
Protein Kinase C Isozymes and Autophagy during Neurodegenerative Disease Progression


Abstract
1. Introduction
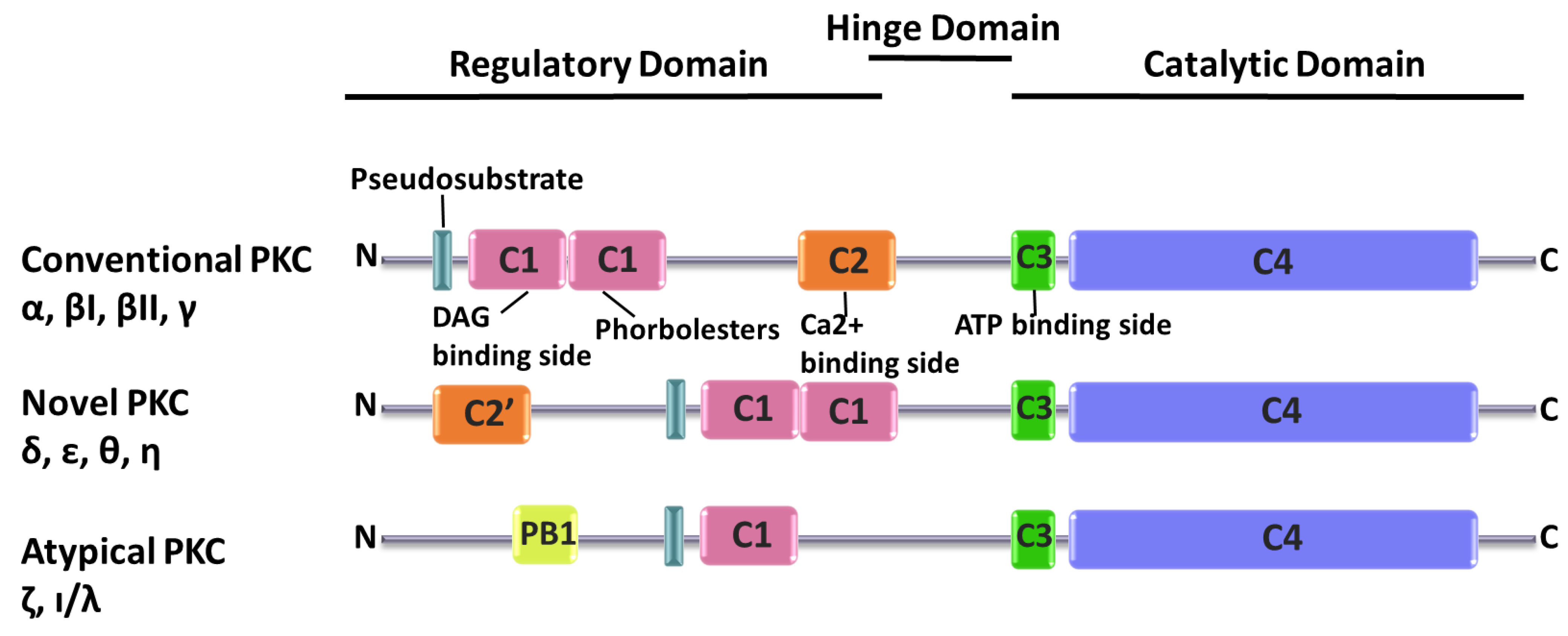
2. PKC Superfamily
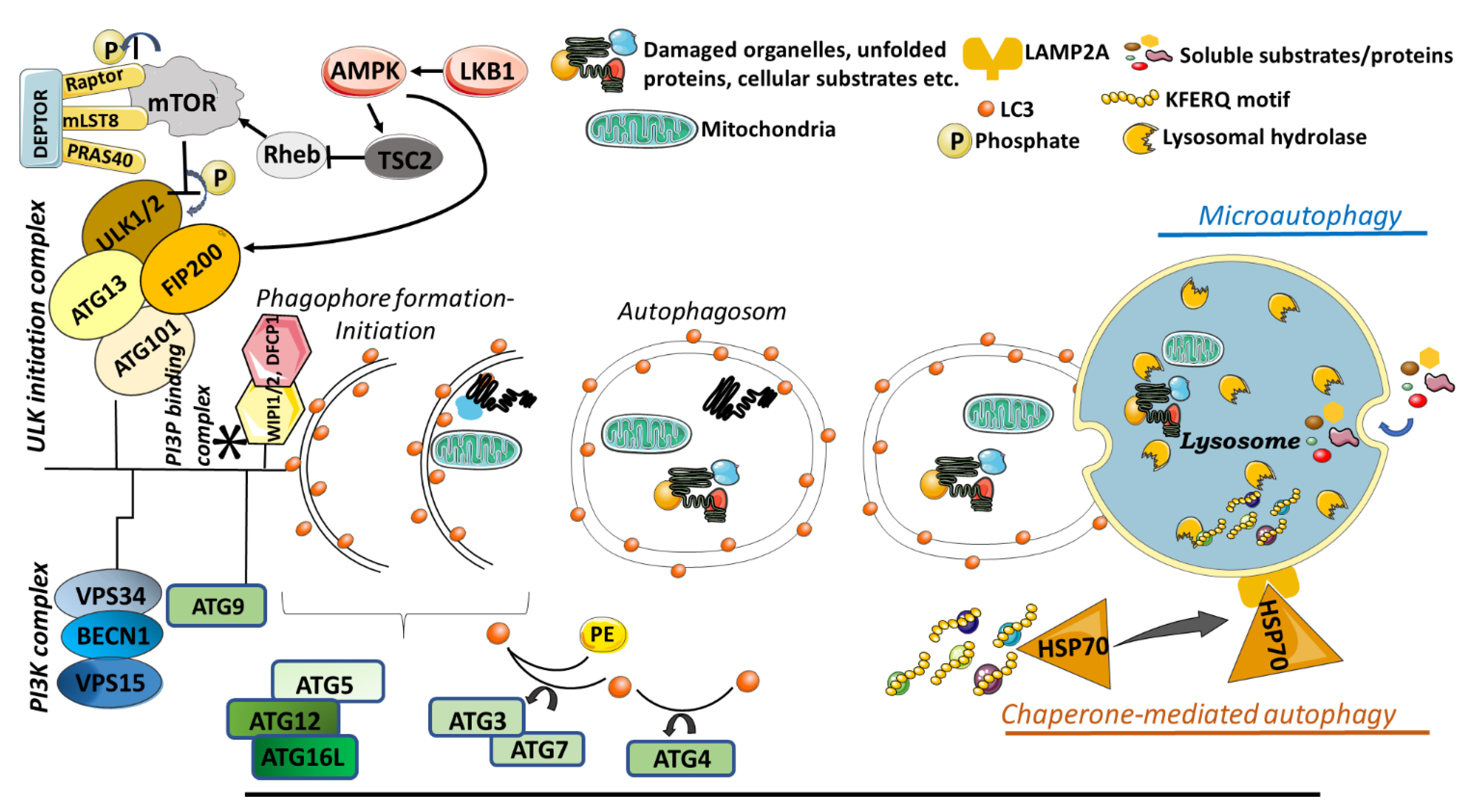
3. Autophagy
3.1. Macroautophagy
3.2. Microautophagy
3.3. Chaperone Mediated Autophagy
3.4. Selective Autophagy
3.4.1. Mitophagy
3.4.2. Lipophagy
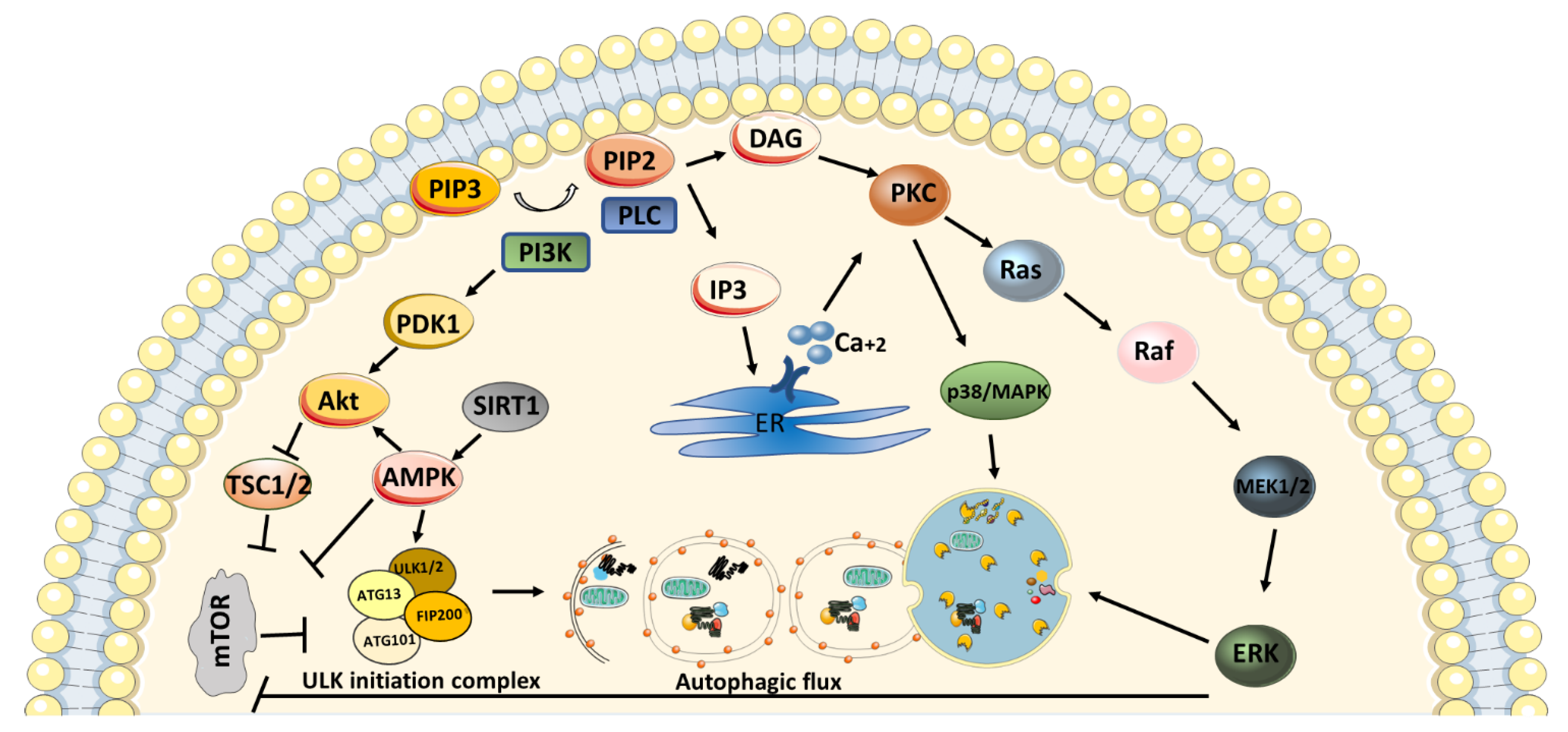
4. PKC in Autophagy Mechanism
5. Autophagy in Neurodegenerative Diseases
5.1. Parkinson’s Disease
5.2. Alzheimer’s Disease
5.3. Amyotrophic Lateral Sclerosis (ALS)
5.4. Huntington’s Disease
5.5. Multiple Sclerosis (MS)
6. Roles of PKC Isozymes in Neurodegenerative Disease Progression via Autophagy Mechanism
7. Polyphenols Act as Neuroprotective Agent During PKC and Autophagy Regulation
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kishimoto, A.; Takai, Y.; Mori, T.; Kikkawa, U.; Nishizuka, Y. Activation of calcium and phospholipid-dependent protein kinase by diacylglycerol, its possible relation to phosphatidylinositol turnover. J. Biol. Chem. 1980, 255, 2273–2276. [Google Scholar] [PubMed]
- Crawley, O.; Giles, A.C.; Desbois, M.; Kashyap, S.; Birnbaum, R.; Grill, B. A MIG-15/JNK-1 MAP kinase cascade opposes RPM-1 signaling in synapse formation and learning. PLoS Genet. 2017, 13, e1007095. [Google Scholar] [CrossRef] [PubMed]
- Do Van, B.; Gouel, F.; Jonneaux, A.; Timmerman, K.; Gelé, P.; Pétrault, M.; Bastide, M.; Laloux, C.; Moreau, C.; Bordet, R. Ferroptosis, a newly characterized form of cell death in Parkinson’s disease that is regulated by PKC. Neurobiol. Dis. 2016, 94, 169–178. [Google Scholar] [CrossRef]
- Gordon, R.; Singh, N.; Lawana, V.; Ghosh, A.; Harischandra, D.S.; Jin, H.; Hogan, C.; Sarkar, S.; Rokad, D.; Panicker, N. Protein kinase Cδ upregulation in microglia drives neuroinflammatory responses and dopaminergic neurodegeneration in experimental models of Parkinson’s disease. Neurobiol. Dis. 2016, 93, 96–114. [Google Scholar] [CrossRef]
- Jha, S.K.; Jha, N.K.; Kar, R.; Ambasta, R.K.; Kumar, P. p38 MAPK and PI3K/AKT signalling cascades inParkinson’s disease. Int. J. Mol. Cell. Med. 2015, 4, 67. [Google Scholar]
- Kirouac, L.; Rajic, A.J.; Cribbs, D.H.; Padmanabhan, J. Activation of Ras-ERK signaling and GSK-3 by amyloid precursor protein and amyloid beta facilitates neurodegeneration in Alzheimer’s disease. Eneuro 2017, 4. [Google Scholar] [CrossRef]
- Zisopoulou, S.; Asimaki, O.; Leondaritis, G.; Vasilaki, A.; Sakellaridis, N.; Pitsikas, N.; Mangoura, D. PKC-epsilon activation is required for recognition memory in the rat. Behav. Brain Res. 2013, 253, 280–289. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Mariño, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Oral, O.; Yedier, O.; Kilic, S.; Gozuacik, D. Involvement of autophagy in T cell biology. Histol. Histopathol. 2017, 32, 11–20. [Google Scholar]
- Bento, C.F.; Ashkenazi, A.; Jimenez-Sanchez, M.; Rubinsztein, D.C. The Parkinson’s disease-associated genes ATP13A2 and SYT11 regulate autophagy via a common pathway. Nat. Commun. 2016, 7, 11803. [Google Scholar] [CrossRef]
- Giaime, E.; Tong, Y.; Wagner, L.K.; Yuan, Y.; Huang, G.; Shen, J. Age-dependent dopaminergic neurodegeneration and impairment of the autophagy-lysosomal pathway in LRRK-deficient mice. Neuron 2017, 96, 796–807. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Yu, W.H.; Kumar, A.; Lee, S.; Mohan, P.S.; Peterhoff, C.M.; Wolfe, D.M.; Martinez-Vicente, M.; Massey, A.C.; Sovak, G. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 2010, 141, 1146–1158. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Han, Q.; Zhao, X.; Song, J.; Cheng, Y.; Fang, Z.; Ouyang, Y.; Yuan, W.E.; Fan, C. 3D melatonin nerve scaffold reduces oxidative stress and inflammation and increases autophagy in peripheral nerve regeneration. J. Pineal Res. 2018, 65, e12516. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.-i.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880. [Google Scholar] [CrossRef]
- Menzies, F.M.; Fleming, A.; Caricasole, A.; Bento, C.F.; Andrews, S.P.; Ashkenazi, A.; Füllgrabe, J.; Jackson, A.; Sanchez, M.J.; Karabiyik, C. Autophagy and neurodegeneration: Pathogenic mechanisms and therapeutic opportunities. Neuron 2017, 93, 1015–1034. [Google Scholar] [CrossRef]
- Adachi, N.; Kobayashi, T.; Takahashi, H.; Kawasaki, T.; Shirai, Y.; Ueyama, T.; Matsuda, T.; Seki, T.; Sakai, N.; Saito, N. Enzymological analysis of mutant protein kinase Cγ causing spinocerebellar ataxia type 14 and dysfunction in Ca2+ homeostasis. J. Biol. Chem. 2008, 283, 19854–19863. [Google Scholar] [CrossRef]
- Alkon, D.L.; Sun, M.-K.; Nelson, T.J. PKC signaling deficits: A mechanistic hypothesis for the origins of Alzheimer’s disease. Trends Pharmacol. Sci. 2007, 28, 51–60. [Google Scholar] [CrossRef]
- Choi, D.-S.; Wang, D.; Yu, G.-Q.; Zhu, G.; Kharazia, V.N.; Paredes, J.P.; Chang, W.S.; Deitchman, J.K.; Mucke, L.; Messing, R.O. PKCε increases endothelin converting enzyme activity and reduces amyloid plaque pathology in transgenic mice. Proc. Natl. Acad. Sci. USA 2006, 103, 8215–8220. [Google Scholar] [CrossRef]
- Chopra, R.; Wasserman, A.H.; Pulst, S.M.; De Zeeuw, C.I.; Shakkottai, V.G. Protein kinase C activity is a protective modifier of Purkinje neuron degeneration in cerebellar ataxia. Hum. Mol. Genet. 2018, 27, 1396–1410. [Google Scholar] [CrossRef]
- Doran, G.; Davies, K.E.; Talbot, K. Activation of mutant protein kinase Cγ leads to aberrant sequestration and impairment of its cellular function. Biochem. Biophys. Res. Commun. 2008, 372, 447–453. [Google Scholar] [CrossRef]
- Hongpaisan, J.; Sun, M.-K.; Alkon, D.L. PKC ε activation prevents synaptic loss, Aβ elevation, and cognitive deficits in Alzheimer’s disease transgenic mice. J. Neurosci. 2011, 31, 630–643. [Google Scholar] [CrossRef] [PubMed]
- Isagawa, T.; Mukai, H.; Oishi, K.; Taniguchi, T.; Hasegawa, H.; Kawamata, T.; Tanaka, C.; Ono, Y. Dual effects of PKNα and protein kinase C on phosphorylation of tau protein by glycogen synthase kinase-3β. Biochem. Biophys. Res. Commun. 2000, 273, 209–212. [Google Scholar] [CrossRef] [PubMed]
- Khan, T.K.; Sen, A.; Hongpaisan, J.; Lim, C.S.; Nelson, T.J.; Alkon, D.L. PKCε deficits in Alzheimer’s disease brains and skin fibroblasts. J. Alzheimer’s Dis. 2015, 43, 491–509. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Weng, Y.-C.; Wu, Y.-C.; Huang, Y.-T.; Chou, W.-H. PKCε phosphorylation regulates the mitochondrial translocation of ATF2 in ischemia-induced neurodegeneration. Bmc Neurosci. 2018, 19, 76. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Boo, J.H.; Jung, M.W.; Dai Park, S.; Kim, Y.H.; Kim, S.U.; Mook-Jung, I. Amyloid beta peptide directly inhibits PKC activation. Mol. Cell. Neurosci. 2004, 26, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Nelson, T.J.; Cui, C.; Luo, Y.; Alkon, D.L. Reduction of β-amyloid levels by novel protein kinase Cϵ activators. J. Biol. Chem. 2009, 284, 34514–34521. [Google Scholar] [CrossRef]
- Sen, A.; Nelson, T.J.; Alkon, D.L.; Hongpaisan, J. Loss in PKC epsilon causes downregulation of MnSOD and BDNF expression in neurons of Alzheimer’s disease hippocampus. J. Alzheimer’s Dis. 2018, 63, 1173–1189. [Google Scholar] [CrossRef]
- Williams, A.D.; Jung, S.; Poolos, N.P. Protein kinase C bidirectionally modulates Ih and hyperpolarization-activated cyclic nucleotide-gated (HCN) channel surface expression in hippocampal pyramidal neurons. J. Physiol. 2015, 593, 2779–2792. [Google Scholar] [CrossRef]
- Wong, M.M.; Hoekstra, S.D.; Vowles, J.; Watson, L.M.; Fuller, G.; Németh, A.H.; Cowley, S.A.; Ansorge, O.; Talbot, K.; Becker, E.B. Neurodegeneration in SCA14 is associated with increased PKCγ kinase activity, mislocalization and aggregation. Acta Neuropathol. Commun. 2018, 6, 99. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.-P. The mechanism of protein kinase C activation. Trends Neurosci. 1989, 12, 425–432. [Google Scholar] [CrossRef]
- Newton, A.C. Protein kinase C: Structure, function, and regulation. J. Biol. Chem. 1995, 270, 28495–28498. [Google Scholar] [CrossRef] [PubMed]
- Schultz, A.; Ling, M.; Larsson, C. Identification of an amino acid residue in the protein kinase C C1b domain crucial for its localization to the Golgi network. J. Biol. Chem. 2004, 279, 31750–31760. [Google Scholar] [CrossRef]
- Johnson, J.E.; Giorgione, J.; Newton, A.C. The C1 and C2 domains of protein kinase C are independent membrane targeting modules, with specificity for phosphatidylserine conferred by the C1 domain. Biochemistry 2000, 39, 11360–11369. [Google Scholar] [CrossRef] [PubMed]
- Ramos, K.; Reyes-Reyes, E.; Nanez, A. Overview of Receptor Systems. In Comprehensive Toxicology; Elsevier Press: Amsterdam, The Netherlands, 2018; pp. 8–17. [Google Scholar]
- Yang, Q.; Langston, J.C.; Tang, Y.; Kiani, M.F.; Kilpatrick, L.E. The Role of Tyrosine Phosphorylation of Protein Kinase C Delta in Infection and Inflammation. Int. J. Mol. Sci. 2019, 20, 1498. [Google Scholar] [CrossRef] [PubMed]
- De Duve, C.; Pressman, B.C.; Gianetto, R.; Wattiaux, R.; Appelmans, F. Tissue fractionation studies. 6. Intracellular distribution patterns of enzymes in rat-liver tissue. Biochem. J. 1955, 60, 604–617. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.E.; Farquhar, M.G. Lysosome function in the regulation of the secretory process in cells of the anterior pituitary gland. J. Cell Biol. 1966, 31, 319–347. [Google Scholar] [CrossRef]
- Majeski, A.E.; Dice, J.F. Mechanisms of chaperone-mediated autophagy. Int. J. Biochem. Cell Biol. 2004, 36, 2435–2444. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Klionsky, D.J. Autophagy wins the 2016 Nobel Prize in Physiology or Medicine: Breakthroughs in baker’s yeast fuel advances in biomedical research. Proc. Natl. Acad. Sci. USA 2017, 114, 201–205. [Google Scholar] [CrossRef]
- Jewell, J.L.; Guan, K.-L. Nutrient signaling to mTOR and cell growth. Trends Biochem. Sci. 2013, 38, 233–242. [Google Scholar] [CrossRef]
- Morita, M.; Gravel, S.-P.; Hulea, L.; Larsson, O.; Pollak, M.; St-Pierre, J.; Topisirovic, I. mTOR coordinates protein synthesis, mitochondrial activity and proliferation. Cell Cycle 2015, 14, 473–480. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR signaling in growth, metabolism, and disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed]
- Angliker, N.; Rüegg, M.A. In vivo evidence for mTORC2-mediated actin cytoskeleton rearrangement in neurons. Bioarchitecture 2013, 3, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Jacinto, E.; Loewith, R.; Schmidt, A.; Lin, S.; Rüegg, M.A.; Hall, A.; Hall, M.N. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat. Cell Biol. 2004, 6, 1122. [Google Scholar] [CrossRef] [PubMed]
- Sen, B.; Xie, Z.; Case, N.; Thompson, W.R.; Uzer, G.; Styner, M.; Rubin, J. mTORC2 regulates mechanically induced cytoskeletal reorganization and lineage selection in marrow-derived mesenchymal stem cells. J. Bone Miner. Res. 2014, 29, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Alers, S.; Löffler, A.S.; Wesselborg, S.; Stork, B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: Cross talk, shortcuts, and feedbacks. Mol. Cell. Biol. 2012, 32, 2–11. [Google Scholar] [CrossRef]
- Kuma, A.; Mizushima, N. Physiological role of autophagy as an intracellular recycling system: With an emphasis on nutrient metabolism. Semin. Cell Dev. Biol. 2010, 21, 683–690. [Google Scholar] [CrossRef]
- Devereaux, K.; Dall’Armi, C.; Alcazar-Roman, A.; Ogasawara, Y.; Zhou, X.; Wang, F.; Yamamoto, A.; De Camilli, P.; Di Paolo, G. Regulation of mammalian autophagy by class II and III PI 3-kinases through PI3P synthesis. PloS ONE 2013, 8, e76405. [Google Scholar] [CrossRef]
- Burman, C.; Ktistakis, N.T. Regulation of autophagy by phosphatidylinositol 3-phosphate. FEBS Lett. 2010, 584, 1302–1312. [Google Scholar] [CrossRef]
- Young, A.R.J.; Chan, E.Y.W.; Hu, X.W.; Köchl, R.; Crawshaw, S.G.; High, S.; Hailey, D.W.; Lippincott-Schwartz, J.; Tooze, S.A. Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J. Cell Sci. 2006, 119, 3888. [Google Scholar] [CrossRef]
- Hanada, T.; Noda, N.N.; Satomi, Y.; Ichimura, Y.; Fujioka, Y.; Takao, T.; Inagaki, F.; Ohsumi, Y. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J. Biol. Chem. 2007, 282, 37298–37302. [Google Scholar] [CrossRef]
- Nakatogawa, H.; Ichimura, Y.; Ohsumi, Y. Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell 2007, 130, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Obara, K.; Noda, T.; Niimi, K.; Ohsumi, Y. Transport of phosphatidylinositol 3-phosphate into the vacuole via autophagic membranes in Saccharomyces cerevisiae. Genes Cells 2008, 13, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Klionsky, D.J. Autophagosome formation: Core machinery and adaptations. Nat. Cell Biol. 2007, 9, 1102. [Google Scholar] [CrossRef] [PubMed]
- Pattingre, S.; Levine, B. Bcl-2 inhibition of autophagy: A new route to cancer? Cancer Res. 2006, 66, 2885–2888. [Google Scholar] [CrossRef]
- Tasdemir, E.; Maiuri, M.C.; Morselli, E.; Criollo, A.; D’Amelio, M.; Djavaheri-Mergny, M.; Cecconi, F.; Tavernarakis, N.; Kroemer, G. A dual role of p53 in the control of autophagy. Autophagy 2008, 4, 810–814. [Google Scholar] [CrossRef]
- Levine, B.; Abrams, J. p53: The Janus of autophagy? Nat. Cell Biol. 2008, 10, 637. [Google Scholar] [CrossRef]
- Tasdemir, E.; Maiuri, M.C.; Galluzzi, L.; Vitale, I.; Djavaheri-Mergny, M.; D’Amelio, M.; Criollo, A.; Morselli, E.; Zhu, C.; Harper, F.; et al. Regulation of autophagy by cytoplasmic p53. Nat. Cell Biol. 2008, 10, 676–687. [Google Scholar] [CrossRef]
- Crighton, D.; Wilkinson, S.; O’Prey, J.; Syed, N.; Smith, P.; Harrison, P.R.; Gasco, M.; Garrone, O.; Crook, T.; Ryan, K.M. DRAM, a p53-Induced Modulator of Autophagy, Is Critical for Apoptosis. Cell 2006, 126, 121–134. [Google Scholar] [CrossRef]
- Feng, Z.; Zhang, H.; Levine, A.J.; Jin, S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc. Natl. Acad. Sci. USA 2005, 102, 8204–8209. [Google Scholar] [CrossRef]
- Lee, D.-F.; Kuo, H.-P.; Chen, C.-T.; Hsu, J.-M.; Chou, C.-K.; Wei, Y.; Sun, H.-L.; Li, L.-Y.; Ping, B.; Huang, W.-C. IKKβ suppression of TSC1 links inflammation and tumor angiogenesis via the mTOR pathway. Cell 2007, 130, 440–455. [Google Scholar] [CrossRef]
- Uttenweiler, A.; Schwarz, H.; Mayer, A. Microautophagic vacuole invagination requires calmodulin in a Ca2+-independent function. J. Biol. Chem. 2005, 280, 33289–33297. [Google Scholar] [CrossRef] [PubMed]
- Müller, O.; Sattler, T.; Flötenmeyer, M.; Schwarz, H.; Plattner, H.; Mayer, A. Autophagic Tubes. J. Cell Biol. 2000, 151, 519. [Google Scholar] [CrossRef] [PubMed]
- Li, W.-w.; Li, J.; Bao, J.-k. Microautophagy: Lesser-known self-eating. Cell. Mol. life Sci. 2012, 69, 1125–1136. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Reviews. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef]
- Park, C.; Suh, Y.; Cuervo, A.M. Regulated degradation of Chk1 by chaperone-mediated autophagy in response to DNA damage. Nat. Commun. 2015, 6, 6823. [Google Scholar] [CrossRef]
- Valdor, R.; Mocholi, E.; Botbol, Y.; Guerrero-Ros, I.; Chandra, D.; Koga, H.; Gravekamp, C.; Cuervo, A.M.; Macian, F. Chaperone-mediated autophagy regulates T cell responses through targeted degradation of negative regulators of T cell activation. Nat. Immunol. 2014, 15, 1046–1054. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.L.; Villarroya, J.; Diaz-Carretero, A.; Patel, B.; Urbanska, A.M.; Thi, M.M.; Villarroya, F.; Santambrogio, L.; Cuervo, A.M. Loss of hepatic chaperone-mediated autophagy accelerates proteostasis failure in aging. Aging Cell 2015, 14, 249–264. [Google Scholar] [CrossRef] [PubMed]
- Finn, P.F.; Dice, J.F. Ketone bodies stimulate chaperone-mediated autophagy. J. Biol. Chem. 2005, 280, 25864–25870. [Google Scholar] [CrossRef] [PubMed]
- Tait, S.W.G.; Green, D.R. Mitochondria and cell signalling. J. Cell Sci. 2012, 125, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Kroemer, G. The Pathophysiology of Mitochondrial Cell Death. Science 2004, 305, 626–629. [Google Scholar] [CrossRef]
- Wang, K.; Klionsky, D.J. Mitochondria removal by autophagy. Autophagy 2011, 7, 297–300. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.-S.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.; Tanaka, A.; Suen, D.-F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Deas, E.; Plun-Favreau, H.; Gandhi, S.; Desmond, H.; Kjaer, S.; Loh, S.H.Y.; Renton, A.E.M.; Harvey, R.J.; Whitworth, A.J.; Martins, L.M.; et al. PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum. Mol. Genet. 2011, 20, 867–879. [Google Scholar] [CrossRef] [PubMed]
- Yamano, K.; Youle, R.J. PINK1 is degraded through the N-end rule pathway. Autophagy 2013, 9, 1758–1769. [Google Scholar] [CrossRef]
- Lazarou, M.; Jin, S.M.; Kane, L.A.; Youle, R.J. Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev. cell 2012, 22, 320–333. [Google Scholar] [CrossRef]
- Strappazzon, F.; Nazio, F.; Corrado, M.; Cianfanelli, V.; Romagnoli, A.; Fimia, G.M.; Campello, S.; Nardacci, R.; Piacentini, M.; Campanella, M.; et al. AMBRA1 is able to induce mitophagy via LC3 binding, regardless of PARKIN and p62/SQSTM1. Cell Death Differ. 2015, 22, 419–432. [Google Scholar] [CrossRef]
- Walther, T.C.; Farese, R.V., Jr. Lipid droplets and cellular lipid metabolism. Annu. Rev. Biochem. 2012, 81, 687–714. [Google Scholar] [CrossRef]
- Mochida, K.; Oikawa, Y.; Kimura, Y.; Kirisako, H.; Hirano, H.; Ohsumi, Y.; Nakatogawa, H. Receptor-mediated selective autophagy degrades the endoplasmic reticulum and the nucleus. Nature 2015, 522, 359. [Google Scholar] [CrossRef]
- Seo, A.Y.; Lau, P.-W.; Feliciano, D.; Sengupta, P.; Gros, M.A.L.; Cinquin, B.; Larabell, C.A.; Lippincott-Schwartz, J. AMPK and vacuole-associated Atg14p orchestrate μ-lipophagy for energy production and long-term survival under glucose starvation. ELife 2017, 6, e21690. [Google Scholar] [CrossRef]
- van Zutphen, T.; Todde, V.; de Boer, R.; Kreim, M.; Hofbauer, H.F.; Wolinski, H.; Veenhuis, M.; van der Klei, I.J.; Kohlwein, S.D. Lipid droplet autophagy in the yeast Saccharomyces cerevisiae. Mol. Biol. Cell 2014, 25, 290–301. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xu, M.; Ding, X.; Yan, C.; Song, Z.; Chen, L.; Huang, X.; Wang, X.; Jian, Y.; Tang, G.; et al. Protein kinase C controls lysosome biogenesis independently of mTORC1. Nat. Cell Biol. 2016, 18, 1065–1077. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Chen, Z.; Wei, X.; Chen, Z.; Fu, Y.; Yang, X.; Chen, D.; Wang, R.; Jenner, P.; Lu, J.-H.; et al. Cystatin C as a potential therapeutic mediator against Parkinson’s disease via VEGF-induced angiogenesis and enhanced neuronal autophagy in neurovascular units. Cell Death Dis. 2017, 8, e2854. [Google Scholar] [CrossRef] [PubMed]
- Ikenoue, T.; Inoki, K.; Yang, Q.; Zhou, X.; Guan, K.L. Essential function of TORC2 in PKC and Akt turn motif phosphorylation, maturation and signalling. Embo J. 2008, 27, 1919–1931. [Google Scholar] [CrossRef] [PubMed]
- Coward, J.; Ambrosini, G.; Musi, E.; Truman, J.-P.; Haimovitz-Friedman, A.; Allegood, J.C.; Wang, E.; Merrill, J.; Alfred, H.; Schwartz, G.K. Safingol (L-threo-sphinganine) induces autophagy in solid tumor cells through inhibition of PKC and the PI3-kinase pathway. Autophagy 2009, 5, 184–193. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Xu, C.; Reece, E.A.; Li, X.; Wu, Y.; Harman, C.; Yu, J.; Dong, D.; Wang, C.; Yang, P.; et al. Protein kinase C-alpha suppresses autophagy and induces neural tube defects via miR-129-2 in diabetic pregnancy. Nat. Commun. 2017, 8, 15182. [Google Scholar] [CrossRef]
- Wong, V.K.W.; Zeng, W.; Chen, J.; Yao, X.J.; Leung, E.L.H.; Wang, Q.Q.; Chiu, P.; Ko, B.C.B.; Law, B.Y.K. Tetrandrine, an Activator of Autophagy, Induces Autophagic Cell Death via PKC-α Inhibition and mTOR-Dependent Mechanisms. Front. Pharmacol. 2017, 8, 351. [Google Scholar] [CrossRef]
- Xue, X.; Ren, J.; Sun, X.; Gui, Y.; Feng, Y.; Shu, B.; Wei, W.; Lu, Q.; Liang, Y.; He, W.; et al. Protein kinase Cα drives fibroblast activation and kidney fibrosis by stimulating autophagic flux. J. Biol. Chem. 2018, 293, 11119–11130. [Google Scholar] [CrossRef]
- Li, N.; Zhang, W. Protein kinase C β inhibits autophagy and sensitizes cervical cancer Hela cells to cisplatin. Biosci. Rep. 2017, 37, BSR20160445. [Google Scholar] [CrossRef]
- Rimessi, A.; Pavan, C.; Ioannidi, E.; Nigro, F.; Morganti, C.; Brugnoli, A.; Longo, F.; Gardin, C.; Ferroni, L.; Morari, M.; et al. Protein Kinase C β: A New Target Therapy to Prevent the Long-Term Atypical Antipsychotic-Induced Weight Gain. Neuropsychopharmacology 2017, 42, 1491–1501. [Google Scholar] [CrossRef]
- Hua, R.; Han, S.; Zhang, N.; Dai, Q.; Liu, T.; Li, J. cPKCγ-Modulated Sequential Reactivation of mTOR Inhibited Autophagic Flux in Neurons Exposed to Oxygen Glucose Deprivation/Reperfusion. Int. J. Mol. Sci. 2018, 19, 1380. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Han, S.; Wang, S.; Luo, Y.; Zhao, L.; Li, J. cPKCγ-mediated down-regulation of UCHL1 alleviates ischaemic neuronal injuries by decreasing autophagy via ERK-mTOR pathway. J. Cell. Mol. Med. 2017, 21, 3641–3657. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Pan, J.; Xiang, X.; Liu, Y.; Dong, G.; Livingston, M.J.; Chen, J.-K.; Yin, X.-M.; Dong, Z. Protein Kinase Cδ Suppresses Autophagy to Induce Kidney Cell Apoptosis in Cisplatin Nephrotoxicity. J. Am. Soc. Nephrol. 2017, 28, 1131–1144. [Google Scholar] [CrossRef] [PubMed]
- So, K.-Y.; Oh, S.-H. Cadmium-induced heme-oxygenase-1 expression plays dual roles in autophagy and apoptosis and is regulated by both PKC-δ and PKB/Akt activation in NRK52E kidney cells. Toxicology 2016, 370, 49–59. [Google Scholar] [CrossRef]
- Huang, S.-P.; Chien, J.-Y.; Tsai, R.-K. Ethambutol induces impaired autophagic flux and apoptosis in the rat retina. Dis. Models Mech. 2015, 8, 977–987. [Google Scholar] [CrossRef]
- Yan, R.; Niu, C.-Y.; Tian, Y. Roles of Autophagy and Protein Kinase C-epsilon in Lipid Metabolism of Nonalcoholic Fatty Liver Cell Models. Arch. Med Res. 2018, 49, 381–390. [Google Scholar] [CrossRef]
- Toton, E.; Romaniuk, A.; Konieczna, N.; Hofmann, J.; Barciszewski, J.; Rybczynska, M. Impact of PKCε downregulation on autophagy in glioblastoma cells. Bmc Cancer 2018, 18, 185. [Google Scholar] [CrossRef]
- Gonnella, R.; Granato, M.; Farina, A.; Santarelli, R.; Faggioni, A.; Cirone, M. PKC theta and p38 MAPK activate the EBV lytic cycle through autophagy induction. BBA-Mol. Cell Res. 2015, 1853, 1586–1595. [Google Scholar] [CrossRef]
- Jin, Y.; Bai, Y.; Ni, H.; Qiang, L.; Ye, L.; Shan, Y.; Zhou, M. Activation of autophagy through calcium-dependent AMPK/mTOR and PKCθ pathway causes activation of rat hepatic stellate cells under hypoxic stress. Febs Lett. 2016, 590, 672–682. [Google Scholar] [CrossRef]
- Micheva-Viteva, S.N.; Shou, Y.; Ganguly, K.; Wu, T.H.; Hong-Geller, E. PKC-η-MARCKS Signaling Promotes Intracellular Survival of Unopsonized Burkholderia thailandensis. Front. Cell. Infect. Microbiol. 2017, 7, 231. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.-S.; Yang, Y.; Yang, H.; Liu, Y.-Z.; Hao, J.-J.; Zhang, Y.; Shi, Z.-Z.; Jia, X.-M.; Zhan, Q.-M.; Wang, M.-R. PKCι counteracts oxidative stress by regulating Hsc70 in an esophageal cancer cell line. Cell Stress Chaperones 2013, 18, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.-S.; Yang, Y.; Lu, H.-Z.; Shang, L.; Zhang, Y.; Hao, J.-J.; Shi, Z.-Z.; Wang, X.-M.; Liu, Y.-Z.; Zhan, Q.-M.; et al. Inhibition of atypical protein kinase Cι induces apoptosis through autophagic degradation of β-catenin in esophageal cancer cells. Mol. Carcinog. 2014, 53, 514–525. [Google Scholar] [CrossRef] [PubMed]
- Qu, L.; Li, G.; Xia, D.; Hongdu, B.; Xu, C.; Lin, X.; Chen, Y. PRKCI negatively regulates autophagy via PIK3CA/AKT–MTOR signaling. Biochem. Biophys. Res. Commun. 2016, 470, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Burchell, V.S.; Nelson, D.E.; Sanchez-Martinez, A.; Delgado-Camprubi, M.; Ivatt, R.M.; Pogson, J.H.; Randle, S.J.; Wray, S.; Lewis, P.A.; Houlden, H.; et al. The Parkinson’s disease-linked proteins Fbxo7 and Parkin interact to mediate mitophagy. Nat. Neurosci. 2013, 16, 1257–1265. [Google Scholar] [CrossRef]
- Park, S.; Han, S.; Choi, I.; Kim, B.; Park, S.P.; Joe, E.-H.; Suh, Y.H. Interplay between Leucine-Rich Repeat Kinase 2 (LRRK2) and p62/SQSTM-1 in Selective Autophagy. PloS ONE 2016, 11, e0163029. [Google Scholar] [CrossRef]
- Ho, D.H.; Kim, H.; Nam, D.; Sim, H.; Kim, J.; Kim, H.G.; Son, I.; Seol, W. LRRK2 impairs autophagy by mediating phosphorylation of leucyl-tRNA synthetase. Cell Biochem. Funct. 2018, 36, 431–442. [Google Scholar] [CrossRef]
- Gómez-Suaga, P.; Luzón-Toro, B.; Churamani, D.; Zhang, L.; Bloor-Young, D.; Patel, S.; Woodman, P.G.; Churchill, G.C.; Hilfiker, S. Leucine-rich repeat kinase 2 regulates autophagy through a calcium-dependent pathway involving NAADP. Hum. Mol. Genet. 2012, 21, 511–525. [Google Scholar] [CrossRef]
- Niu, J.; Yu, M.; Wang, C.; Xu, Z. Leucine-rich repeat kinase 2 disturbs mitochondrial dynamics via Dynamin-like protein. J. Neurochem. 2012, 122, 650–658. [Google Scholar] [CrossRef]
- Bravo-San Pedro, J.M.; Niso-Santano, M.; Gómez-Sánchez, R.; Pizarro-Estrella, E.; Aiastui-Pujana, A.; Gorostidi, A.; Climent, V.; De Maturana, R.L.; Sanchez-Pernaute, R.; De Munain, A.L. The LRRK2 G2019S mutant exacerbates basal autophagy through activation of the MEK/ERK pathway. Cell. Mol. Life Sci. 2013, 70, 121–136. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Stefanis, L.; Fredenburg, R.; Lansbury, P.T.; Sulzer, D. Impaired Degradation of Mutant α-Synuclein by Chaperone-Mediated Autophagy. Science 2004, 305, 1292–1295. [Google Scholar] [CrossRef]
- Ren, H.; Fu, K.; Mu, C.; Li, B.; Wang, D.; Wang, G. DJ-1, a cancer and Parkinson’s disease associated protein, regulates autophagy through JNK pathway in cancer cells. Cancer Lett. 2010, 297, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Thomas, K.J.; McCoy, M.K.; Blackinton, J.; Beilina, A.; van der Brug, M.; Sandebring, A.; Miller, D.; Maric, D.; Cedazo-Minguez, A.; Cookson, M.R. DJ-1 acts in parallel to the PINK1/parkin pathway to control mitochondrial function and autophagy. Hum. Mol. Genet. 2011, 20, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Petrie, T.G.; Liu, Y.; Liu, J.; Fujioka, H.; Zhu, X. Parkinson’s disease-associated DJ-1 mutations impair mitochondrial dynamics and cause mitochondrial dysfunction. J. Neurochem. 2012, 121, 830–839. [Google Scholar] [CrossRef] [PubMed]
- Nash, Y.; Schmukler, E.; Trudler, D.; Pinkas-Kramarski, R.; Frenkel, D. DJ-1 deficiency impairs autophagy and reduces alpha-synuclein phagocytosis by microglia. J. Neurochem. 2017, 143, 584–594. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.-Y.; Kang, W.-Y.; Chen, Y.-M.; Jiang, T.-F.; Zhang, J.; Zhang, L.-N.; Ding, J.-Q.; Liu, J.; Chen, S.-D. DJ-1 Inhibits α-Synuclein Aggregation by Regulating Chaperone-Mediated Autophagy. Front. Aging Neurosci. 2017, 9. [Google Scholar] [CrossRef]
- Murphy, K.E.; Gysbers, A.M.; Abbott, S.K.; Tayebi, N.; Kim, W.S.; Sidransky, E.; Cooper, A.; Garner, B.; Halliday, G.M. Reduced glucocerebrosidase is associated with increased α-synuclein in sporadic Parkinson’s disease. Brain: A J. Neurol. 2014, 137, 834–848. [Google Scholar] [CrossRef]
- Urano, Y.; Mori, C.; Fuji, A.; Konno, K.; Yamamoto, T.; Yashirogi, S.; Ando, M.; Saito, Y.; Noguchi, N. 6-Hydroxydopamine induces secretion of PARK7/DJ-1 via autophagy-based unconventional secretory pathway. Autophagy 2018, 14, 1943–1958. [Google Scholar] [CrossRef]
- Ivatt, R.M.; Sanchez-Martinez, A.; Godena, V.K.; Brown, S.; Ziviani, E.; Whitworth, A.J. Genome-wide RNAi screen identifies the Parkinson disease GWAS risk locus SREBF1 as a regulator of mitophagy. Proc. Natl. Acad. Sci. USA 2014, 111, 8494–8499. [Google Scholar] [CrossRef]
- Guven, G.; Vurgun, E.; Bilgic, B.; Hanagasi, H.; Gurvit, H.; Ozer, E.; Lohmann, E.; Erginel-Unaltuna, N. Association between selected cholesterol-related gene polymorphisms and Alzheimer’s disease in a Turkish cohort. Mol. Biol. Rep. 2019, 46, 1701–1707. [Google Scholar] [CrossRef]
- Edler, M.K.; Sherwood, C.C.; Meindl, R.S.; Hopkins, W.D.; Ely, J.J.; Erwin, J.M.; Mufson, E.J.; Hof, P.R.; Raghanti, M.A. Aged chimpanzees exhibit pathologic hallmarks of Alzheimer’s disease. Neurobiol. Aging 2017, 59, 107–120. [Google Scholar] [CrossRef]
- Espuny-Camacho, I.; Arranz, A.M.; Fiers, M.; Snellinx, A.; Ando, K.; Munck, S.; Bonnefont, J.; Lambot, L.; Corthout, N.; Omodho, L.; et al. Hallmarks of Alzheimer’s Disease in Stem-Cell-Derived Human Neurons Transplanted into Mouse Brain. Neuron 2017, 93, 1066–1081. [Google Scholar] [CrossRef] [PubMed]
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Martinez-Vicente, M.; Krüger, U.; Kaushik, S.; Wong, E.; Mandelkow, E.-M.; Cuervo, A.M.; Mandelkow, E. Tau fragmentation, aggregation and clearance: The dual role of lysosomal processing. Hum. Mol. Genet. 2009, 18, 4153–4170. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef]
- Tammineni, P.; Ye, X.; Feng, T.; Aikal, D.; Cai, Q. Impaired retrograde transport of axonal autophagosomes contributes to autophagic stress in Alzheimer’s disease neurons. Elife 2017, 6, e21776. [Google Scholar] [CrossRef]
- Caccamo, A.; De Pinto, V.; Messina, A.; Branca, C.; Oddo, S. Genetic reduction of mammalian target of rapamycin ameliorates Alzheimer’s disease-like cognitive and pathological deficits by restoring hippocampal gene expression signature. J. Neurosci. 2014, 34, 7988–7998. [Google Scholar] [CrossRef]
- Lee, H.R.; Shin, H.K.; Park, S.Y.; Kim, H.Y.; Lee, W.S.; Rhim, B.Y.; Hong, K.W.; Kim, C.D. Attenuation of β-amyloid-induced tauopathy via activation of CK2α/SIRT1: Targeting for cilostazol. J. Neurosci. Res. 2014, 92, 206–217. [Google Scholar] [CrossRef]
- Lee, H.R.; Shin, H.K.; Park, S.Y.; Kim, H.Y.; Bae, S.S.; Lee, W.S.; Rhim, B.Y.; Hong, K.W.; Kim, C.D. Cilostazol Upregulates Autophagy via SIRT1 Activation: Reducing Amyloid-β Peptide and APP-CTFβ Levels in Neuronal Cells. PloS ONE 2015, 10, e0134486. [Google Scholar] [CrossRef]
- Rocchi, A.; Yamamoto, S.; Ting, T.; Fan, Y.; Sadleir, K.; Wang, Y.; Zhang, W.; Huang, S.; Levine, B.; Vassar, R. A Becn1 mutation mediates hyperactive autophagic sequestration of amyloid oligomers and improved cognition in Alzheimer’s disease. PLoS Genetics 2017, 13, e1006962. [Google Scholar] [CrossRef]
- Xie, Y.; Zhou, B.; Lin, M.-Y.; Wang, S.; Foust, K.D.; Sheng, Z.-H. Endolysosomal Deficits Augment Mitochondria Pathology in Spinal Motor Neurons of Asymptomatic fALS Mice. Neuron 2015, 87, 355–370. [Google Scholar] [CrossRef]
- Rudnick, N.D.; Griffey, C.J.; Guarnieri, P.; Gerbino, V.; Wang, X.; Piersaint, J.A.; Tapia, J.C.; Rich, M.M.; Maniatis, T. Distinct roles for motor neuron autophagy early and late in the SOD1(G93A) mouse model of ALS. Proc. Natl. Acad. Sci. USA 2017, 114, E8294–E8303. [Google Scholar] [CrossRef] [PubMed]
- Fabbrizio, P.; Amadio, S.; Apolloni, S.; Volonté, C. P2X7 Receptor Activation Modulates Autophagy in SOD1-G93A Mouse Microglia. Front. Cell. Neurosci. 2017, 11, 249. [Google Scholar] [CrossRef]
- Perera, N.D.; Sheean, R.K.; Lau, C.L.; Shin, Y.S.; Beart, P.M.; Horne, M.K.; Turner, B.J. Rilmenidine promotes MTOR-independent autophagy in the mutant SOD1 mouse model of amyotrophic lateral sclerosis without slowing disease progression. Autophagy 2018, 14, 534–551. [Google Scholar] [CrossRef]
- Zheng, S.; Clabough, E.B.; Sarkar, S.; Futter, M.; Rubinsztein, D.C.; Zeitlin, S.O. Deletion of the huntingtin polyglutamine stretch enhances neuronal autophagy and longevity in mice. PLoS Genetics 2010, 6, e1000838. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.D.; Ladha, S.; Ehrnhoefer, D.E.; Hayden, M.R. Autophagy in Huntington disease and huntingtin in autophagy. Trends Neurosci. 2015, 38, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Steffan, J.S. Does Huntingtin play a role in selective macroautophagy? Cell Cycle 2010, 9, 3401–3413. [Google Scholar] [CrossRef]
- Rui, Y.-N.; Xu, Z.; Patel, B.; Chen, Z.; Chen, D.; Tito, A.; David, G.; Sun, Y.; Stimming, E.F.; Bellen, H.J.; et al. Huntingtin functions as a scaffold for selective macroautophagy. Nat. Cell Biol. 2015, 17, 262. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.C.; Holzbaur, E.L.F. The Regulation of Autophagosome Dynamics by Huntingtin and HAP1 Is Disrupted by Expression of Mutant Huntingtin, Leading to Defective Cargo Degradation. J. Euroscience 2014, 34, 1293. [Google Scholar] [CrossRef]
- White, J.A., II; Anderson, E.; Zimmerman, K.; Zheng, K.H.; Rouhani, R.; Gunawardena, S. Huntingtin differentially regulates the axonal transport of a sub-set of Rab-containing vesicles in vivo. Hum. Mol. Genet. 2015, 24, 7182–7195. [Google Scholar] [CrossRef]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef]
- Saitoh, T.; Fujita, N.; Hayashi, T.; Takahara, K.; Satoh, T.; Lee, H.; Matsunaga, K.; Kageyama, S.; Omori, H.; Noda, T.; et al. Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. Proc. Natl. Acad. Sci. USA 2009, 106, 20842–20846. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, T.; Akira, S. Regulation of innate immune responses by autophagy-related proteins. J. Cell Biol. 2010, 189, 925–935. [Google Scholar] [CrossRef] [PubMed]
- Nedjic, J.; Aichinger, M.; Emmerich, J.; Mizushima, N.; Klein, L. Autophagy in thymic epithelium shapes the T-cell repertoire and is essential for tolerance. Nature 2008, 455, 396. [Google Scholar] [CrossRef] [PubMed]
- Motori, E.; Puyal, J.; Toni, N.; Ghanem, A.; Angeloni, C.; Malaguti, M.; Cantelli-Forti, G.; Berninger, B.; Conzelmann, K.-K.; Götz, M.; et al. Inflammation-Induced Alteration of Astrocyte Mitochondrial Dynamics Requires Autophagy for Mitochondrial Network Maintenance. Cell Metab. 2013, 18, 844–859. [Google Scholar] [CrossRef] [PubMed]
- Rangaraju, S.; Verrier, J.D.; Madorsky, I.; Nicks, J.; Dunn, W.A., Jr.; Notterpek, L. Rapamycin activates autophagy and improves myelination in explant cultures from neuropathic mice. J. Neurosci. 2010, 30, 11388–11397. [Google Scholar] [CrossRef]
- Mudo, G.; Mäkelä, J.; Di Liberto, V.; Tselykh, T.V.; Olivieri, M.; Piepponen, P.; Eriksson, O.; Mälkiä, A.; Bonomo, A.; Kairisalo, M. Transgenic expression and activation of PGC-1α protect dopaminergic neurons in the MPTP mouse model of Parkinson’s disease. Cell. Mol. Life Sci. 2012, 69, 1153–1165. [Google Scholar] [CrossRef]
- Jason, J.Y.; Barnes, A.P.; Hand, R.; Polleux, F.; Ehlers, M.D. TGF-β signaling specifies axons during brain development. Cell 2010, 142, 144–157. [Google Scholar]
- Gunaratne, A.; Thai, B.L.; Di Guglielmo, G.M. Atypical protein kinase C phosphorylates Par6 and facilitates transforming growth factor β-induced epithelial-to-mesenchymal transition. Mol. Cell. Biol. 2013, 33, 874–886. [Google Scholar] [CrossRef]
- Hapak, S.M.; Rothlin, C.V.; Ghosh, S. PAR3–PAR6–atypical PKC polarity complex proteins in neuronal polarization. Cell. Mol. Life Sci. 2018, 75, 2735–2761. [Google Scholar] [CrossRef]
- Facchinetti, V.; Ouyang, W.; Wei, H.; Soto, N.; Lazorchak, A.; Gould, C.; Lowry, C.; Newton, A.C.; Mao, Y.; Miao, R.Q.; et al. The mammalian target of rapamycin complex 2 controls folding and stability of Akt and protein kinase C. EMBO J. 2008, 27, 1932–1943. [Google Scholar] [CrossRef]
- Nagai, K. Bovine milk phospholipid fraction protects Neuro2a cells from endoplasmic reticulum stress via PKC activation and autophagy. J. Biosci. Bioeng. 2012, 114, 466–471. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Yin, Y.; Han, S.; Jiang, J.; Yang, W.; Bu, X.; Li, J. Hypoxic preconditioning induced neuroprotection against cerebral ischemic injuries and its cPKCγ-mediated molecular mechanism. Neurochem. Int. 2011, 58, 684–692. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Li, Y.; Han, S.; Liu, S.; Zhang, N.; Zhao, L.; Li, S.; Li, J. cPKCγ-Modulated Autophagy in Neurons Alleviates Ischemic Injury in Brain of Mice with Ischemic Stroke Through Akt-mTOR Pathway. Transl. Stroke Res. 2016, 7, 497–511. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Blanco, J.; Martín, V.; García-Santos, G.; Herrera, F.; Casado-Zapico, S.; Antolín, I.; Rodriguez, C. Cooperative action of JNK and AKT/mTOR in 1-methyl-4-phenylpyridinium-induced autophagy of neuronal PC12 cells. J. Neurosci. Res. 2012, 90, 1850–1860. [Google Scholar] [CrossRef] [PubMed]
- Liang, P.; Le, W. Role of autophagy in the pathogenesis of multiple sclerosis. Neurosci. Bull. 2015, 31, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Mao, P.; Reddy, P.H. Is multiple sclerosis a mitochondrial disease? Biochim. et Biophys. Acta 2010, 1802, 66–79. [Google Scholar] [CrossRef]
- Giorgio, M.; Migliaccio, E.; Orsini, F.; Paolucci, D.; Moroni, M.; Contursi, C.; Pelliccia, G.; Luzi, L.; Minucci, S.; Marcaccio, M.; et al. Electron Transfer between Cytochrome c and p66Shc Generates Reactive Oxygen Species that Trigger Mitochondrial Apoptosis. Cell 2005, 122, 221–233. [Google Scholar] [CrossRef]
- Su, K.; Bourdette, D.; Forte, M. Mitochondrial dysfunction and neurodegeneration in multiple sclerosis. Front. Physiol. 2013, 4, 169. [Google Scholar] [CrossRef]
- Farese, R.V.; Sajan, M.P. Atypical PKC: Therapeutic target for Alzheimer’s? Aging 2018, 11, 13–14. [Google Scholar] [CrossRef]
- Vijayakumaran, S.; Pountney, D.L. SUMOylation, aging and autophagy in neurodegeneration. NeuroToxicology 2018, 66, 53–57. [Google Scholar] [CrossRef]
- Choi, J.-H.; Park, J.-Y.; Park, S.P.; Lee, H.; Han, S.; Park, K.H.; Suh, Y.H. Regulation of mGluR7 trafficking by SUMOylation in neurons. Neuropharmacology 2016, 102, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Pelkey, K.A.; Lavezzari, G.; Racca, C.; Roche, K.W.; McBain, C.J. mGluR7 Is a Metaplastic Switch Controlling Bidirectional Plasticity of Feedforward Inhibition. Neuron 2005, 46, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhao, J.; Chen, Y.; Shi, H.; Huang, X.; Wang, Y.; Wang, Y.; Wei, Y.; Xue, W.; Han, J. Effect of mGluR7 on proliferation of human embryonic neural stem cells. Medicine 2019, 98, e14683. [Google Scholar] [CrossRef] [PubMed]
- Soobrattee, M.A.; Neergheen, V.S.; Luximon-Ramma, A.; Aruoma, O.I.; Bahorun, T. Phenolics as potential antioxidant therapeutic agents: Mechanism and actions. Mutat. Res. /Fundam. Mol. Mech. Mutagenesis 2005, 579, 200–213. [Google Scholar] [CrossRef] [PubMed]
- Costa, S.L.; Silva, V.D.A.; dos Santos Souza, C.; Santos, C.C.; Paris, I.; Muñoz, P.; Segura-Aguilar, J. Impact of plant-derived flavonoids on neurodegenerative diseases. Neurotox. Res. 2016, 30, 41–52. [Google Scholar] [CrossRef]
- Flanagan, E.; Müller, M.; Hornberger, M.; Vauzour, D. Impact of flavonoids on cellular and molecular mechanisms underlying age-related cognitive decline and neurodegeneration. Curr. Nutr. Rep. 2018, 7, 49–57. [Google Scholar] [CrossRef]
- Singh, N.A.; Mandal, A.K.A.; Khan, Z.A. Potential neuroprotective properties of epigallocatechin-3-gallate (EGCG). Nutr. J. 2015, 15, 60. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, Y.; Quan, Z.; Wong, W.; Guo, J.; Zhang, R.; Yang, Q.; Dai, R.; McGeer, P.L.; Qing, H. Epigallocatechin gallate (EGCG) inhibits alpha-synuclein aggregation: A potential agent for Parkinson’s disease. Neurochem. Res. 2016, 41, 2788–2796. [Google Scholar] [CrossRef]
- Xicota, L.; Rodriguez-Morato, J.; Dierssen, M.; de la Torre, R. Potential role of (-)-epigallocatechin-3-gallate (EGCG) in the secondary prevention of Alzheimer disease. Curr. Drug Targets 2017, 18, 174–195. [Google Scholar] [CrossRef]
- Chen, D.; Kanthasamy, A.G.; Reddy, M.B. EGCG protects against 6-OHDA-induced neurotoxicity in a cell culture model. Parkinson’s Disease 2015, 2015. [Google Scholar] [CrossRef]
- Holczer, M.; Besze, B.; Zámbó, V.; Csala, M.; Bánhegyi, G.; Kapuy, O. Epigallocatechin-3-gallate (EGCG) promotes autophagy-dependent survival via influencing the balance of mTOR-AMPK pathways upon endoplasmic reticulum stress. Oxidative Med. Cell. Longev. 2018. [Google Scholar] [CrossRef]
- Lee, J.-H.; Moon, J.-H.; Kim, S.-W.; Jeong, J.-K.; Nazim, U.M.; Lee, Y.-J.; Seol, J.-W.; Park, S.-Y. EGCG-mediated autophagy flux has a neuroprotection effect via a class III histone deacetylase in primary neuron cells. Oncotarget 2015, 6, 9701–9717. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Huang, L.; Zhang, H.; Diao, X.; Zhao, S.; Zhou, W. Reduction in autophagy by (-)-epigallocatechin-3-gallate (EGCG): A potential mechanism of prevention of mitochondrial dysfunction after subarachnoid hemorrhage. Mol. Neurobiol. 2017, 54, 392–405. [Google Scholar] [CrossRef]
- Levites, Y.; Amit, T.; Mandel, S.; Youdim, M.B. Neuroprotection and neurorescue against Aβ toxicity and PKC-dependent release of nonamyloidogenic soluble precursor protein by green tea polyphenol (-)-epigallocatechin-3-gallate. FASEB J. 2003, 17, 952–954. [Google Scholar] [CrossRef] [PubMed]
- Levites, Y.; Amit, T.; Youdim, M.B.; Mandel, S. Involvement of protein kinase C activation and cell survival/cell cycle genes in green tea polyphenol (−)-epigallocatechin 3-gallate neuroprotective action. J. Biol. Chem. 2002, 277, 30574–30580. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Liu, F.; Jin, H.; Li, R.; Wang, Y.; Zhang, W.; Wang, H.; Chen, W. Involvement of PKCα and ERK1/2 signaling pathways in EGCG’s protection against stress-induced neural injuries in Wistar rats. Neuroscience 2017, 346, 226–237. [Google Scholar] [CrossRef]
- Feng, Y.; Wang, X.-p.; Yang, S.-g.; Wang, Y.-j.; Zhang, X.; Du, X.-t.; Sun, X.-x.; Zhao, M.; Huang, L.; Liu, R.-t. Resveratrol inhibits beta-amyloid oligomeric cytotoxicity but does not prevent oligomer formation. Neurotoxicology 2009, 30, 986–995. [Google Scholar] [CrossRef]
- Anastácio, J.R.; Netto, C.A.; Castro, C.C.; Sanches, E.F.; Ferreira, D.C.; Noschang, C.; Krolow, R.; Dalmaz, C.; Pagnussat, A. Resveratrol treatment has neuroprotective effects and prevents cognitive impairment after chronic cerebral hypoperfusion. Neurol. Res. 2014, 36, 627–633. [Google Scholar] [CrossRef]
- Wu, Y.; Li, X.; Zhu, J.X.; Xie, W.; Le, W.; Fan, Z.; Jankovic, J.; Pan, T. Resveratrol-activated AMPK/SIRT1/autophagy in cellular models of Parkinson’s disease. Neurosignals 2011, 19, 163–174. [Google Scholar] [CrossRef]
- Pineda-Ramírez, N.; Alquisiras-Burgos, I.; Ortiz-Plata, A.; Ruiz-Tachiquín, M.-E.; Espinoza-Rojo, M.; Aguilera, P. Resveratrol Activates Neuronal Autophagy Through AMPK in the Ischemic Brain. Mol. Neurobiol. 2019, 1–15. [Google Scholar] [CrossRef]
- Park, D.; Jeong, H.; Lee, M.N.; Koh, A.; Kwon, O.; Yang, Y.R.; Noh, J.; Suh, P.-G.; Park, H.; Ryu, S.H. Resveratrol induces autophagy by directly inhibiting mTOR through ATP competition. Sci. Rep. 2016, 6, 21772. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.S.; Zheng, W.H.; Bastianetto, S.; Chabot, J.G.; Quirion, R. Neuroprotective effects of resveratrol against β-amyloid-induced neurotoxicity in rat hippocampal neurons: Involvement of protein kinase C. Br. J. Pharmacol. 2004, 141, 997–1005. [Google Scholar] [CrossRef] [PubMed]
- Pany, S.; Ghosh, A.; You, Y.; Nguyen, N.; Das, J. Resveratrol inhibits phorbol ester-induced membrane translocation of presynaptic Munc13-1. Biochim. et Biophys. Acta 2017, 1861, 2640–2651. [Google Scholar] [CrossRef] [PubMed]
- Du, G.; Zhao, Z.; Chen, Y.; Li, Z.; Tian, Y.; Liu, Z.; Liu, B.; Song, J. Quercetin attenuates neuronal autophagy and apoptosis in rat traumatic brain injury model via activation of PI3K/Akt signaling pathway. Neurol. Res. 2016, 38, 1012–1019. [Google Scholar] [CrossRef]
- El-Horany, H.E.; El-latif, R.N.A.; ElBatsh, M.M.; Emam, M.N. Ameliorative effect of quercetin on neurochemical and behavioral deficits in rotenone rat model of Parkinson’s disease: Modulating autophagy (quercetin on experimental Parkinson’s disease). J. Biochem. Mol. Toxicol. 2016, 30, 360–369. [Google Scholar] [CrossRef]
- Lee, B.K.; Jung, Y.-S. Allium cepa extract and quercetin protect neuronal cells from oxidative stress via PKC-ε inactivation/ERK1/2 activation. Oxidative Med. Cell. Longev. 2016, 2016. [Google Scholar] [CrossRef]
- Liu, Y.-W.; Liu, X.-L.; Kong, L.; Zhang, M.-Y.; Chen, Y.-J.; Zhu, X.; Hao, Y.-C. Neuroprotection of quercetin on central neurons against chronic high glucose through enhancement of Nrf2/ARE/glyoxalase-1 pathway mediated by phosphorylation regulation. Biomed. Pharmacother. 2019, 109, 2145–2154. [Google Scholar] [CrossRef]
- Gao, W.; Zan, Y.; Wang, Z.-j.J.; Hu, X.-y.; Huang, F. Quercetin ameliorates paclitaxel-induced neuropathic pain by stabilizing mast cells, and subsequently blocking PKCε-dependent activation of TRPV1. Acta Pharmacol. Sin. 2016, 37, 1166. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, J.; Ren, X.; Liu, Q.; Yang, X. The mechanism of quercetin in regulating osteoclast activation and the PAR2/TRPV1 signaling pathway in the treatment of bone cancer pain. Int. J. Clin. Exp. Pathol. 2018, 11, 5149–5156. [Google Scholar]
- Jaroonwitchawan, T.; Chaicharoenaudomrung, N.; Namkaew, J.; Noisa, P. Curcumin attenuates paraquat-induced cell death in human neuroblastoma cells through modulating oxidative stress and autophagy. Neurosci. Lett. 2017, 636, 40–47. [Google Scholar] [CrossRef]
- Huang, L.; Chen, C.; Zhang, X.; Li, X.; Chen, Z.; Yang, C.; Liang, X.; Zhu, G.; Xu, Z. Neuroprotective effect of curcumin against cerebral ischemia-reperfusion via mediating autophagy and inflammation. J. Mol. Neurosci. 2018, 64, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Gui, B.; Sun, Y.; Shi, N.; Gu, Z.; Zhang, T.; Sun, X. Inhibition of L-type Ca2+ channels by curcumin requires a novel protein kinase-theta isoform in rat hippocampal neurons. Cell Calcium 2013, 53, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, K.; Hiyama, A.; Arai, F.; Nukaga, T.; Sakai, D.; Mochida, J. C-Fos regulation by the MAPK and PKC pathways in intervertebral disc cells. PloS ONE 2013, 8, e73210. [Google Scholar] [CrossRef] [PubMed]
- Carelli-Alinovi, C.; Pirolli, D.; Giardina, B.; Misiti, F. Protein kinase C mediates caspase 3 activation: A role for erythrocyte morphology changes. Clin. Hemorheol. Microcirc. 2015, 59, 345–354. [Google Scholar] [CrossRef]
- Gao, J.; Guo, J.; Li, H.; Bai, S.; Li, H.; Wu, B.; Wang, L.; Xi, Y.; Tian, Y.; Yang, G.; et al. Involvement of dopamine D2 receptors activation in ischemic post-conditioning-induced cardioprotection through promoting PKC-ε particulate translocation in isolated rat hearts. Mol. Cell. Biochem. 2013, 379, 267–276. [Google Scholar] [CrossRef]
- Sadeghi, H.; Parishani, M.; Akbartabar Touri, M.; Ghavamzadeh, M.; Jafari Barmak, M.; Zarezade, V.; Delaviz, H.; Sadeghi, H. Pramipexole reduces inflammation in the experimental animal models of inflammation. Immunopharmacol. and Immunotoxicol. 2017, 39, 80–86. [Google Scholar] [CrossRef]
- Inoue, T.; Itoh, S.; Kobayashi, M.; Kang, Y.; Matsuo, R.; Wakisaka, S.; Morimoto, T. Serotonergic modulation of the hyperpolarizing spike afterpotential in rat jaw-closing motoneurons by PKA and PKC. J. Neurophysiol. 1999, 82, 626–637. [Google Scholar] [CrossRef]
- Shigeto, M.; Ramracheya, R.; Tarasov, A.I.; Cha, C.Y.; Chibalina, M.V.; Hastoy, B.; Philippaert, K.; Reinbothe, T.; Rorsman, N.; Salehi, A.; et al. GLP-1 stimulates insulin secretion by PKC-dependent TRPM4 and TRPM5 activation. J. Clin. Investig. 2015, 125, 4714–4728. [Google Scholar] [CrossRef]
- Roth, B.L.; Nakaki, T.; Chuang, D.M.; Costa, E. Aortic recognition sites for serotonin (5HT) are coupled to phospholipase C and modulate phosphatidylinositol turnover. Neuropharmacology 1984, 23, 1223–1225. [Google Scholar] [CrossRef]
- Lu, C.W.; Lin, T.Y.; Wang, S.J. Memantine depresses glutamate release through inhibition of voltage-dependent Ca2+ entry and protein kinase C in rat cerebral cortex nerve terminals: An NMDA receptor-independent mechanism. Neurochem. Int. 2010, 57, 168–176. [Google Scholar] [CrossRef]
- Moriguchi, S.; Shioda, N.; Han, F.; Yeh, J.Z.; Narahashi, T.; Fukunaga, K. Galantamine enhancement of long-term potentiation is mediated by calcium/calmodulin-dependent protein kinase II and protein kinase C activation. Hippocampus 2009, 19, 844–854. [Google Scholar] [CrossRef] [PubMed]
- Yogev-Falach, M.; Amit, T.; Bar-Am, O.; Weinstock, M.; YOUDIM, M.B. Involvement of MAP kinase in the regulation of amyloid precursor protein processing by novel cholinesterase inhibitors derived from rasagiline. FASEB J. 2002, 16, 1674–1676. [Google Scholar] [CrossRef] [PubMed]
- del Puerto, A.; Díaz-Hernández, J.I.; Tapia, M.; Gomez-Villafuertes, R.; Benitez, M.J.; Zhang, J.; Miras-Portugal, M.T.; Wandosell, F.; Díaz-Hernández, M.; Garrido, J.J. Adenylate cyclase 5 coordinates the action of ADP, P2Y1, P2Y13 and ATP-gated P2X7 receptors on axonal elongation. J. cell Sci. 2012, 125, 176–188. [Google Scholar] [CrossRef] [PubMed]
- Cutuli, D.; De Bartolo, P.; Caporali, P.; Tartaglione, A.M.; Oddi, D.; D’Amato, F.R.; Nobili, A.; D’Amelio, M.; Petrosini, L. Neuroprotective effects of donepezil against cholinergic depletion. Alzheimer’s Res. Therapy 2013, 5, 50. [Google Scholar]
- Li, M.; Bermak, J.C.; Wang, Z.W.; Zhou, Q.Y. Modulation of dopamine D2 receptor signaling by actin-binding protein (ABP-280). Mol. Pharmacol. 2000, 57, 446–452. [Google Scholar] [CrossRef]
- Shim, K.H.; Kim, S.H.; Hur, J.; Kim, D.H.; Demirev, A.V.; Yoon, S.Y. Small-molecule drug screening identifies drug Ro 31-8220 that reduces toxic phosphorylated tau in Drosophila melanogaster. Neurobiol. Dis. 2019, 130, 104519. [Google Scholar] [CrossRef]
- Wang, D.; Papp, A.C.; Binkley, P.F.; Johnson, J.A.; Sadée, W. Highly variable mRNA expression and splicing of L-type voltage-dependent calcium channel alpha subunit 1C in human heart tissues. Pharm. Genom. 2006, 16, 735. [Google Scholar] [CrossRef]
- Fão, L.; Mota, S.I.; Rego, A.C. c-Src regulates Nrf2 activity through PKCδ after oxidant stimulus. Biochim. et Biophys. Acta 2019, 1866, 686–698. [Google Scholar] [CrossRef]
- Zhang, G.; Stackman Jr, R.W. The role of serotonin 5-HT2A receptors in memory and cognition. Front. Pharmacol. 2015, 6, 225. [Google Scholar] [CrossRef]
- Mize, A.L.; Alper, R.H. Rapid uncoupling of serotonin-1A receptors in rat hippocampus by 17β-estradiol in vitro requires protein kinases A and C. Neuroendocrinology 2002, 76, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Sun, W.; Gao, L.; Liu, S. Therapeutic targets of hypercholesterolemia: HMGCR and LDLR. Diabetes, metabolic syndrome and obesity: Targets and therapy. Diabetes Metab. Syndr. Obes 2019, 12, 1543. [Google Scholar] [CrossRef] [PubMed]
- Behjati, M.; Etemadifar, M.; Esfahani, M.A. Cardiovascular effects of fingolimod: A review article. Iran. J. Neurol. 2014, 13, 119. [Google Scholar] [PubMed]
- Busquets-Garcia, A.; Gomis-González, M.; Salgado-Mendialdúa, V.; Galera-López, L.; Puighermanal, E.; Martín-García, E.; Maldonado, R.; Ozaita, A. Hippocampal protein kinase C signaling mediates the short-term memory impairment induced by delta9-tetrahydrocannabinol. Neuropsychopharmacology 2018, 43, 1021–1031. [Google Scholar] [CrossRef]
- Ringshausen, I.; Oelsner, M.; Bogner, C.; Peschel, C.; Decker, T. The immunomodulatory drug Leflunomide inhibits cell cycle progression of B-CLL cells. Leukemia 2008, 22, 635–638. [Google Scholar] [CrossRef] [PubMed]
- Sensken, S.C.; Gräler, M.H. Down-regulation of S1P1 receptor surface expression by protein kinase C inhibition. J. Biol. Chem. 2010, 285, 6298–6307. [Google Scholar] [CrossRef] [PubMed]
- Gorshkova, I.; He, D.; Berdyshev, E.; Usatuyk, P.; Burns, M.; Kalari, S.; Zhao, Y.; Pendyala, S.; Garcia, J.G.; Pyne, N.J.; et al. Protein kinase C-ϵ regulates sphingosine 1-phosphate-mediated migration of human lung endothelial cells through activation of phospholipase D2, protein kinase C-ζ, and Rac 1. J. Biol. Chem. 2008, 283, 11794–11806. [Google Scholar] [CrossRef]
- Onuţu, A.H. Duloxetine, an antidepressant with analgesic properties–a preliminary analysis. Rom. J. Anaesth. Intensive Care 2015, 22, 123. [Google Scholar]
- Gerrow, K.; Triller, A. GABAA receptor subunit composition and competition at synapses are tuned by GABAB receptor activity. Mol. Cell. Neurosci. 2014, 60, 97–107. [Google Scholar] [CrossRef]
- Torres, B.; Ruoho, A.E. N-terminus regulation of VMAT2 mediates methamphetamine-stimulated efflux. Neuroscience 2014, 259, 194–202. [Google Scholar] [CrossRef]
- Luderman, K.D.; Chen, R.; Ferris, M.J.; Jones, S.R.; Gnegy, M.E. Protein kinase C beta regulates the D2-Like dopamine autoreceptor. Neuropharmacology 2015, 89, 335–341. [Google Scholar] [CrossRef]
- Choi, J.A.; Chung, Y.R.; Byun, H.R.; Park, H.; Koh, J.Y.; Yoon, Y.H. The anti-ALS drug riluzole attenuates pericyte loss in the diabetic retinopathy of streptozotocin-treated mice. Toxicol. Appl. Pharmacol. 2017, 315, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Abou-Lovergne, A.; Collado-Hilly, M.; Monnet, F.P.; Koukoui, O.; Prigent, S.; Coquil, J.F.; Dupont, G.; Combettes, L. Investigation of the role of sigma1-receptors in inositol 1, 4, 5-trisphosphate dependent calcium signaling in hepatocytes. Cell calcium 2011, 50, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Nikodemova, M.; Watters, J.J.; Jackson, S.J.; Yang, S.K.; Duncan, I.D. Minocycline down-regulates MHC II expression in microglia and macrophages through inhibition of IRF-1 and protein kinase C (PKC) α/βII. J. Biol. Chem. 2007, 282, 15208–15216. [Google Scholar] [CrossRef]
- Lee, J.G.; Cho, H.Y.; Park, S.W.; Seo, M.K.; Kim, Y.H. Effects of olanzapine on brain-derived neurotrophic factor gene promoter activity in SH-SY5Y neuroblastoma cells. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2010, 34, 1001–1006. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Oishi, K.; Uchida, M.; Echizen, H. Edaravone, a radical scavenger, may enhance or produce antiproliferative effects of fluvastatin, amlodipine, ozagrel, GF109203X and Y27632 on cultured basilar artery smooth muscle cells. Biol. Pharm. Bull. 2003, 26, 1706–1710. [Google Scholar] [CrossRef] [PubMed]
- Kimoto, Y.; Kinoshita, H.; Nakahata, K.; Dojo, M.; Hatano, Y. Inhibitory Effects of Lidocaine and Mexiletine on Vasorelaxation Mediated by Adenosine Triphosphate–sensitive K+ Channels and the c. Anesthesiology 2005, 102, 581–587. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PKC Isozymes | Compound | Function |
|---|---|---|
| PKCα PKCε | Epigallocatechin 3-gallate (EGCG) [169,170,171,172,173,174,175,176,177] | Neuroprotection against Aβ toxicity Promoting autophagy |
| PKCδ PKCα | Resveratrol [182,183,184] | Attenuating cellular toxicity Inducing autophagy mechanism via AMPK/SIRT1 and mTOR |
| PKCε PKCδ PKCγ | Quercetin [185,186,187,188,189] | Inducing neuronal autophagy and inhibiting cell death through PI3K/Akt signaling Decreases neuropathic pain |
| PKCθ | Curcumin [191,192,193] | Increase autophagy in neuroblastoma Decrease autophagy under cerebral ischemia- reperfusion via PI3K/Akt/mTOR |
| Disease | Molecule | Class/SubClass | Target | Mode of Action | Targeted Pathway | Affected PKCs Isozyme | PKC Related Action | Clinical Trial Status |
|---|---|---|---|---|---|---|---|---|
| Parkinson’s | Levodopa [194] | Carboxylic acids—Amino acids, peptides | Dopamine D1, D2, D3, D4, D5 receptors | Agonist | Dopaminergic synapse | PKC-α, PKC-β, PKC-γ | Induced phosphatidylinositol signaling | FDA Approved 2008 |
| Rasagiline [195] | Benzenoids—Indanes | Amine oxidase (flavin-containing) B, Apoptosis regulator Bcl-2 | Inhibitor | Serotonergic synapse | PKC-α, PKC-β, PKC-γ | Neuroprotection by inhibiting Caspase 3 | FDA Approved 2006 | |
| Bromocriptine [196] | Ergoline—Lysergic acids | D2 dopamine receptor | Agonist | Neuroactive ligand-receptor interaction | PKC-α, PKC-β, PKC-γ, PKC-ε | Enhanced MAPK phosphorylation and PKC activity | FDA Approved 2004 | |
| Pramipexole [197] | Organonitrogen—Amines | Dopamine D2, D3, D4 receptors | Agonist | Dopaminergic synapse | PKC-α, PKC-β, PKC-γ | PKC/MAPK pathways interference | FDA Approved 1997 | |
| Quetiapine [198] | Benzothiazepines—Dibenzothiazepines | D2 dopamine receptor, 5-hydroxytryptamine receptor 2A | Antagonist | Neuroactive ligand-receptor interaction | PKC-α, PKC-β, PKC-γ | PKC activation through cAMP and calcium pathways | Phase IV Completed | |
| Isradipine [199] | Benzoxadiazoles | Calcium voltage-gated channel subunit alpha1 C | Inhibitor | GABAergic synapse | PKC-α, PKC-β, PKC-γ | Increased PKC activity | Phase III Completed | |
| Pimavanserin [200] | Phenol ethers | D2 dopamine receptor, 5-hydroxytryptamine receptor 2A | Inverse Agonist | Neuroactive ligand-receptor interaction | PKC-α, PKC-β, PKC-γ | PKC activation and phosphatidylinositol signaling | Phase III Completed | |
| Alzheimer’s | Memantine [201] | Organonitrogen—Amines | Glutamate ionotropic receptor NMDA type subunit 3A | Antagonist | Neuroactive ligand-receptor interaction | PKC-α, PKC-β, PKC-γ | Reduced depolarization-induced phosphorylation of PKC | FDA Approved 2003 |
| Galantamine [202] | Piperidines—Benzylpiperidines | Acetylcholinesterase | Inhibitor | Cholinergic synapse | PKC-α, PKC-β, PKC-γ | Calmodulin-dependent PKC activation | FDA Approved 2001 | |
| Rivastigmine [203] | Benzene—Phenoxy compounds | Acetylcholinesterase | Inhibitor | Cholinergic synapse | PKC-α, PKC-β, PKC-γ | Stimulation of sAPPα increases PKC activity | FDA Approved 2000 | |
| Rosiglitazone [204] | Phenol ethers | Peroxisome proliferator-activated receptor gamma | Agonist | PPAR signaling pathway | PKC-α, PKC-β, PKC-γ | Downstream modulation of PKC through adenylyl cyclase | FDA Approved 1999 | |
| Donepezil [205] | Piperidines—Benzylpiperidines | Acetylcholinesterase | Modulator | Cholinergic synapse | PKC-α, PKC-β, PKC-γ | Activation of phospholipase C / PKC | FDA Approved 1996 | |
| Aripiprazole [206] | Diazinanes—Piperazines | D2 dopamine receptor, 5-hydroxytryptamine receptor 2A | Antagonist | Dopaminergic synapse | PKC-α, PKC-β, PKC-γ | Reduced phosphorylation of DRD2 | Phase IV Completed | |
| TRx0237 [207] | Benzothiazines | Microtubule-associated protein tau | Aggregation Inhibitor | MAPK signaling pathway | PKC-α, PKC-β, PKC-γ | Decreased activity of PKC through tau inhibition | Phase III Completed | |
| Nilvadipine [208] | Pyridines—Hydropyridines | Voltage-dependent L-type calcium channel subunit alpha-1C | Inhibitor | GABAergic synapse | PKC-α, PKC-β, PKC-γ | Reduced activation via PKC | Phase III Completed | |
| Intepirdine [209] | Diazinanes—Piperazines | 5-hydroxytryptamine receptor 4 | Antagonist | Neuroactive ligand-receptor interaction | PKC-δ | Decreased activity of PKCδ through c-src kinase | Phase III Completed | |
| Idalopirdine [210] | Indoles—Tryptamines | 5-hydroxytryptamine receptor 2A | Antagonist | Neuroactive ligand-receptor interaction | PKC-α, PKC-β, PKC-γ | Reduced calmodulin mediated phosphorylation | Phase III Completed | |
| Brexpiprazole [211] | Diazinanes—Piperazines | 5-hydroxytryptamine receptor 1A, D2 dopamine receptor | Agonist/Partial agonist | Neuroactive ligand-receptor interaction | PKC-α, PKC-β, PKC-γ | Increased phosphorylation activity | Phase III Completed | |
| Atorvastatin [212] | Pyrroles—Substituted pyrroles | 3-hydroxy-3-methylglutaryl-coenzyme A reductase | Inhibitor | AMPK signaling pathway | PKC-α, PKC-β, PKC-γ | Reduced phosphorylation of HMGCR | Phase III Completed | |
| Multiple Sclerosis | Fingolimod [213] | Organonitrogen—Amimes | Sphingosine 1-phosphate receptor 5 | Modulator | Neuroactive ligand-receptor interaction | PKC-βII | Increased PKC activity | FDA Approved 2010 |
| Cannabidiol [214] | Prenol lipids—Monoterpenoids | Cannabinoid receptors | Agonist | Neuroactive ligand-receptor interaction | PKC-α, PKC-β, PKC-γ | Hippocampal PKC/neurogranin signaling | Phase IV Completed | |
| Teriflunomide [215] | Benzene—Trifluoromethylbenzenes | Dihydroorotate dehydrogenase | Inhibitor | Metabolic pathways | PKC-δ | PI3-kinase/PKC-δ and nuclear factor-kappa B signaling | Phase IV Completed | |
| Ponesimod [216] | Phenol ethers | Sphingosine-1-phosphate receptor 1 | Modulator | Neuroactive ligand-receptor interaction | PKC-α, PKC-β, PKC-γ | Inhibited PKC signaling | Phase III Completed | |
| Ozanimod [217] | Azoles—Oxadiazoles | Sphingosine-1-phosphate receptor 5 | Modulator | Neuroactive ligand-receptor interaction | PKC-ε | Increased PKC-ε activity | Phase III Completed | |
| Duloxetine [218] | Naphthalenes | Sodium-dependent dopamine transporter | Inhibitor | Dopaminergic synapse | PKC-ε | PKC-ε activity through cytokines release | Phase III Completed | |
| Arbaclofen [219] | Carboxylic acids—Amino acids, peptides | GABA type A receptor associated protein like 1 | Inhibitor | GABAergic synapse | PKC-α, PKC-β, PKC-γ | Reduced PKC phosphorylation | Phase III Completed | |
| Huntington’s | Deutetrabenazine [220] | Tetrahydroisoquinolines | Solute carrier family 18 member A2 | Inhibitor | Dopaminergic synapse | PKC-α, PKC-β, PKC-γ | Reduced PKC phosphorylation | FDA Approved 2017 |
| Tetrabenazine [221] | Tetrahydroisoquinolines | D2 dopamine receptor | Inhibitor | Dopaminergic synapse | PKC-β | Reduced dopaminergic signaling | FDA Approved 2015 | |
| Riluzole [222] | Benzothiazoles | Glutamate metabotropic receptor 1 | Inhibitor | Neuroactive ligand-receptor interaction | PKC-α, PKC-β, PKC-γ | Inhibited PKC signaling | Phase III Completed | |
| Tiapride [221] | Benzene—Benzenesulfonyl | D2 dopamine receptor | Inhibitor | Dopaminergic synapse | PKC-β | Reduced dopaminergic signaling | Phase III Completed | |
| Pridopidine [223] | Piperidines—Phenylpiperidines | Superoxide Dismutase-1 | Modulator | Neuroactive ligand-receptor interaction | PKC-α, PKC-β, PKC-γ | Stimulation of PKC activity | Phase III Completed | |
| Minocycline [224] | Tetracyclines | Caspase-3 | Inhibitor | Serotonergic synapse | PKC-α, PKC-βII | Downregulated MHC II through PKC Inhibition | Phase III Completed | |
| Olanzapine [225] | Benzodiazepines | D2 dopamine receptor | Agonist | Dopaminergic synapse | PKC-α, PKC-β, PKC-γ | Enhanced PKC activity | Phase III Completed | |
| ALS | Edaravone [226] | Azolines—Pyrazolines | Peroxyl radicals | Inhibitor | ROS-Triggered Intracellular Signaling | PKC-α, PKC-β, PKC-γ | Enhanced PKC activity | FDA Approved 2017 |
| Riluzole [222] | Benzothiazoles | Glutamate metabotropic receptor 1 | Inhibitor | Neuroactive ligand-receptor interaction | PKC-α, PKC-β, PKC-γ | Inhibited PKC signaling | FDA Approved 1995 | |
| Mexiletine [227] | Phenol ethers | Sodium channel protein type 5 subunit alpha | Inhibitor | Adrenergic signaling in cardiomyocytes | PKC-α, PKC-βI | Enhanced PKC activity | Phase IV Completed |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaleli, H.N.; Ozer, E.; Kaya, V.O.; Kutlu, O. Protein Kinase C Isozymes and Autophagy during Neurodegenerative Disease Progression. Cells 2020, 9, 553. https://doi.org/10.3390/cells9030553
Kaleli HN, Ozer E, Kaya VO, Kutlu O. Protein Kinase C Isozymes and Autophagy during Neurodegenerative Disease Progression. Cells. 2020; 9(3):553. https://doi.org/10.3390/cells9030553
Chicago/Turabian StyleKaleli, Humeyra Nur, Ebru Ozer, Veysel Ogulcan Kaya, and Ozlem Kutlu. 2020. "Protein Kinase C Isozymes and Autophagy during Neurodegenerative Disease Progression" Cells 9, no. 3: 553. https://doi.org/10.3390/cells9030553
APA StyleKaleli, H. N., Ozer, E., Kaya, V. O., & Kutlu, O. (2020). Protein Kinase C Isozymes and Autophagy during Neurodegenerative Disease Progression. Cells, 9(3), 553. https://doi.org/10.3390/cells9030553