Translational Regulations in Response to Endoplasmic Reticulum Stress in Cancers

 , ,
, , {kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Endoplasmic Reticulum Stress Signalling in Cancer
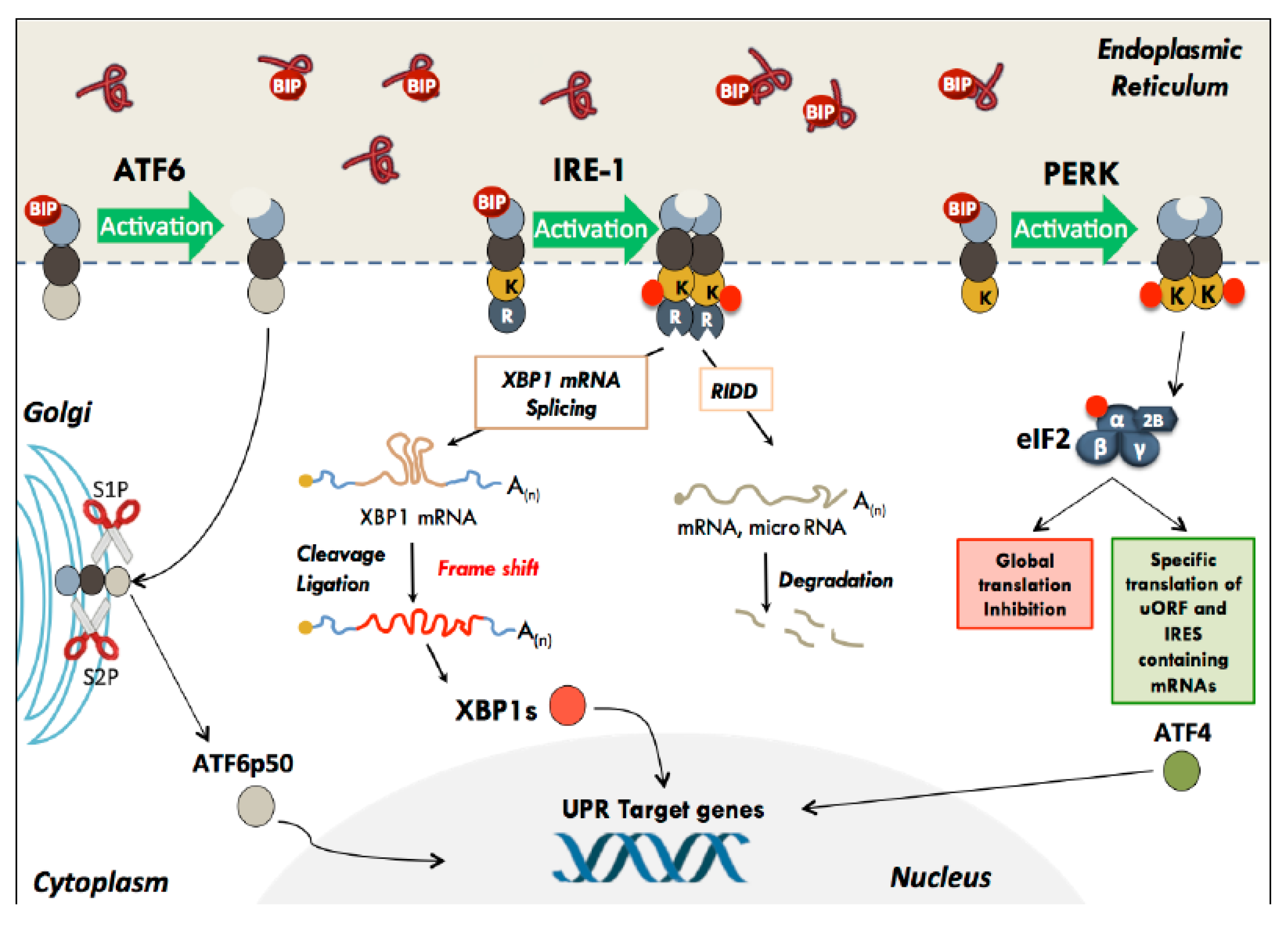
2.1. The Unfolded Protein Response
2.2. ATF6
2.3. IRE-1
2.4. PERK
3. Translational Regulation: Dealing with Endoplasmic Reticulum Stress
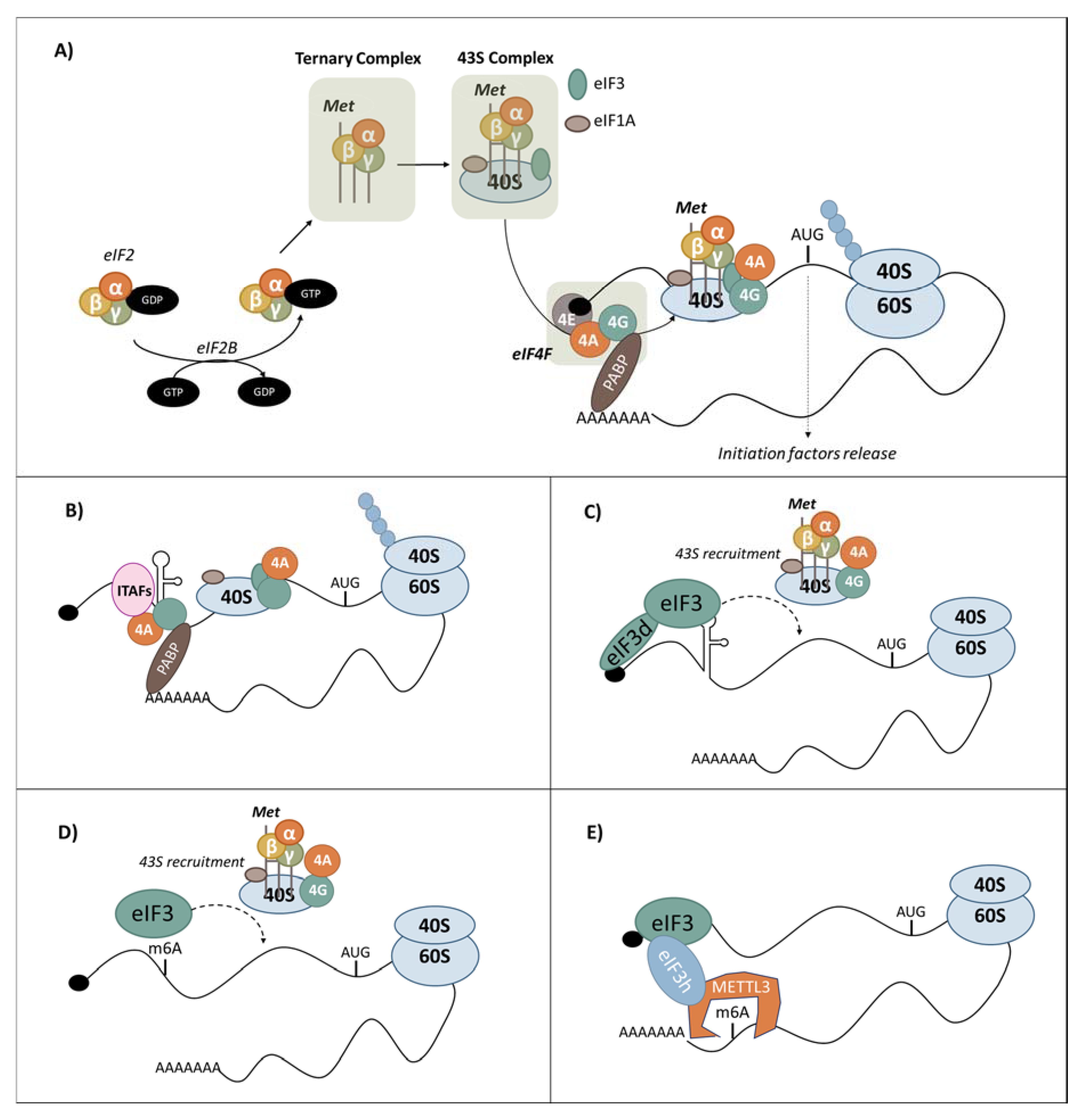
3.1. Canonical Cap-Dependent Translation Initiation
3.2. Translational Control PERK-Mediated under ER Stress
3.3. Selective mRNA Translation during eIF2 Phosphorylation:
3.3.1. Regulation by uORF
Upstream Open Reading Frames: uORFs
uORFs Translation upon ER Stress
3.3.2. Cap-Independent Translation Regulation
IRES-Dependent Translation Initiation
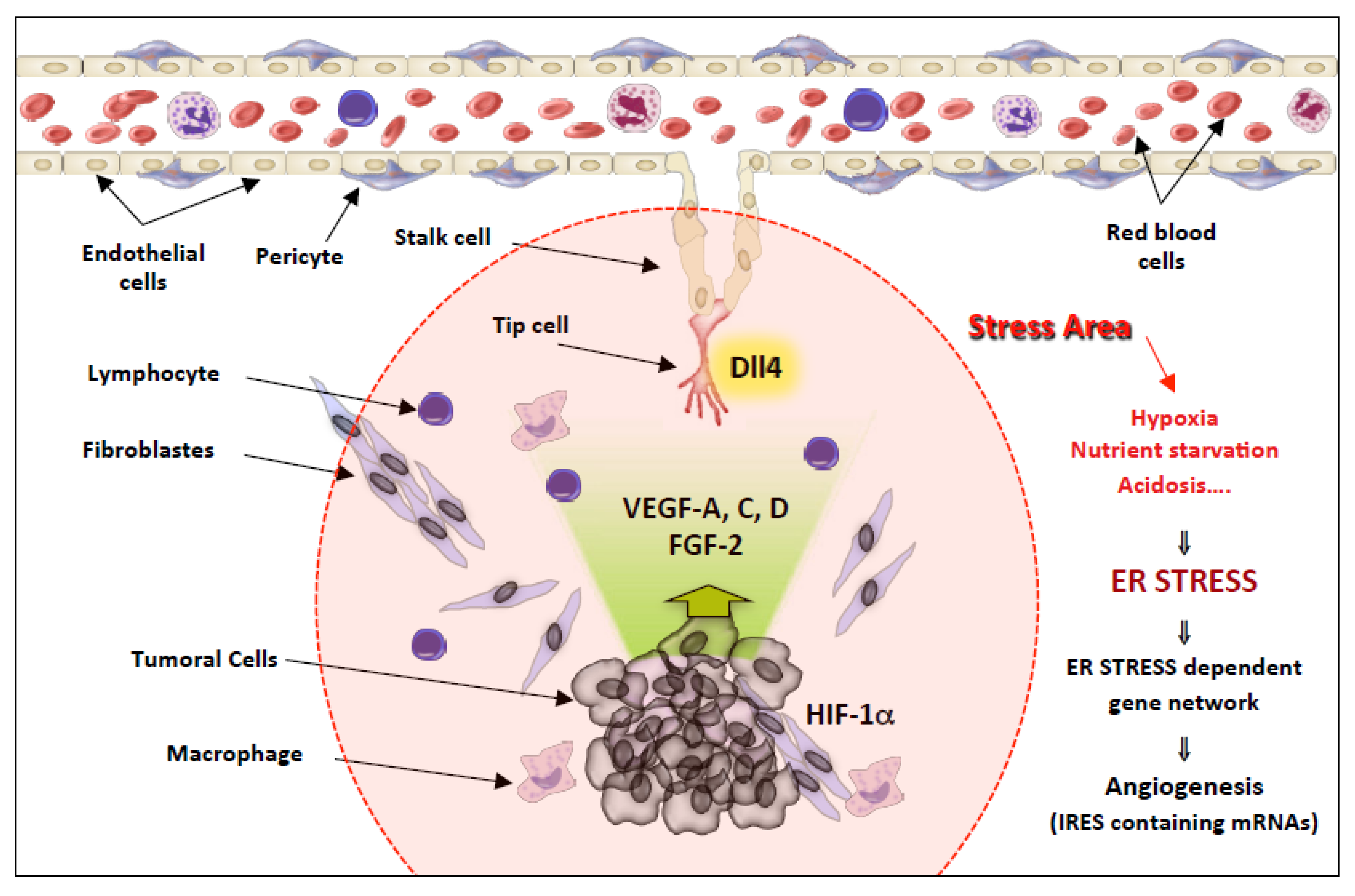
IRES Regulation in Response to Stress
The Angiogenesis Paradigm
3.4. mRNA Translational Control by IRE1 under ER Stress
3.5. mRNA Structures and Modifications Regulate Translation:
3.5.1. eIF3 Recruitment by RNA Structures
3.5.2. m6A-Dependent Translation Initiation
4. Conclusions
Funding
Conflicts of Interest
References
- Reid, D.W.; Nicchitta, C.V. Diversity and selectivity in mRNA translation on the endoplasmic reticulum. Nat. Rev. Mol. Cell Biol. 2015, 16, 221–231. [Google Scholar] [CrossRef]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef]
- Dejeans, N.; Manie, S.; Hetz, C.; Bard, F.; Hupp, T.; Agostinis, P.; Samali, A.; Chevet, E. Addicted to secrete —Novel concepts and targets in cancer therapy. Trends Mol. Med. 2014, 20, 242–250. [Google Scholar] [CrossRef]
- Khan, M.M.; Nomura, T.; Chiba, T.; Tanaka, K.; Yoshida, H.; Mori, K.; Ishii, S. The fusion oncoprotein PML-RARalpha induces endoplasmic reticulum (ER)-associated degradation of N-CoR and ER stress. J. Biol. Chem. 2004, 279, 11814–11824. [Google Scholar] [CrossRef]
- Kriss, C.L.; Pinilla-Ibarz, J.A.; Mailloux, A.W.; Powers, J.J.; Tang, C.H.; Kang, C.W.; Zanesi, N.; Epling-Burnette, P.K.; Sotomayor, E.M.; Croce, C.M.; et al. Overexpression of TCL1 activates the endoplasmic reticulum stress response: A novel mechanism of leukemic progression in mice. Blood 2012, 120, 1027–1038. [Google Scholar] [CrossRef]
- Senovilla, L.; Vitale, I.; Martins, I.; Tailler, M.; Pailleret, C.; Michaud, M.; Galluzzi, L.; Adjemian, S.; Kepp, O.; Niso-Santano, M.; et al. An immunosurveillance mechanism controls cancer cell ploidy. Science 2012, 337, 1678–1684. [Google Scholar] [CrossRef]
- Hetz, C.; Glimcher, L.H. Fine-tuning of the unfolded protein response: Assembling the IRE1alpha interactome. Mol. Cell 2009, 35, 551–561. [Google Scholar] [CrossRef]
- Kaufman, R.J. Stress signaling from the lumen of the endoplasmic reticulum: Coordination of gene transcriptional and translational controls. Genes Dev. 1999, 13, 1211–1233. [Google Scholar] [CrossRef]
- Higa, A.; Taouji, S.; Lhomond, S.; Jensen, D.; Fernandez-Zapico, M.E.; Simpson, J.C.; Pasquet, J.M.; Schekman, R.; Chevet, E. Endoplasmic reticulum stress-activated transcription factor ATF6alpha requires the disulfide isomerase PDIA5 to modulate chemoresistance. Mol. Cell Biol. 2014, 34, 1839–1849. [Google Scholar] [CrossRef]
- Eletto, D.; Eletto, D.; Dersh, D.; Gidalevitz, T.; Argon, Y. Protein disulfide isomerase A6 controls the decay of IRE1alpha signaling via disulfide-dependent association. Mol. Cell 2014, 53, 562–576. [Google Scholar] [CrossRef] [PubMed]
- Sepulveda, D.; Rojas-Rivera, D.; Rodriguez, D.A.; Groenendyk, J.; Kohler, A.; Lebeaupin, C.; Ito, S.; Urra, H.; Carreras-Sureda, A.; Hazari, Y.; et al. Interactome Screening Identifies the ER Luminal Chaperone Hsp47 as a Regulator of the Unfolded Protein Response Transducer IRE1alpha. Mol. Cell 2018, 69, 238–252 e237. [Google Scholar] [CrossRef] [PubMed]
- Karagoz, G.E.; Acosta-Alvear, D.; Nguyen, H.T.; Lee, C.P.; Chu, F.; Walter, P. An unfolded protein-induced conformational switch activates mammalian IRE1. eLife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Papa, F.R. The Unfolded Protein Response and Cell Fate Control. Mol. Cell 2018, 69, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Chevet, E.; Hetz, C.; Samali, A. Endoplasmic reticulum stress-activated cell reprogramming in oncogenesis. Cancer Discov. 2015, 5, 586–597. [Google Scholar] [CrossRef]
- Thuerauf, D.J.; Marcinko, M.; Belmont, P.J.; Glembotski, C.C. Effects of the isoform-specific characteristics of ATF6 alpha and ATF6 beta on endoplasmic reticulum stress response gene expression and cell viability. J. Biol. Chem. 2007, 282, 22865–22878. [Google Scholar] [CrossRef]
- Yamamoto, K.; Sato, T.; Matsui, T.; Sato, M.; Okada, T.; Yoshida, H.; Harada, A.; Mori, K. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev. Cell 2007, 13, 365–376. [Google Scholar] [CrossRef]
- Shen, J.; Chen, X.; Hendershot, L.; Prywes, R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev. Cell 2002, 3, 99–111. [Google Scholar] [CrossRef]
- Yamamoto, K.; Yoshida, H.; Kokame, K.; Kaufman, R.J.; Mori, K. Differential contributions of ATF6 and XBP1 to the activation of endoplasmic reticulum stress-responsive cis-acting elements ERSE, UPRE and ERSE-II. J. Biochem. 2004, 136, 343–350. [Google Scholar] [CrossRef]
- Adachi, Y.; Yamamoto, K.; Okada, T.; Yoshida, H.; Harada, A.; Mori, K. ATF6 is a transcription factor specializing in the regulation of quality control proteins in the endoplasmic reticulum. Cell Struct. Funct. 2008, 33, 75–89. [Google Scholar] [CrossRef]
- Yoshida, H.; Okada, T.; Haze, K.; Yanagi, H.; Yura, T.; Negishi, M.; Mori, K. ATF6 activated by proteolysis binds in the presence of NF-Y (CBF) directly to the cis-acting element responsible for the mammalian unfolded protein response. Mol. Cell Biol. 2000, 20, 6755–6767. [Google Scholar] [CrossRef]
- Ma, Y.; Brewer, J.W.; Diehl, J.A.; Hendershot, L.M. Two distinct stress signaling pathways converge upon the CHOP promoter during the mammalian unfolded protein response. J. Mol. Biol. 2002, 318, 1351–1365. [Google Scholar] [CrossRef]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef]
- Schewe, D.M.; Aguirre-Ghiso, J.A. ATF6alpha-Rheb-mTOR signaling promotes survival of dormant tumor cells in vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 10519–10524. [Google Scholar] [CrossRef]
- Iwawaki, T.; Akai, R.; Yamanaka, S.; Kohno, K. Function of IRE1 alpha in the placenta is essential for placental development and embryonic viability. Proc. Natl. Acad. Sci. USA 2009, 106, 16657–16662. [Google Scholar] [CrossRef]
- Bertolotti, A.; Wang, X.; Novoa, I.; Jungreis, R.; Schlessinger, K.; Cho, J.H.; West, A.B.; Ron, D. Increased sensitivity to dextran sodium sulfate colitis in IRE1beta-deficient mice. J. Clin. Investig. 2001, 107, 585–593. [Google Scholar] [CrossRef]
- Martino, M.B.; Jones, L.; Brighton, B.; Ehre, C.; Abdulah, L.; Davis, C.W.; Ron, D.; O’Neal, W.K.; Ribeiro, C.M. The ER stress transducer IRE1beta is required for airway epithelial mucin production. Mucosal Immunol. 2013, 6, 639–654. [Google Scholar] [CrossRef]
- Urano, F.; Bertolotti, A.; Ron, D. IRE1 and efferent signaling from the endoplasmic reticulum. J. Cell Sci. 2000, 113, 3697–3702. [Google Scholar]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef]
- Peschek, J.; Acosta-Alvear, D.; Mendez, A.S.; Walter, P. A conformational RNA zipper promotes intron ejection during non-conventional XBP1 mRNA splicing. EMBO Rep. 2015, 16, 1688–1698. [Google Scholar] [CrossRef]
- Lu, Y.; Liang, F.X.; Wang, X. A synthetic biology approach identifies the mammalian UPR RNA ligase RtcB. Mol. Cell 2014, 55, 758–770. [Google Scholar] [CrossRef]
- Plumb, R.; Zhang, Z.R.; Appathurai, S.; Mariappan, M. A functional link between the co-translational protein translocation pathway and the UPR. eLife 2015, 4. [Google Scholar] [CrossRef]
- Yanagitani, K.; Imagawa, Y.; Iwawaki, T.; Hosoda, A.; Saito, M.; Kimata, Y.; Kohno, K. Cotranslational targeting of XBP1 protein to the membrane promotes cytoplasmic splicing of its own mRNA. Mol. Cell 2009, 34, 191–200. [Google Scholar] [CrossRef]
- Yanagitani, K.; Kimata, Y.; Kadokura, H.; Kohno, K. Translational pausing ensures membrane targeting and cytoplasmic splicing of XBP1u mRNA. Science 2011, 331, 586–589. [Google Scholar] [CrossRef]
- Acosta-Alvear, D.; Zhou, Y.; Blais, A.; Tsikitis, M.; Lents, N.H.; Arias, C.; Lennon, C.J.; Kluger, Y.; Dynlacht, B.D. XBP1 controls diverse cell type- and condition-specific transcriptional regulatory networks. Mol. Cell 2007, 27, 53–66. [Google Scholar] [CrossRef]
- Hollien, J.; Weissman, J.S. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 2006, 313, 104–107. [Google Scholar] [CrossRef]
- Han, D.; Lerner, A.G.; Vande Walle, L.; Upton, J.P.; Xu, W.; Hagen, A.; Backes, B.J.; Oakes, S.A.; Papa, F.R. IRE1alpha kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell 2009, 138, 562–575. [Google Scholar] [CrossRef]
- So, J.S.; Hur, K.Y.; Tarrio, M.; Ruda, V.; Frank-Kamenetsky, M.; Fitzgerald, K.; Koteliansky, V.; Lichtman, A.H.; Iwawaki, T.; Glimcher, L.H.; et al. Silencing of lipid metabolism genes through IRE1alpha-mediated mRNA decay lowers plasma lipids in mice. Cell Metab. 2012, 16, 487–499. [Google Scholar] [CrossRef]
- Upton, J.P.; Wang, L.; Han, D.; Wang, E.S.; Huskey, N.E.; Lim, L.; Truitt, M.; McManus, M.T.; Ruggero, D.; Goga, A.; et al. IRE1alpha cleaves select microRNAs during ER stress to derepress translation of proapoptotic Caspase-2. Science 2012, 338, 818–822. [Google Scholar] [CrossRef]
- Pluquet, O.; Dejeans, N.; Bouchecareilh, M.; Lhomond, S.; Pineau, R.; Higa, A.; Delugin, M.; Combe, C.; Loriot, S.; Cubel, G.; et al. Posttranscriptional regulation of PER1 underlies the oncogenic function of IREalpha. Cancer Res. 2013, 73, 4732–4743. [Google Scholar] [CrossRef]
- Dejeans, N.; Pluquet, O.; Lhomond, S.; Grise, F.; Bouchecareilh, M.; Juin, A.; Meynard-Cadars, M.; Bidaud-Meynard, A.; Gentil, C.; Moreau, V.; et al. Autocrine control of glioma cells adhesion and migration through IRE1alpha-mediated cleavage of SPARC mRNA. J. Cell Sci. 2012, 125, 4278–4287. [Google Scholar] [CrossRef] [PubMed]
- Bright, M.D.; Itzhak, D.N.; Wardell, C.P.; Morgan, G.J.; Davies, F.E. Cleavage of BLOC1S1 mRNA by IRE1 Is Sequence Specific, Temporally Separate from XBP1 Splicing, and Dispensable for Cell Viability under Acute Endoplasmic Reticulum Stress. Mol. Cell Biol. 2015, 35, 2186–2202. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Lawrence, D.A.; Marsters, S.; Acosta-Alvear, D.; Kimmig, P.; Mendez, A.S.; Paton, A.W.; Paton, J.C.; Walter, P.; Ashkenazi, A. Opposing unfolded-protein-response signals converge on death receptor 5 to control apoptosis. Science 2014, 345, 98–101. [Google Scholar] [CrossRef]
- Maurel, M.; Chevet, E.; Tavernier, J.; Gerlo, S. Getting RIDD of RNA: IRE1 in cell fate regulation. Trends Biochem. Sci. 2014, 39, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Korennykh, A.; Walter, P. Structural basis of the unfolded protein response. Annu. Rev. Cell Dev. Biol. 2012, 28, 251–277. [Google Scholar] [CrossRef] [PubMed]
- Korennykh, A.V.; Egea, P.F.; Korostelev, A.A.; Finer-Moore, J.; Zhang, C.; Shokat, K.M.; Stroud, R.M.; Walter, P. The unfolded protein response signals through high-order assembly of Ire1. Nature 2009, 457, 687–693. [Google Scholar] [CrossRef]
- Tam, A.B.; Koong, A.C.; Niwa, M. Ire1 has distinct catalytic mechanisms for XBP1/HAC1 splicing and RIDD. Cell Rep. 2014, 9, 850–858. [Google Scholar] [CrossRef]
- Ghosh, R.; Wang, L.; Wang, E.S.; Perera, B.G.; Igbaria, A.; Morita, S.; Prado, K.; Thamsen, M.; Caswell, D.; Macias, H.; et al. Allosteric inhibition of the IRE1alpha RNase preserves cell viability and function during endoplasmic reticulum stress. Cell 2014, 158, 534–548. [Google Scholar] [CrossRef]
- Obacz, J.; Avril, T.; Le Reste, P.J.; Urra, H.; Quillien, V.; Hetz, C.; Chevet, E. Endoplasmic reticulum proteostasis in glioblastoma-From molecular mechanisms to therapeutic perspectives. Sci. Signal. 2017, 10. [Google Scholar] [CrossRef]
- Auf, G.; Jabouille, A.; Guerit, S.; Pineau, R.; Delugin, M.; Bouchecareilh, M.; Magnin, N.; Favereaux, A.; Maitre, M.; Gaiser, T.; et al. Inositol-requiring enzyme 1alpha is a key regulator of angiogenesis and invasion in malignant glioma. Proc. Natl. Acad. Sci. USA 2010, 107, 15553–15558. [Google Scholar] [CrossRef]
- Hong, S.Y.; Hagen, T. Multiple myeloma Leu167Ile (c.499C>A) mutation prevents XBP1 mRNA splicing. Br. J. Haematol. 2013, 161, 898–901. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Iliopoulos, D.; Zhang, Q.; Tang, Q.; Greenblatt, M.B.; Hatziapostolou, M.; Lim, E.; Tam, W.L.; Ni, M.; Chen, Y.; et al. XBP1 promotes triple-negative breast cancer by controlling the HIF1alpha pathway. Nature 2014, 508, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Bagratuni, T.; Wu, P.; Gonzalez de Castro, D.; Davenport, E.L.; Dickens, N.J.; Walker, B.A.; Boyd, K.; Johnson, D.C.; Gregory, W.; Morgan, G.J.; et al. XBP1s levels are implicated in the biology and outcome of myeloma mediating different clinical outcomes to thalidomide-based treatments. Blood 2010, 116, 250–253. [Google Scholar] [CrossRef] [PubMed]
- Gambella, M.; Rocci, A.; Passera, R.; Gay, F.; Omede, P.; Crippa, C.; Corradini, P.; Romano, A.; Rossi, D.; Ladetto, M.; et al. High XBP1 expression is a marker of better outcome in multiple myeloma patients treated with bortezomib. Haematologica 2014, 99, e14–e16. [Google Scholar] [CrossRef]
- Chapman, M.A.; Lawrence, M.S.; Keats, J.J.; Cibulskis, K.; Sougnez, C.; Schinzel, A.C.; Harview, C.L.; Brunet, J.P.; Ahmann, G.J.; Adli, M.; et al. Initial genome sequencing and analysis of multiple myeloma. Nature 2011, 471, 467–472. [Google Scholar] [CrossRef]
- Leung-Hagesteijn, C.; Erdmann, N.; Cheung, G.; Keats, J.J.; Stewart, A.K.; Reece, D.E.; Chung, K.C.; Tiedemann, R.E. Xbp1s-Negative Tumor B Cells and Pre-Plasmablasts Mediate Therapeutic Proteasome Inhibitor Resistance in Multiple Myeloma. Cancer Cell 2015, 28, 541–542. [Google Scholar] [CrossRef]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef]
- Harding, H.P.; Novoa, I.; Zhang, Y.; Zeng, H.; Wek, R.; Schapira, M.; Ron, D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 2000, 6, 1099–1108. [Google Scholar] [CrossRef]
- Marciniak, S.J.; Garcia-Bonilla, L.; Hu, J.; Harding, H.P.; Ron, D. Activation-dependent substrate recruitment by the eukaryotic translation initiation factor 2 kinase PERK. J. Cell Biol. 2006, 172, 201–209. [Google Scholar] [CrossRef]
- Bobrovnikova-Marjon, E.; Grigoriadou, C.; Pytel, D.; Zhang, F.; Ye, J.; Koumenis, C.; Cavener, D.; Diehl, J.A. PERK promotes cancer cell proliferation and tumor growth by limiting oxidative DNA damage. Oncogene 2010, 29, 3881–3895. [Google Scholar] [CrossRef]
- Zhang, W.; Hietakangas, V.; Wee, S.; Lim, S.C.; Gunaratne, J.; Cohen, S.M. ER stress potentiates insulin resistance through PERK-mediated FOXO phosphorylation. Genes Dev. 2013, 27, 441–449. [Google Scholar] [CrossRef]
- Bobrovnikova-Marjon, E.; Pytel, D.; Riese, M.J.; Vaites, L.P.; Singh, N.; Koretzky, G.A.; Witze, E.S.; Diehl, J.A. PERK utilizes intrinsic lipid kinase activity to generate phosphatidic acid, mediate Akt activation, and promote adipocyte differentiation. Mol. Cell Biol. 2012, 32, 2268–2278. [Google Scholar] [CrossRef]
- Nanduri, S.; Rahman, F.; Williams, B.R.; Qin, J. A dynamically tuned double-stranded RNA binding mechanism for the activation of antiviral kinase PKR. EMBO J. 2000, 19, 5567–5574. [Google Scholar] [CrossRef]
- Ung, T.L.; Cao, C.; Lu, J.; Ozato, K.; Dever, T.E. Heterologous dimerization domains functionally substitute for the double-stranded RNA binding domains of the kinase PKR. EMBO J. 2001, 20, 3728–3737. [Google Scholar] [CrossRef]
- Zhang, F.; Romano, P.R.; Nagamura-Inoue, T.; Tian, B.; Dever, T.E.; Mathews, M.B.; Ozato, K.; Hinnebusch, A.G. Binding of double-stranded RNA to protein kinase PKR is required for dimerization and promotes critical autophosphorylation events in the activation loop. J. Biol. Chem. 2001, 276, 24946–24958. [Google Scholar] [CrossRef]
- Dong, J.; Qiu, H.; Garcia-Barrio, M.; Anderson, J.; Hinnebusch, A.G. Uncharged tRNA activates GCN2 by displacing the protein kinase moiety from a bipartite tRNA-binding domain. Mol. Cell 2000, 6, 269–279. [Google Scholar] [CrossRef]
- Deng, J.; Harding, H.P.; Raught, B.; Gingras, A.C.; Berlanga, J.J.; Scheuner, D.; Kaufman, R.J.; Ron, D.; Sonenberg, N. Activation of GCN2 in UV-irradiated cells inhibits translation. Curr. Biol. 2002, 12, 1279–1286. [Google Scholar] [CrossRef]
- Hao, S.; Sharp, J.W.; Ross-Inta, C.M.; McDaniel, B.J.; Anthony, T.G.; Wek, R.C.; Cavener, D.R.; McGrath, B.C.; Rudell, J.B.; Koehnle, T.J.; et al. Uncharged tRNA and sensing of amino acid deficiency in mammalian piriform cortex. Science 2005, 307, 1776–1778. [Google Scholar] [CrossRef]
- Chen, J.J.; Crosby, J.S.; London, I.M. Regulation of heme-regulated eIF-2 alpha kinase and its expression in erythroid cells. Biochimie 1994, 76, 761–769. [Google Scholar] [CrossRef]
- Dey, S.; Tameire, F.; Koumenis, C. PERK-ing up autophagy during MYC-induced tumorigenesis. Autophagy 2013, 9, 612–614. [Google Scholar] [CrossRef]
- Hart, L.S.; Cunningham, J.T.; Datta, T.; Dey, S.; Tameire, F.; Lehman, S.L.; Qiu, B.; Zhang, H.; Cerniglia, G.; Bi, M.; et al. ER stress-mediated autophagy promotes Myc-dependent transformation and tumor growth. J. Clin. Investig. 2012, 122, 4621–4634. [Google Scholar] [CrossRef]
- Salaroglio, I.C.; Panada, E.; Moiso, E.; Buondonno, I.; Provero, P.; Rubinstein, M.; Kopecka, J.; Riganti, C. PERK induces resistance to cell death elicited by endoplasmic reticulum stress and chemotherapy. Mol. Cancer 2017, 16, 91. [Google Scholar] [CrossRef]
- Zhu, H.; Chen, X.; Chen, B.; Chen, B.; Song, W.; Sun, D.; Zhao, Y. Activating transcription factor 4 promotes esophageal squamous cell carcinoma invasion and metastasis in mice and is associated with poor prognosis in human patients. PloS ONE 2014, 9, e103882. [Google Scholar] [CrossRef]
- Atkins, C.; Liu, Q.; Minthorn, E.; Zhang, S.Y.; Figueroa, D.J.; Moss, K.; Stanley, T.B.; Sanders, B.; Goetz, A.; Gaul, N.; et al. Characterization of a novel PERK kinase inhibitor with antitumor and antiangiogenic activity. Cancer Res. 2013, 73, 1993–2002. [Google Scholar] [CrossRef]
- Spriggs, K.A.; Bushell, M.; Willis, A.E. Translational regulation of gene expression during conditions of cell stress. Mol. Cell 2010, 40, 228–237. [Google Scholar] [CrossRef]
- Yamasaki, S.; Anderson, P. Reprogramming mRNA translation during stress. Curr. Opin. Cell Biol. 2008, 20, 222–226. [Google Scholar] [CrossRef]
- Furuichi, Y. Discovery of m(7)G-cap in eukaryotic mRNAs. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2015, 91, 394–409. [Google Scholar] [CrossRef]
- Haimov, O.; Sinvani, H.; Dikstein, R. Cap-dependent, scanning-free translation initiation mechanisms. Biochim. Et Biophys. Acta 2015, 1849, 1313–1318. [Google Scholar] [CrossRef]
- Hinnebusch, A.G. The scanning mechanism of eukaryotic translation initiation. Annu. Rev. Biochem. 2014, 83, 779–812. [Google Scholar] [CrossRef]
- Jackson, R.J.; Hellen, C.U.; Pestova, T.V. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol. 2010, 11, 113–127. [Google Scholar] [CrossRef]
- Sonenberg, N.; Hinnebusch, A.G. Regulation of translation initiation in eukaryotes: Mechanisms and biological targets. Cell 2009, 136, 731–745. [Google Scholar] [CrossRef]
- Cho, P.F.; Poulin, F.; Cho-Park, Y.A.; Cho-Park, I.B.; Chicoine, J.D.; Lasko, P.; Sonenberg, N. A new paradigm for translational control: Inhibition via 5′–3′ mRNA tethering by Bicoid and the eIF4E cognate 4EHP. Cell 2005, 121, 411–423. [Google Scholar] [CrossRef]
- Chen, H.H.; Tarn, W.Y. uORF-mediated translational control: Recently elucidated mechanisms and implications in cancer. RNA Biol. 2019, 16, 1327–1338. [Google Scholar] [CrossRef]
- Lin, Y.; May, G.E.; Kready, H.; Nazzaro, L.; Mao, M.; Spealman, P.; Creeger, Y.; McManus, C.J. Impacts of uORF codon identity and position on translation regulation. Nucleic Acids Res. 2019, 47, 9358–9367. [Google Scholar] [CrossRef]
- Young, S.K.; Wek, R.C. Upstream Open Reading Frames Differentially Regulate Gene-specific Translation in the Integrated Stress Response. J. Biol. Chem. 2016, 291, 16927–16935. [Google Scholar] [CrossRef]
- Baird, T.D.; Palam, L.R.; Fusakio, M.E.; Willy, J.A.; Davis, C.M.; McClintick, J.N.; Anthony, T.G.; Wek, R.C. Selective mRNA translation during eIF2 phosphorylation induces expression of IBTKalpha. Mol. Biol. Cell 2014, 25, 1686–1697. [Google Scholar] [CrossRef]
- Hussain, T.; Llacer, J.L.; Fernandez, I.S.; Munoz, A.; Martin-Marcos, P.; Savva, C.G.; Lorsch, J.R.; Hinnebusch, A.G.; Ramakrishnan, V. Structural changes enable start codon recognition by the eukaryotic translation initiation complex. Cell 2014, 159, 597–607. [Google Scholar] [CrossRef]
- Young, S.K.; Willy, J.A.; Wu, C.; Sachs, M.S.; Wek, R.C. Ribosome Reinitiation Directs Gene-specific Translation and Regulates the Integrated Stress Response. J. Biol. Chem. 2015, 290, 28257–28271. [Google Scholar] [CrossRef]
- Palam, L.R.; Baird, T.D.; Wek, R.C. Phosphorylation of eIF2 facilitates ribosomal bypass of an inhibitory upstream ORF to enhance CHOP translation. J. Biol. Chem. 2011, 286, 10939–10949. [Google Scholar] [CrossRef]
- Young, S.K.; Palam, L.R.; Wu, C.; Sachs, M.S.; Wek, R.C. Ribosome Elongation Stall Directs Gene-specific Translation in the Integrated Stress Response. J. Biol. Chem. 2016, 291, 6546–6558. [Google Scholar] [CrossRef]
- Kurosaki, T.; Popp, M.W.; Maquat, L.E. Quality and quantity control of gene expression by nonsense-mediated mRNA decay. Nat. Rev. Mol. Cell Biol. 2019, 20, 406–420. [Google Scholar] [CrossRef]
- Starck, S.R.; Tsai, J.C.; Chen, K.; Shodiya, M.; Wang, L.; Yahiro, K.; Martins-Green, M.; Shastri, N.; Walter, P. Translation from the 5′ untranslated region shapes the integrated stress response. Science 2016, 351, aad3867. [Google Scholar] [CrossRef]
- Schulz, J.; Mah, N.; Neuenschwander, M.; Kischka, T.; Ratei, R.; Schlag, P.M.; Castanos-Velez, E.; Fichtner, I.; Tunn, P.U.; Denkert, C.; et al. Loss-of-function uORF mutations in human malignancies. Sci. Rep. 2018, 8, 2395. [Google Scholar] [CrossRef]
- Fawcett, T.W.; Martindale, J.L.; Guyton, K.Z.; Hai, T.; Holbrook, N.J. Complexes containing activating transcription factor (ATF)/cAMP-responsive-element-binding protein (CREB) interact with the CCAAT/enhancer-binding protein (C/EBP)-ATF composite site to regulate Gadd153 expression during the stress response. Biochem. J. 1999, 339, 135–141. [Google Scholar]
- Harding, H.P.; Zhang, Y.; Bertolotti, A.; Zeng, H.; Ron, D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol. Cell 2000, 5, 897–904. [Google Scholar] [CrossRef]
- Ma, Y.; Hendershot, L.M. Delineation of a negative feedback regulatory loop that controls protein translation during endoplasmic reticulum stress. J. Biol. Chem. 2003, 278, 34864–34873. [Google Scholar] [CrossRef]
- Marciniak, S.J.; Yun, C.Y.; Oyadomari, S.; Novoa, I.; Zhang, Y.; Jungreis, R.; Nagata, K.; Harding, H.P.; Ron, D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004, 18, 3066–3077. [Google Scholar] [CrossRef]
- Han, J.; Back, S.H.; Hur, J.; Lin, Y.H.; Gildersleeve, R.; Shan, J.; Yuan, C.L.; Krokowski, D.; Wang, S.; Hatzoglou, M.; et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 2013, 15, 481–490. [Google Scholar] [CrossRef]
- Lu, P.D.; Harding, H.P.; Ron, D. Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J. Cell Biol. 2004, 167, 27–33. [Google Scholar] [CrossRef]
- Vattem, K.M.; Wek, R.C. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc. Natl. Acad. Sci. USA 2004, 101, 11269–11274. [Google Scholar] [CrossRef]
- Lee, Y.Y.; Cevallos, R.C.; Jan, E. An upstream open reading frame regulates translation of GADD34 during cellular stresses that induce eIF2alpha phosphorylation. J. Biol. Chem. 2009, 284, 6661–6673. [Google Scholar] [CrossRef]
- Chiribau, C.B.; Gaccioli, F.; Huang, C.C.; Yuan, C.L.; Hatzoglou, M. Molecular symbiosis of CHOP and C/EBP beta isoform LIP contributes to endoplasmic reticulum stress-induced apoptosis. Mol. Cell Biol. 2010, 30, 3722–3731. [Google Scholar] [CrossRef]
- Wethmar, K.; Begay, V.; Smink, J.J.; Zaragoza, K.; Wiesenthal, V.; Dorken, B.; Calkhoven, C.F.; Leutz, A. C/EBPbetaDeltauORF mice--a genetic model for uORF-mediated translational control in mammals. Genes Dev. 2010, 24, 15–20. [Google Scholar] [CrossRef]
- Young, S.K.; Baird, T.D.; Wek, R.C. Translation Regulation of the Glutamyl-prolyl-tRNA Synthetase Gene EPRS through Bypass of Upstream Open Reading Frames with Noncanonical Initiation Codons. J. Biol. Chem. 2016, 291, 10824–10835. [Google Scholar] [CrossRef]
- Huang, C.C.; Li, Y.; Lopez, A.B.; Chiang, C.M.; Kaufman, R.J.; Snider, M.D.; Hatzoglou, M. Temporal regulation of Cat-1 (cationic amino acid transporter-1) gene transcription during endoplasmic reticulum stress. Biochem. J. 2010, 429, 215–224. [Google Scholar] [CrossRef][Green Version]
- Yaman, I.; Fernandez, J.; Liu, H.; Caprara, M.; Komar, A.A.; Koromilas, A.E.; Zhou, L.; Snider, M.D.; Scheuner, D.; Kaufman, R.J.; et al. The zipper model of translational control: A small upstream ORF is the switch that controls structural remodeling of an mRNA leader. Cell 2003, 113, 519–531. [Google Scholar] [CrossRef]
- Sarnow, P. Translation of glucose-regulated protein 78/immunoglobulin heavy-chain binding protein mRNA is increased in poliovirus-infected cells at a time when cap-dependent translation of cellular mRNAs is inhibited. Proc. Natl. Acad. Sci. USA 1989, 86, 5795–5799. [Google Scholar] [CrossRef]
- Pelletier, J.; Kaplan, G.; Racaniello, V.R.; Sonenberg, N. Cap-independent translation of poliovirus mRNA is conferred by sequence elements within the 5′ noncoding region. Mol. Cell Biol. 1988, 8, 1103–1112. [Google Scholar] [CrossRef]
- Jang, S.K.; Krausslich, H.G.; Nicklin, M.J.; Duke, G.M.; Palmenberg, A.C.; Wimmer, E. A segment of the 5′ nontranslated region of encephalomyocarditis virus RNA directs internal entry of ribosomes during in vitro translation. J. Virol. 1988, 62, 2636–2643. [Google Scholar] [CrossRef]
- Trono, D.; Andino, R.; Baltimore, D. An RNA sequence of hundreds of nucleotides at the 5′ end of poliovirus RNA is involved in allowing viral protein synthesis. J. Virol. 1988, 62, 2291–2299. [Google Scholar] [CrossRef]
- Pelletier, J.; Sonenberg, N. Internal initiation of translation of eukaryotic mRNA directed by a sequence derived from poliovirus RNA. Nature 1988, 334, 320–325. [Google Scholar] [CrossRef]
- Macejak, D.G.; Sarnow, P. Internal initiation of translation mediated by the 5′ leader of a cellular mRNA. Nature 1991, 353, 90–94. [Google Scholar] [CrossRef]
- Johannes, G.; Sarnow, P. Cap-independent polysomal association of natural mRNAs encoding c-myc, BiP, and eIF4G conferred by internal ribosome entry sites. RNA-A Publ. RNA Soc. 1998, 4, 1500–1513. [Google Scholar] [CrossRef]
- Mokrejs, M.; Masek, T.; Vopalensky, V.; Hlubucek, P.; Delbos, P.; Pospisek, M. IRESite—A tool for the examination of viral and cellular internal ribosome entry sites. Nucleic Acids Res. 2010, 38, D131–D136. [Google Scholar] [CrossRef]
- Weingarten-Gabbay, S.; Elias-Kirma, S.; Nir, R.; Gritsenko, A.A.; Stern-Ginossar, N.; Yakhini, Z.; Weinberger, A.; Segal, E. Comparative genetics. Systematic discovery of cap-independent translation sequences in human and viral genomes. Science 2016, 351. [Google Scholar] [CrossRef]
- Holcik, M.; Sonenberg, N. Translational control in stress and apoptosis. Nat. Rev. Mol. Cell Biol. 2005, 6, 318–327. [Google Scholar] [CrossRef]
- Stoneley, M.; Chappell, S.A.; Jopling, C.L.; Dickens, M.; MacFarlane, M.; Willis, A.E. c-Myc protein synthesis is initiated from the internal ribosome entry segment during apoptosis. Mol. Cell. Biol. 2000, 20, 1162–1169. [Google Scholar] [CrossRef]
- Nevins, T.A.; Harder, Z.M.; Korneluk, R.G.; Holcik, M. Distinct regulation of internal ribosome entry site-mediated translation following cellular stress is mediated by apoptotic fragments of eIF4G translation initiation factor family members eIF4GI and p97/DAP5/NAT1. J. Biol. Chem. 2003, 278, 3572–3579. [Google Scholar] [CrossRef]
- Lewis, S.M.; Cerquozzi, S.; Graber, T.E.; Ungureanu, N.H.; Andrews, M.; Holcik, M. The eIF4G homolog DAP5/p97 supports the translation of select mRNAs during endoplasmic reticulum stress. Nucleic Acids Res. 2008, 36, 168–178. [Google Scholar] [CrossRef]
- Spriggs, K.A.; Cobbold, L.C.; Jopling, C.L.; Cooper, R.E.; Wilson, L.A.; Stoneley, M.; Coldwell, M.J.; Poncet, D.; Shen, Y.C.; Morley, S.J.; et al. Canonical initiation factor requirements of the Myc family of internal ribosome entry segments. Mol. Cell Biol. 2009, 29, 1565–1574. [Google Scholar] [CrossRef]
- Thoma, C.; Bergamini, G.; Galy, B.; Hundsdoerfer, P.; Hentze, M.W. Enhancement of IRES-mediated translation of the c-myc and BiP mRNAs by the poly(A) tail is independent of intact eIF4G and PABP. Mol. Cell 2004, 15, 925–935. [Google Scholar] [CrossRef]
- Gerlitz, G.; Jagus, R.; Elroy-Stein, O. Phosphorylation of initiation factor-2 alpha is required for activation of internal translation initiation during cell differentiation. Eur. J. Biochem. 2002, 269, 2810–2819. [Google Scholar] [CrossRef]
- Jaud, M.; Philippe, C.; Van Den Berghe, L.; Segura, C.; Mazzolini, L.; Pyronnet, S.; Laurell, H.; Touriol, C. The PERK Branch of the Unfolded Protein Response Promotes DLL4 Expression by Activating an Alternative Translation Mechanism. Cancers 2019, 11, 142. [Google Scholar] [CrossRef]
- Philippe, C.; Dubrac, A.; Quelen, C.; Desquesnes, A.; Van Den Berghe, L.; Segura, C.; Filleron, T.; Pyronnet, S.; Prats, H.; Brousset, P.; et al. PERK mediates the IRES-dependent translational activation of mRNAs encoding angiogenic growth factors after ischemic stress. Sci. Signal. 2016, 9, ra44. [Google Scholar] [CrossRef]
- Subkhankulova, T.; Mitchell, S.A.; Willis, A.E. Internal ribosome entry segment-mediated initiation of c-Myc protein synthesis following genotoxic stress. Biochem. J. 2001, 359, 183–192. [Google Scholar] [CrossRef]
- Thakor, N.; Holcik, M. IRES-mediated translation of cellular messenger RNA operates in eIF2alpha- independent manner during stress. Nucleic Acids Res. 2012, 40, 541–552. [Google Scholar] [CrossRef]
- Tinton, S.A.; Schepens, B.; Bruynooghe, Y.; Beyaert, R.; Cornelis, S. Regulation of the cell-cycle-dependent internal ribosome entry site of the PITSLRE protein kinase: Roles of Unr (upstream of N-ras) protein and phosphorylated translation initiation factor eIF-2alpha. Biochem. J. 2005, 385, 155–163. [Google Scholar] [CrossRef]
- Lewis, S.M.; Holcik, M. For IRES trans-acting factors, it is all about location. Oncogene 2008, 27, 1033–1035. [Google Scholar] [CrossRef]
- Kim, Y.K.; Back, S.H.; Rho, J.; Lee, S.H.; Jang, S.K. La autoantigen enhances translation of BiP mRNA. Nucleic Acids Res. 2001, 29, 5009–5016. [Google Scholar] [CrossRef]
- Holcik, M.; Gordon, B.W.; Korneluk, R.G. The internal ribosome entry site-mediated translation of antiapoptotic protein XIAP is modulated by the heterogeneous nuclear ribonucleoproteins C1 and C2. Mol. Cell Biol. 2003, 23, 280–288. [Google Scholar] [CrossRef]
- Damiano, F.; Rochira, A.; Tocci, R.; Alemanno, S.; Gnoni, A.; Siculella, L. hnRNP A1 mediates the activation of the IRES-dependent SREBP-1a mRNA translation in response to endoplasmic reticulum stress. Biochem. J. 2013, 449, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Chappell, S.A.; LeQuesne, J.P.; Paulin, F.E.; deSchoolmeester, M.L.; Stoneley, M.; Soutar, R.L.; Ralston, S.H.; Helfrich, M.H.; Willis, A.E. A mutation in the c-myc-IRES leads to enhanced internal ribosome entry in multiple myeloma: A novel mechanism of oncogene de-regulation. Oncogene 2000, 19, 4437–4440. [Google Scholar] [CrossRef] [PubMed]
- Walters, B.; Thompson, S.R. Cap-Independent Translational Control of Carcinogenesis. Front. Oncol. 2016, 6, 128. [Google Scholar] [CrossRef] [PubMed]
- Muranen, T.; Selfors, L.M.; Worster, D.T.; Iwanicki, M.P.; Song, L.; Morales, F.C.; Gao, S.; Mills, G.B.; Brugge, J.S. Inhibition of PI3K/mTOR leads to adaptive resistance in matrix-attached cancer cells. Cancer Cell 2012, 21, 227–239. [Google Scholar] [CrossRef]
- Silvera, D.; Arju, R.; Darvishian, F.; Levine, P.H.; Zolfaghari, L.; Goldberg, J.; Hochman, T.; Formenti, S.C.; Schneider, R.J. Essential role for eIF4GI overexpression in the pathogenesis of inflammatory breast cancer. Nat. Cell Biol. 2009, 11, 903–908. [Google Scholar] [CrossRef]
- Halaby, M.J.; Harris, B.R.; Miskimins, W.K.; Cleary, M.P.; Yang, D.Q. Deregulation of Internal Ribosome Entry Site-Mediated p53 Translation in Cancer Cells with Defective p53 Response to DNA Damage. Mol. Cell Biol. 2015, 35, 4006–4017. [Google Scholar] [CrossRef]
- Sharathchandra, A.; Katoch, A.; Das, S. IRES mediated translational regulation of p53 isoforms. Wiley Interdiscip. Rev. RNA 2014, 5, 131–139. [Google Scholar] [CrossRef]
- Halaby, M.J.; Li, Y.; Harris, B.R.; Jiang, S.; Miskimins, W.K.; Cleary, M.P.; Yang, D.Q. Translational Control Protein 80 Stimulates IRES-Mediated Translation of p53 mRNA in Response to DNA Damage. Biomed. Res. Int. 2015, 2015, 708158. [Google Scholar] [CrossRef]
- Takagi, M.; Absalon, M.J.; McLure, K.G.; Kastan, M.B. Regulation of p53 translation and induction after DNA damage by ribosomal protein L26 and nucleolin. Cell 2005, 123, 49–63. [Google Scholar] [CrossRef]
- Weingarten-Gabbay, S.; Khan, D.; Liberman, N.; Yoffe, Y.; Bialik, S.; Das, S.; Oren, M.; Kimchi, A. The translation initiation factor DAP5 promotes IRES-driven translation of p53 mRNA. Oncogene 2014, 33, 611–618. [Google Scholar] [CrossRef]
- Halaby, M.J.; Yang, D.Q. p53 translational control: A new facet of p53 regulation and its implication for tumorigenesis and cancer therapeutics. Gene 2007, 395, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Arcondeguy, T.; Lacazette, E.; Millevoi, S.; Prats, H.; Touriol, C. VEGF-A mRNA processing, stability and translation: A paradigm for intricate regulation of gene expression at the post-transcriptional level. Nucleic Acids Res. 2013, 41, 7997–8010. [Google Scholar] [CrossRef] [PubMed]
- Arnaud, E.; Touriol, C.; Boutonnet, C.; Gensac, M.C.; Vagner, S.; Prats, H.; Prats, A.C. A new 34-kilodalton isoform of human fibroblast growth factor 2 is cap dependently synthesized by using a non-AUG start codon and behaves as a survival factor. Mol. Cell Biol. 1999, 19, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Bastide, A.; Karaa, Z.; Bornes, S.; Hieblot, C.; Lacazette, E.; Prats, H.; Touriol, C. An upstream open reading frame within an IRES controls expression of a specific VEGF-A isoform. Nucleic Acids Res. 2008, 36, 2434–2445. [Google Scholar] [CrossRef]
- Bonnet-Magnaval, F.; Philippe, C.; Van Den Berghe, L.; Prats, H.; Touriol, C.; Lacazette, E. Hypoxia and ER stress promote Staufen1 expression through an alternative translation mechanism. Biochem. Biophys. Res. Commun. 2016, 479, 365–371. [Google Scholar] [CrossRef]
- Bornes, S.; Boulard, M.; Hieblot, C.; Zanibellato, C.; Iacovoni, J.S.; Prats, H.; Touriol, C. Control of the vascular endothelial growth factor internal ribosome entry site (IRES) activity and translation initiation by alternatively spliced coding sequences. J. Biol. Chem. 2004, 279, 18717–18726. [Google Scholar] [CrossRef]
- Li, W.; Thakor, N.; Xu, E.Y.; Huang, Y.; Chen, C.; Yu, R.; Holcik, M.; Kong, A.N. An internal ribosomal entry site mediates redox-sensitive translation of Nrf2. Nucleic Acids Res. 2010, 38, 778–788. [Google Scholar] [CrossRef]
- Morfoisse, F.; Kuchnio, A.; Frainay, C.; Gomez-Brouchet, A.; Delisle, M.B.; Marzi, S.; Helfer, A.C.; Hantelys, F.; Pujol, F.; Guillermet-Guibert, J.; et al. Hypoxia induces VEGF-C expression in metastatic tumor cells via a HIF-1alpha-independent translation-mediated mechanism. Cell Rep. 2014, 6, 155–167. [Google Scholar] [CrossRef]
- Rubtsova, M.P.; Sizova, D.V.; Dmitriev, S.E.; Ivanov, D.S.; Prassolov, V.S.; Shatsky, I.N. Distinctive properties of the 5′-untranslated region of human hsp70 mRNA. J. Biol. Chem. 2003, 278, 22350–22356. [Google Scholar] [CrossRef]
- Yang, Q.; Sarnow, P. Location of the internal ribosome entry site in the 5′ non-coding region of the immunoglobulin heavy-chain binding protein (BiP) mRNA: Evidence for specific RNA-protein interactions. Nucleic Acids Res. 1997, 25, 2800–2807. [Google Scholar] [CrossRef]
- Karaa, Z.S.; Iacovoni, J.S.; Bastide, A.; Lacazette, E.; Touriol, C.; Prats, H. The VEGF IRESes are differentially susceptible to translation inhibition by miR-16. RNA 2009, 15, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.P.; Kok, K.H.; Tang, H.M.; Wong, C.M.; Jin, D.Y. Internal ribosome entry site-mediated translational regulation of ATF4 splice variant in mammalian unfolded protein response. Biochim. Et Biophys. Acta 2013, 1833, 2165–2175. [Google Scholar] [CrossRef] [PubMed]
- Bourougaa, K.; Naski, N.; Boularan, C.; Mlynarczyk, C.; Candeias, M.M.; Marullo, S.; Fahraeus, R. Endoplasmic reticulum stress induces G2 cell-cycle arrest via mRNA translation of the p53 isoform p53/47. Mol. Cell 2010, 38, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Ray, P.S.; Grover, R.; Das, S. Two internal ribosome entry sites mediate the translation of p53 isoforms. EMBO Rep. 2006, 7, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Andreev, D.E.; O’Connor, P.B.; Loughran, G.; Dmitriev, S.E.; Baranov, P.V.; Shatsky, I.N. Insights into the mechanisms of eukaryotic translation gained with ribosome profiling. Nucleic Acids Res. 2017, 45, 513–526. [Google Scholar] [CrossRef] [PubMed]
- Jopling, C.L.; Spriggs, K.A.; Mitchell, S.A.; Stoneley, M.; Willis, A.E. L-Myc protein synthesis is initiated by internal ribosome entry. RNA 2004, 10, 287–298. [Google Scholar] [CrossRef]
- Le Quesne, J.P.; Stoneley, M.; Fraser, G.A.; Willis, A.E. Derivation of a structural model for the c-myc IRES. J. Mol. Biol. 2001, 310, 111–126. [Google Scholar] [CrossRef]
- Mitchell, S.A.; Spriggs, K.A.; Coldwell, M.J.; Jackson, R.J.; Willis, A.E. The Apaf-1 internal ribosome entry segment attains the correct structural conformation for function via interactions with PTB and unr. Mol. Cell 2003, 11, 757–771. [Google Scholar] [CrossRef]
- King, H.A.; Cobbold, L.C.; Willis, A.E. The role of IRES trans-acting factors in regulating translation initiation. Biochem. Soc. Trans. 2010, 38, 1581–1586. [Google Scholar] [CrossRef]
- Henis-Korenblit, S.; Strumpf, N.L.; Goldstaub, D.; Kimchi, A. A novel form of DAP5 protein accumulates in apoptotic cells as a result of caspase cleavage and internal ribosome entry site-mediated translation. Mol. Cell Biol. 2000, 20, 496–506. [Google Scholar] [CrossRef]
- Henis-Korenblit, S.; Shani, G.; Sines, T.; Marash, L.; Shohat, G.; Kimchi, A. The caspase-cleaved DAP5 protein supports internal ribosome entry site-mediated translation of death proteins. Proc. Natl. Acad. Sci. USA 2002, 99, 5400–5405. [Google Scholar] [CrossRef] [PubMed]
- Marash, L.; Kimchi, A. DAP5 and IRES-mediated translation during programmed cell death. Cell Death Differ. 2005, 12, 554–562. [Google Scholar] [CrossRef] [PubMed]
- Warnakulasuriyarachchi, D.; Cerquozzi, S.; Cheung, H.H.; Holcik, M. Translational induction of the inhibitor of apoptosis protein HIAP2 during endoplasmic reticulum stress attenuates cell death and is mediated via an inducible internal ribosome entry site element. J. Biol. Chem. 2004, 279, 17148–17157. [Google Scholar] [CrossRef] [PubMed]
- Bevilacqua, E.; Wang, X.; Majumder, M.; Gaccioli, F.; Yuan, C.L.; Wang, C.; Zhu, X.; Jordan, L.E.; Scheuner, D.; Kaufman, R.J.; et al. eIF2alpha phosphorylation tips the balance to apoptosis during osmotic stress. J. Biol. Chem. 2010, 285, 17098–17111. [Google Scholar] [CrossRef]
- Shi, Y.; Yang, Y.; Hoang, B.; Bardeleben, C.; Holmes, B.; Gera, J.; Lichtenstein, A. Therapeutic potential of targeting IRES-dependent c-myc translation in multiple myeloma cells during ER stress. Oncogene 2016, 35, 1015–1024. [Google Scholar] [CrossRef]
- Dobbyn, H.C.; Hill, K.; Hamilton, T.L.; Spriggs, K.A.; Pickering, B.M.; Coldwell, M.J.; de Moor, C.H.; Bushell, M.; Willis, A.E. Regulation of BAG-1 IRES-mediated translation following chemotoxic stress. Oncogene 2008, 27, 1167–1174. [Google Scholar] [CrossRef]
- Morfoisse, F.; Tatin, F.; Hantelys, F.; Adoue, A.; Helfer, A.C.; Cassant-Sourdy, S.; Pujol, F.; Gomez-Brouchet, A.; Ligat, L.; Lopez, F.; et al. Nucleolin Promotes Heat Shock-Associated Translation of VEGF-D to Promote Tumor Lymphangiogenesis. Cancer Res. 2016, 76, 4394–4405. [Google Scholar] [CrossRef]
- Bornes, S.; Prado-Lourenco, L.; Bastide, A.; Zanibellato, C.; Iacovoni, J.S.; Lacazette, E.; Prats, A.C.; Touriol, C.; Prats, H. Translational induction of VEGF internal ribosome entry site elements during the early response to ischemic stress. Circ. Res. 2007, 100, 305–308. [Google Scholar] [CrossRef]
- Creancier, L.; Morello, D.; Mercier, P.; Prats, A.C. Fibroblast growth factor 2 internal ribosome entry site (IRES) activity ex vivo and in transgenic mice reveals a stringent tissue-specific regulation. J. Cell Biol. 2000, 150, 275–281. [Google Scholar] [CrossRef]
- Laklai, H.; Laval, S.; Dumartin, L.; Rochaix, P.; Hagedorn, M.; Bikfalvi, A.; Le Guellec, S.; Delisle, M.B.; Schally, A.V.; Susini, C.; et al. Thrombospondin-1 is a critical effector of oncosuppressive activity of sst2 somatostatin receptor on pancreatic cancer. Proc. Natl. Acad. Sci. USA 2009, 106, 17769–17774. [Google Scholar] [CrossRef]
- Lang, K.J.; Kappel, A.; Goodall, G.J. Hypoxia-inducible factor-1alpha mRNA contains an internal ribosome entry site that allows efficient translation during normoxia and hypoxia. Mol. Biol. Cell 2002, 13, 1792–1801. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Ferreira, V.; Breier, G.; Pollefeyt, S.; Kieckens, L.; Gertsenstein, M.; Fahrig, M.; Vandenhoeck, A.; Harpal, K.; Eberhardt, C.; et al. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature 1996, 380, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Carver-Moore, K.; Chen, H.; Dowd, M.; Lu, L.; O’Shea, K.S.; Powell-Braxton, L.; Hillan, K.J.; Moore, M.W. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature 1996, 380, 439–442. [Google Scholar] [CrossRef] [PubMed]
- Gale, N.W.; Dominguez, M.G.; Noguera, I.; Pan, L.; Hughes, V.; Valenzuela, D.M.; Murphy, A.J.; Adams, N.C.; Lin, H.C.; Holash, J.; et al. Haploinsufficiency of delta-like 4 ligand results in embryonic lethality due to major defects in arterial and vascular development. Proc. Natl. Acad. Sci. USA 2004, 101, 15949–15954. [Google Scholar] [CrossRef]
- Blais, J.D.; Addison, C.L.; Edge, R.; Falls, T.; Zhao, H.; Wary, K.; Koumenis, C.; Harding, H.P.; Ron, D.; Holcik, M.; et al. Perk-dependent translational regulation promotes tumor cell adaptation and angiogenesis in response to hypoxic stress. Mol. Cell Biol. 2006, 26, 9517–9532. [Google Scholar] [CrossRef]
- Hur, K.Y.; So, J.S.; Ruda, V.; Frank-Kamenetsky, M.; Fitzgerald, K.; Koteliansky, V.; Iwawaki, T.; Glimcher, L.H.; Lee, A.H. IRE1alpha activation protects mice against acetaminophen-induced hepatotoxicity. J. Exp. Med. 2012, 209, 307–318. [Google Scholar] [CrossRef]
- Oikawa, D.; Tokuda, M.; Hosoda, A.; Iwawaki, T. Identification of a consensus element recognized and cleaved by IRE1 alpha. Nucleic Acids Res. 2010, 38, 6265–6273. [Google Scholar] [CrossRef]
- Gaddam, D.; Stevens, N.; Hollien, J. Comparison of mRNA localization and regulation during endoplasmic reticulum stress in Drosophila cells. Mol. Biol. Cell 2013, 24, 14–20. [Google Scholar] [CrossRef]
- Jagannathan, S.; Reid, D.W.; Cox, A.H.; Nicchitta, C.V. De novo translation initiation on membrane-bound ribosomes as a mechanism for localization of cytosolic protein mRNAs to the endoplasmic reticulum. RNA 2014, 20, 1489–1498. [Google Scholar] [CrossRef]
- Reid, D.W.; Chen, Q.; Tay, A.S.; Shenolikar, S.; Nicchitta, C.V. The unfolded protein response triggers selective mRNA release from the endoplasmic reticulum. Cell 2014, 158, 1362–1374. [Google Scholar] [CrossRef]
- Reid, D.W.; Nicchitta, C.V. Primary role for endoplasmic reticulum-bound ribosomes in cellular translation identified by ribosome profiling. J. Biol. Chem. 2012, 287, 5518–5527. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.; Hollien, J. Ire1-mediated decay in mammalian cells relies on mRNA sequence, structure, and translational status. Mol. Biol. Cell 2015, 26, 2873–2884. [Google Scholar] [CrossRef] [PubMed]
- Iwawaki, T.; Hosoda, A.; Okuda, T.; Kamigori, Y.; Nomura-Furuwatari, C.; Kimata, Y.; Tsuru, A.; Kohno, K. Translational control by the ER transmembrane kinase/ribonuclease IRE1 under ER stress. Nat. Cell Biol. 2001, 3, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Imagawa, Y.; Hosoda, A.; Sasaka, S.; Tsuru, A.; Kohno, K. RNase domains determine the functional difference between IRE1alpha and IRE1beta. FEBS Lett. 2008, 582, 656–660. [Google Scholar] [CrossRef] [PubMed]
- Khajuria, R.K.; Munschauer, M.; Ulirsch, J.C.; Fiorini, C.; Ludwig, L.S.; McFarland, S.K.; Abdulhay, N.J.; Specht, H.; Keshishian, H.; Mani, D.R.; et al. Ribosome Levels Selectively Regulate Translation and Lineage Commitment in Human Hematopoiesis. Cell 2018, 173, 90–103 e119. [Google Scholar] [CrossRef] [PubMed]
- Acosta-Alvear, D.; Karagoz, G.E.; Frohlich, F.; Li, H.; Walther, T.C.; Walter, P. The unfolded protein response and endoplasmic reticulum protein targeting machineries converge on the stress sensor IRE1. eLife 2018, 7. [Google Scholar] [CrossRef]
- Leppek, K.; Das, R.; Barna, M. Functional 5′ UTR mRNA structures in eukaryotic translation regulation and how to find them. Nat. Rev. Mol. Cell Biol. 2018, 19, 158–174. [Google Scholar] [CrossRef]
- Lee, A.S.; Kranzusch, P.J.; Cate, J.H. eIF3 targets cell-proliferation messenger RNAs for translational activation or repression. Nature 2015, 522, 111–114. [Google Scholar] [CrossRef]
- Lee, A.S.; Kranzusch, P.J.; Doudna, J.A.; Cate, J.H. eIF3d is an mRNA cap-binding protein that is required for specialized translation initiation. Nature 2016, 536, 96–99. [Google Scholar] [CrossRef]
- Fraser, C.S.; Doudna, J.A. Structural and mechanistic insights into hepatitis C viral translation initiation. Nat. Rev. Microbiol. 2007, 5, 29–38. [Google Scholar] [CrossRef]
- Bhardwaj, U.; Powell, P.; Goss, D.J. Eukaryotic initiation factor (eIF) 3 mediates Barley Yellow Dwarf Viral mRNA 3′-5′ UTR interactions and 40S ribosomal subunit binding to facilitate cap-independent translation. Nucleic Acids Res. 2019, 47, 6225–6235. [Google Scholar] [CrossRef] [PubMed]
- Cattie, D.J.; Richardson, C.E.; Reddy, K.C.; Ness-Cohn, E.M.; Droste, R.; Thompson, M.K.; Gilbert, W.V.; Kim, D.H. Mutations in Nonessential eIF3k and eIF3l Genes Confer Lifespan Extension and Enhanced Resistance to ER Stress in Caenorhabditis elegans. PLoS Genet. 2016, 12, e1006326. [Google Scholar] [CrossRef] [PubMed]
- Guan, B.J.; van Hoef, V.; Jobava, R.; Elroy-Stein, O.; Valasek, L.S.; Cargnello, M.; Gao, X.H.; Krokowski, D.; Merrick, W.C.; Kimball, S.R.; et al. A Unique ISR Program Determines Cellular Responses to Chronic Stress. Mol. Cell 2017, 68, 885–900 e886. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.D.; Patil, D.P.; Zhou, J.; Zinoviev, A.; Skabkin, M.A.; Elemento, O.; Pestova, T.V.; Qian, S.B.; Jaffrey, S.R. 5′ UTR m(6)A Promotes Cap-Independent Translation. Cell 2015, 163, 999–1010. [Google Scholar] [CrossRef]
- Desrosiers, R.; Friderici, K.; Rottman, F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc. Natl. Acad. Sci. USA 1974, 71, 3971–3975. [Google Scholar] [CrossRef]
- Rottman, F.; Shatkin, A.J.; Perry, R.P. Sequences containing methylated nucleotides at the 5′ termini of messenger RNAs: Possible implications for processing. Cell 1974, 3, 197–199. [Google Scholar] [CrossRef]
- Meyer, K.D.; Saletore, Y.; Zumbo, P.; Elemento, O.; Mason, C.E.; Jaffrey, S.R. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 2012, 149, 1635–1646. [Google Scholar] [CrossRef]
- Zhou, J.; Wan, J.; Gao, X.; Zhang, X.; Jaffrey, S.R.; Qian, S.B. Dynamic m(6)A mRNA methylation directs translational control of heat shock response. Nature 2015, 526, 591–594. [Google Scholar] [CrossRef]
- Shi, H.; Wei, J.; He, C. Where, When, and How: Context-Dependent Functions of RNA Methylation Writers, Readers, and Erasers. Mol. Cell 2019, 74, 640–650. [Google Scholar] [CrossRef]
- Hsu, P.J.; Zhu, Y.; Ma, H.; Guo, Y.; Shi, X.; Liu, Y.; Qi, M.; Lu, Z.; Shi, H.; Wang, J.; et al. Ythdc2 is an N(6)-methyladenosine binding protein that regulates mammalian spermatogenesis. Cell Res. 2017, 27, 1115–1127. [Google Scholar] [CrossRef]
- Shi, H.; Wang, X.; Lu, Z.; Zhao, B.S.; Ma, H.; Hsu, P.J.; Liu, C.; He, C. YTHDF3 facilitates translation and decay of N(6)-methyladenosine-modified RNA. Cell Res. 2017, 27, 315–328. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhao, B.S.; Roundtree, I.A.; Lu, Z.; Han, D.; Ma, H.; Weng, X.; Chen, K.; Shi, H.; He, C. N(6)-methyladenosine Modulates Messenger RNA Translation Efficiency. Cell 2015, 161, 1388–1399. [Google Scholar] [CrossRef] [PubMed]
- Choe, J.; Lin, S.; Zhang, W.; Liu, Q.; Wang, L.; Ramirez-Moya, J.; Du, P.; Kim, W.; Tang, S.; Sliz, P.; et al. mRNA circularization by METTL3-eIF3h enhances translation and promotes oncogenesis. Nature 2018, 561, 556–560. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Choe, J.; Du, P.; Triboulet, R.; Gregory, R.I. The m(6)A Methyltransferase METTL3 Promotes Translation in Human Cancer Cells. Mol. Cell 2016, 62, 335–345. [Google Scholar] [CrossRef]
- Barbieri, I.; Tzelepis, K.; Pandolfini, L.; Shi, J.; Millan-Zambrano, G.; Robson, S.C.; Aspris, D.; Migliori, V.; Bannister, A.J.; Han, N.; et al. Promoter-bound METTL3 maintains myeloid leukaemia by m(6)A-dependent translation control. Nature 2017, 552, 126–131. [Google Scholar] [CrossRef]
- Zhou, J.; Wan, J.; Shu, X.E.; Mao, Y.; Liu, X.M.; Yuan, X.; Zhang, X.; Hess, M.E.; Bruning, J.C.; Qian, S.B. N(6)-Methyladenosine Guides mRNA Alternative Translation during Integrated Stress Response. Mol. Cell 2018, 69, 636–647 e637. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jaud, M.; Philippe, C.; Di Bella, D.; Tang, W.; Pyronnet, S.; Laurell, H.; Mazzolini, L.; Rouault-Pierre, K.; Touriol, C. Translational Regulations in Response to Endoplasmic Reticulum Stress in Cancers. Cells 2020, 9, 540. https://doi.org/10.3390/cells9030540
Jaud M, Philippe C, Di Bella D, Tang W, Pyronnet S, Laurell H, Mazzolini L, Rouault-Pierre K, Touriol C. Translational Regulations in Response to Endoplasmic Reticulum Stress in Cancers. Cells. 2020; 9(3):540. https://doi.org/10.3390/cells9030540
Chicago/Turabian StyleJaud, Manon, Céline Philippe, Doriana Di Bella, Weiwei Tang, Stéphane Pyronnet, Henrik Laurell, Laurent Mazzolini, Kevin Rouault-Pierre, and Christian Touriol. 2020. "Translational Regulations in Response to Endoplasmic Reticulum Stress in Cancers" Cells 9, no. 3: 540. https://doi.org/10.3390/cells9030540
APA StyleJaud, M., Philippe, C., Di Bella, D., Tang, W., Pyronnet, S., Laurell, H., Mazzolini, L., Rouault-Pierre, K., & Touriol, C. (2020). Translational Regulations in Response to Endoplasmic Reticulum Stress in Cancers. Cells, 9(3), 540. https://doi.org/10.3390/cells9030540