, A.; Sousa, A.; Fernandez-Borges, N.; Consiglio, A.; Gavin, R.; et al. iPS Cell Cultures from a Gerstmann-Straussler-Scheinker Patient with the Y218N PRNP Mutation Recapitulate tau Pathology. Mol. Neurobiol. 2017. [Google Scholar] [CrossRef]
- Jansen, C.; Parchi, P.; Capellari, S.; Strammiello, R.; Dopper, E.G.; van Swieten, J.C.; Kamphorst, W.; Rozemuller, A.J. A second case of Gerstmann-Straussler-Scheinker disease linked to the G131V mutation in the prion protein gene in a Dutch patient. J. Neuropathol. Exp. Neurol. 2011, 70, 698–702. [Google Scholar] [CrossRef]
- Llorens, F.; Ansoleaga, B.; Garcia-Esparcia, P.; Zafar, S.; Grau-Rivera, O.; Lopez-Gonzalez, I.; Blanco, R.; Carmona, M.; Yague, J.; Nos, C.; et al. PrP mRNA and protein expression in brain and PrP in CSF in Creutzfeldt-Jakob disease MM1 and VV2. Prion 2013, 7. [Google Scholar] [CrossRef] [PubMed]
- Bradley, C.A.; Peineau, S.; Taghibiglou, C.; Nicolas, C.S.; Whitcomb, D.J.; Bortolotto, Z.A.; Kaang, B.K.; Cho, K.; Wang, Y.T.; Collingridge, G.L. A pivotal role of GSK-3 in synaptic plasticity. Front. Mol. Neurosci. 2012, 5, 13. [Google Scholar] [CrossRef] [PubMed]
- Guglielmotto, M.; Giliberto, L.; Tamagno, E.; Tabaton, M. Oxidative stress mediates the pathogenic effect of different Alzheimer’s disease risk factors. Front. Aging Neurosci. 2010, 2, 3. [Google Scholar] [CrossRef] [PubMed]
- Wadsworth, J.D.; Hill, A.F.; Beck, J.A.; Collinge, J. Molecular and clinical classification of human prion disease. Br. Med. Bull. 2003, 66, 241–254. [Google Scholar] [CrossRef]
- McNeill, A. A molecular analysis of prion protein expression in Alzheimer’s disease. Mcgill J. Med. 2004, 8, 7–14. [Google Scholar]
- Rezaie, P.; Pontikis, C.C.; Hudson, L.; Cairns, N.J.; Lantos, P.L. Expression of cellular prion protein in the frontal and occipital lobe in Alzheimer’s disease, diffuse Lewy body disease, and in normal brain: An immunohistochemical study. J. Histochem. Cytochem. 2005, 53, 929–940. [Google Scholar] [CrossRef]
- Watt, N.T.; Taylor, D.R.; Kerrigan, T.L.; Griffiths, H.H.; Rushworth, J.V.; Whitehouse, I.J.; Hooper, N.M. Prion protein facilitates uptake of zinc into neuronal cells. Nat. Commun. 2012, 3, 1134. [Google Scholar] [CrossRef]
- Lee, J.Y.; Cole, T.B.; Palmiter, R.D.; Suh, S.W.; Koh, J.Y. Contribution by synaptic zinc to the gender-disparate plaque formation in human Swedish mutant APP transgenic mice. Proc. Natl. Acad. Sci. USA 2002, 99, 7705–7710. [Google Scholar] [CrossRef]
- Bush, A.I.; Pettingell, W.H.; Multhaup, G.; d Paradis, M.; Vonsattel, J.P.; Gusella, J.F.; Beyreuther, K.; Masters, C.L.; Tanzi, R.E. Rapid induction of Alzheimer A beta amyloid formation by zinc. Science 1994, 265, 1464–1467. [Google Scholar] [CrossRef]
- Deshpande, A.; Kawai, H.; Metherate, R.; Glabe, C.G.; Busciglio, J. A role for synaptic zinc in activity-dependent Abeta oligomer formation and accumulation at excitatory synapses. J. Neurosci. 2009, 29, 4004–4015. [Google Scholar] [CrossRef]
- Whitehouse, I.J.; Miners, J.S.; Glennon, E.B.; Kehoe, P.G.; Love, S.; Kellett, K.A.; Hooper, N.M. Prion protein is decreased in Alzheimer’s brain and inversely correlates with BACE1 activity, amyloid-beta levels and Braak stage. PLoS ONE 2013, 8, e59554. [Google Scholar] [CrossRef]
- Beyer, N.; Coulson, D.T.; Heggarty, S.; Ravid, R.; Hellemans, J.; Irvine, G.B.; Johnston, J.A. Zinc transporter mRNA levels in Alzheimer’s disease postmortem brain. J. Alzheimers Dis. 2012, 29, 863–873. [Google Scholar] [CrossRef]
- Li, S.H.; Li, X.J. Huntingtin-protein interactions and the pathogenesis of Huntington’s disease. Trends Genet. 2004, 20, 146–154. [Google Scholar] [CrossRef]
- Landles, C.; Bates, G.P. Huntingtin and the molecular pathogenesis of Huntington’s disease. Fourth in molecular medicine review series. EMBO Rep. 2004, 5, 958–963. [Google Scholar] [CrossRef]
- Saudou, F.; Humbert, S. The Biology of Huntingtin. Neuron 2016, 89, 910–926. [Google Scholar] [CrossRef]
- De la Monte, S.M.; Vonsattel, J.P.; Richardson, E.P., Jr. Morphometric demonstration of atrophic changes in the cerebral cortex, white matter, and neostriatum in Huntington’s disease. J. Neuropathol. Exp. Neurol. 1988, 47, 516–525. [Google Scholar] [CrossRef]
- Vonsattel, J.P.; DiFiglia, M. Huntington disease. J. Neuropathol. Exp. Neurol. 1998, 57, 369–384. [Google Scholar] [CrossRef]
- Strong, T.V.; Tagle, D.A.; Valdes, J.M.; Elmer, L.W.; Boehm, K.; Swaroop, M.; Kaatz, K.W.; Collins, F.S.; Albin, R.L. Widespread expression of the human and rat Huntington’s disease gene in brain and nonneural tissues. Nat. Genet. 1993, 5, 259–265. [Google Scholar] [CrossRef]
- Lee, F.J.; Liu, F. Genetic factors involved in the pathogenesis of Parkinson’s disease. Brain Res. Rev. 2008, 58, 354–364. [Google Scholar] [CrossRef]
- Bartels, A.L.; Leenders, K.L. Parkinson’s disease: The syndrome, the pathogenesis and pathophysiology. Cortex 2009, 45, 915–921. [Google Scholar] [CrossRef]
- Goedert, M.; Spillantini, M.G.; Del Tredici, K.; Braak, H. 100 years of Lewy pathology. Nat. Rev. Neurol. 2013, 9, 13–24. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Perluigi, M.; Sultana, R. Oxidative stress in Alzheimer’s disease brain: New insights from redox proteomics. Eur. J. Pharm. 2006, 545, 39–50. [Google Scholar] [CrossRef]
- Toulorge, D.; Schapira, A.H.; Hajj, R. Molecular changes in the postmortem parkinsonian brain. J. Neurochem. 2016. [Google Scholar] [CrossRef]
- Ribeiro, F.M.; Vieira, L.B.; Pires, R.G.; Olmo, R.P.; Ferguson, S.S. Metabotropic glutamate receptors and neurodegenerative diseases. Pharm. Res. 2017, 115, 179–191. [Google Scholar] [CrossRef]
- Lewerenz, J.; Maher, P. Chronic Glutamate Toxicity in Neurodegenerative Diseases-What is the Evidence? Front. Neurosci. 2015, 9, 469. [Google Scholar] [CrossRef]
- Choi, D.W. Glutamate neurotoxicity and diseases of the nervous system. Neuron 1988, 1, 623–634. [Google Scholar] [CrossRef]
- Balazs, R.; Miller, S.; Romano, C.; de Vries, A.; Chun, Y.; Cotman, C.W. Metabotropic glutamate receptor mGluR5 in astrocytes: Pharmacological properties and agonist regulation. J. Neurochem. 1997, 69, 151–163. [Google Scholar] [CrossRef]
- Amalric, M. Targeting metabotropic glutamate receptors (mGluRs) in Parkinson’s disease. Curr. Opin. Pharm. 2015, 20, 29–34. [Google Scholar] [CrossRef]
- Breysse, N.; Baunez, C.; Spooren, W.; Gasparini, F.; Amalric, M. Chronic but not acute treatment with a metabotropic glutamate 5 receptor antagonist reverses the akinetic deficits in a rat model of parkinsonism. J. Neurosci. 2002, 22, 5669–5678. [Google Scholar] [CrossRef]
- Coccurello, R.; Breysse, N.; Amalric, M. Simultaneous blockade of adenosine A2A and metabotropic glutamate mGlu5 receptors increase their efficacy in reversing Parkinsonian deficits in rats. Neuropsychopharmacology 2004, 29, 1451–1461. [Google Scholar] [CrossRef]
- Phillips, J.M.; Lam, H.A.; Ackerson, L.C.; Maidment, N.T. Blockade of mGluR glutamate receptors in the subthalamic nucleus ameliorates motor asymmetry in an animal model of Parkinson’s disease. Eur. J. Neurosci. 2006, 23, 151–160. [Google Scholar] [CrossRef]
- Ossowska, K.; Konieczny, J.; Wardas, J.; Pietraszek, M.; Kuter, K.; Wolfarth, S.; Pilc, A. An influence of ligands of metabotropic glutamate receptor subtypes on parkinsonian-like symptoms and the striatopallidal pathway in rats. Amino Acids 2007, 32, 179–188. [Google Scholar] [CrossRef]
- Thakur, A.K.; Jayaraman, M.; Mishra, R.; Thakur, M.; Chellgren, V.M.; Byeon, I.J.; Anjum, D.H.; Kodali, R.; Creamer, T.P.; Conway, J.F.; et al. Polyglutamine disruption of the huntingtin exon 1 N terminus triggers a complex aggregation mechanism. Nat. Struct Mol. Biol. 2009, 16, 380–389. [Google Scholar] [CrossRef]
- Fernandez-Nogales, M.; Cabrera, J.R.; Santos-Galindo, M.; Hoozemans, J.J.; Ferrer, I.; Rozemuller, A.J.; Hernandez, F.; Avila, J.; Lucas, J.J. Huntington’s disease is a four-repeat tauopathy with tau nuclear rods. Nat. Med. 2014, 20, 881–885. [Google Scholar] [CrossRef]
- Carmichael, J.; Sugars, K.L.; Bao, Y.P.; Rubinsztein, D.C. Glycogen synthase kinase-3beta inhibitors prevent cellular polyglutamine toxicity caused by the Huntington’s disease mutation. J. Biol. Chem. 2002, 277, 33791–33798. [Google Scholar] [CrossRef]
- Surgucheva, I.; Sharov, V.S.; Surguchov, A. gamma-Synuclein: Seeding of alpha-synuclein aggregation and transmission between cells. Biochemistry 2012, 51, 4743–4754. [Google Scholar] [CrossRef]
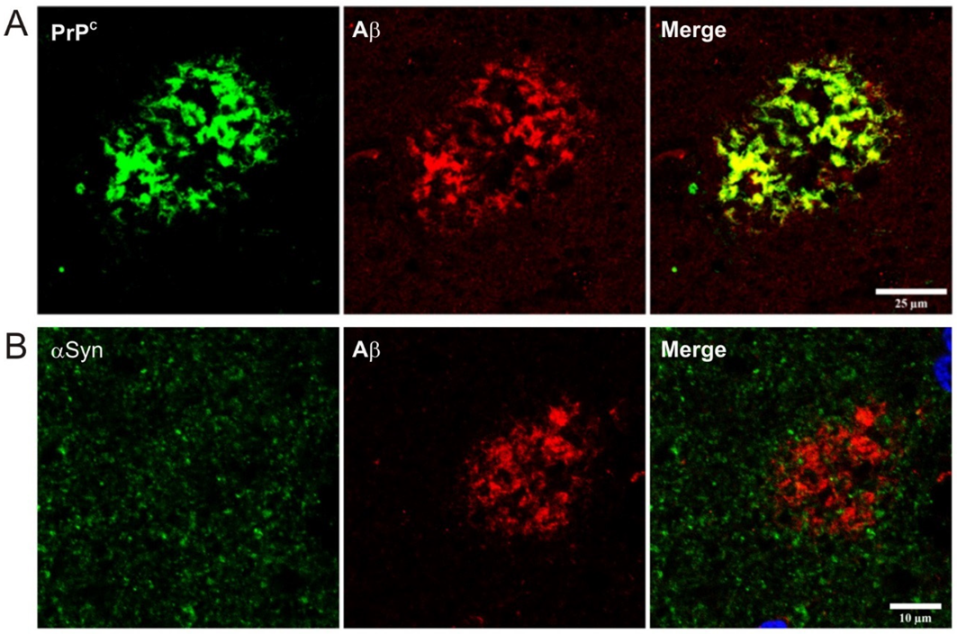
- Urrea, L.; Ferrer, I.; Gavin, R.; Del Rio, J.A. The cellular prion protein (PrPC) as neuronal receptor for alpha-synuclein. Prion 2017, 11, 226–233. [Google Scholar] [CrossRef][Green Version]
- Mao, X.; Ou, M.T.; Karuppagounder, S.S.; Kam, T.I.; Yin, X.; Xiong, Y.; Ge, P.; Umanah, G.E.; Brahmachari, S.; Shin, J.H.; et al. Pathological alpha-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science 2016, 353. [Google Scholar] [CrossRef]
- De Cecco, E.; Legname, G. The role of the prion protein in the internalization of alpha-synuclein amyloids. Prion 2018, 12, 23–27. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Finding | Model | Role of PrPC | Key Reference(s) |
|---|---|---|---|---|
| Alzheimer’s disease | Inhibition of BACE1 | In vitro | Decreases Aβ production | [158,159] |
| Binding of PrPN1 to Aβ | In vitro | Blocks transformation into ADDLs | [160,161] | |
| Binding to STI1 | In vitro | Decreases ADDLs toxicity | [162] | |
| Binding to Zn2+ | In vitro | Decreases Aβ aggregation | [163] | |
| Binding of PrPN1 to ADDLs | In vivo | Decreases ADDLs toxicity | [164] | |
| Prevention of cell death by Aβ | In vivo | Decreases caspase-3 and Bax/Bcl2 levels | [165] | |
| Increase in PrPN1 production in brain patients | Human samples | Blocks transformation into ADDLs | [160] | |
| Increase in brain regions prone to oxidative stress | Human samples | SOD and GR activity regulation | [166] | |
| Increase in initial stages of the disease | Human samples | Downregulates tau levels | [167] | |
| Huntington’s disease | Increase in proteasome activity | In vitro | Decreases HTT aggregation and toxicity | [139] |
| Amyotrophic lateral sclerosis | Induction of neuronal and glial survival signaling | In vivo | Antioxidant | [168] |
| Nonspecific disorder | Binding to Cu2+ | In vitro | Antioxidant | [75] |
| Modulation of SOD | In vitro | Antioxidant | [86] | |
| Modulation of GR | In vitro | Antioxidant | [83] | |
| Modulation of Bax function | In vitro | Antiapoptotic | [95] | |
| Regulation of Ca2+ homeostasis | In vitro | Reduces excitotoxicity | [101] | |
| Inhibition of NMDAR | In vitro | Reduces excitotoxicity | [108,109,169] | |
| PrP113-128 peptide | In vitro | Activates cAMP/PKA and MEK/Erk pathways | [116] | |
| PrP-Fc signaling | In vitro | Activates PI3K/Akt pathway | [75] | |
| Binding to STI1 | In vivo | Inhibits GSK3β activity and activates 7nAChR. All together induces neuroprotective signals. | [120,122,123,170] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gavín, R.; Lidón, L.; Ferrer, I.; del Río, J.A. The Quest for Cellular Prion Protein Functions in the Aged and Neurodegenerating Brain. Cells 2020, 9, 591. https://doi.org/10.3390/cells9030591
Gavín R, Lidón L, Ferrer I, del Río JA. The Quest for Cellular Prion Protein Functions in the Aged and Neurodegenerating Brain. Cells. 2020; 9(3):591. https://doi.org/10.3390/cells9030591
Chicago/Turabian StyleGavín, Rosalina, Laia Lidón, Isidre Ferrer, and José Antonio del Río. 2020. "The Quest for Cellular Prion Protein Functions in the Aged and Neurodegenerating Brain" Cells 9, no. 3: 591. https://doi.org/10.3390/cells9030591
APA StyleGavín, R., Lidón, L., Ferrer, I., & del Río, J. A. (2020). The Quest for Cellular Prion Protein Functions in the Aged and Neurodegenerating Brain. Cells, 9(3), 591. https://doi.org/10.3390/cells9030591