
Modulation of Fatty Acid-Related Genes in the Response of H9c2 Cardiac Cells to Palmitate and n-3 Polyunsaturated Fatty Acids


Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatments
2.2. Cell Viability and Caspase Activity
2.3. Mitochondrial Potential Assay
2.4. Cell Size And Lipid Content Measurement
2.5. Real-Time RT-PCR
2.6. Western Blotting
2.7. Cell Transfection
2.8. Statistical Analysis
3. Results
3.1. EPA and DHA Prevent Apoptosis and Hypertrophy Induced by Palmitate in H9c2 Cardiac Cells
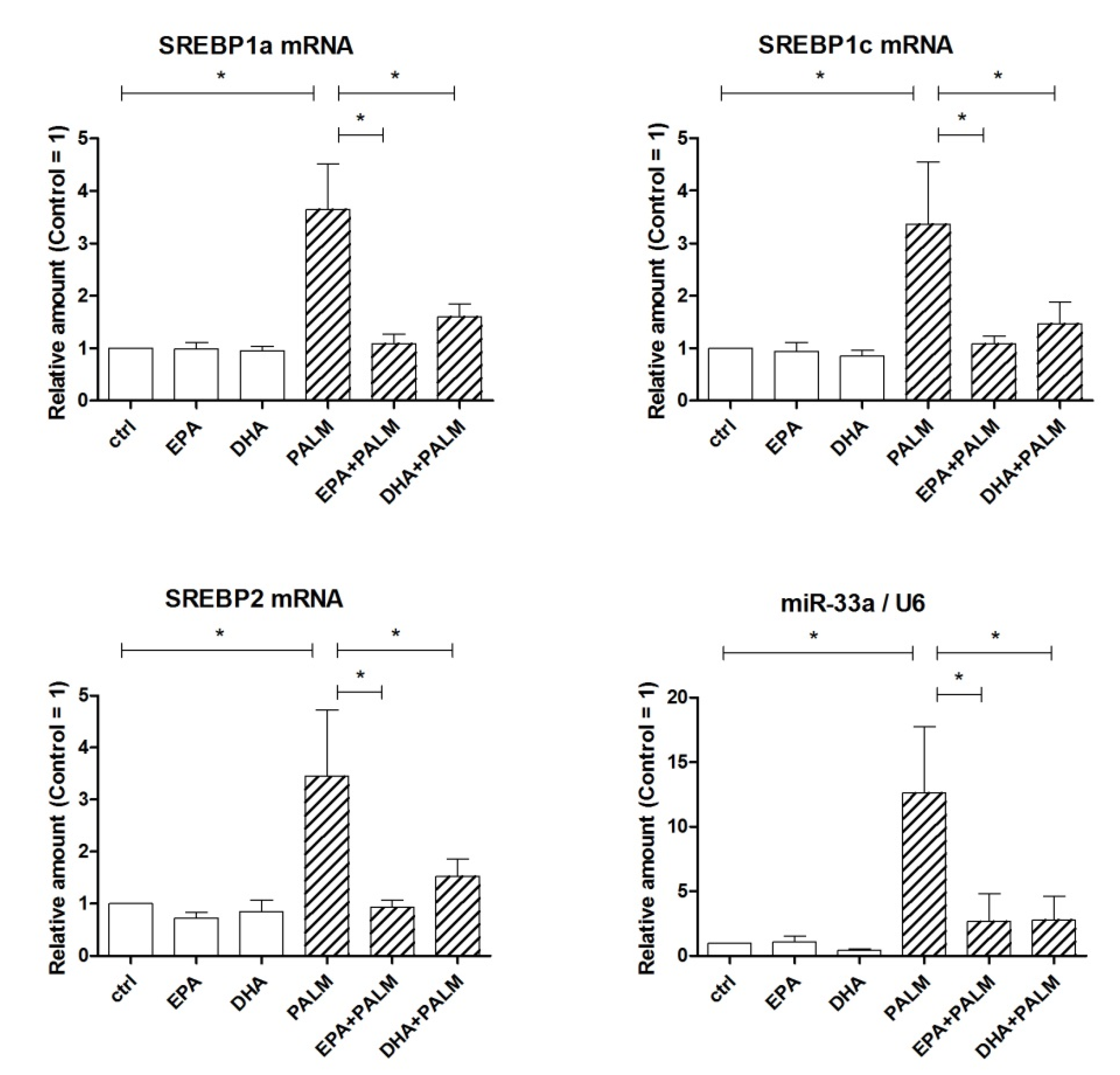
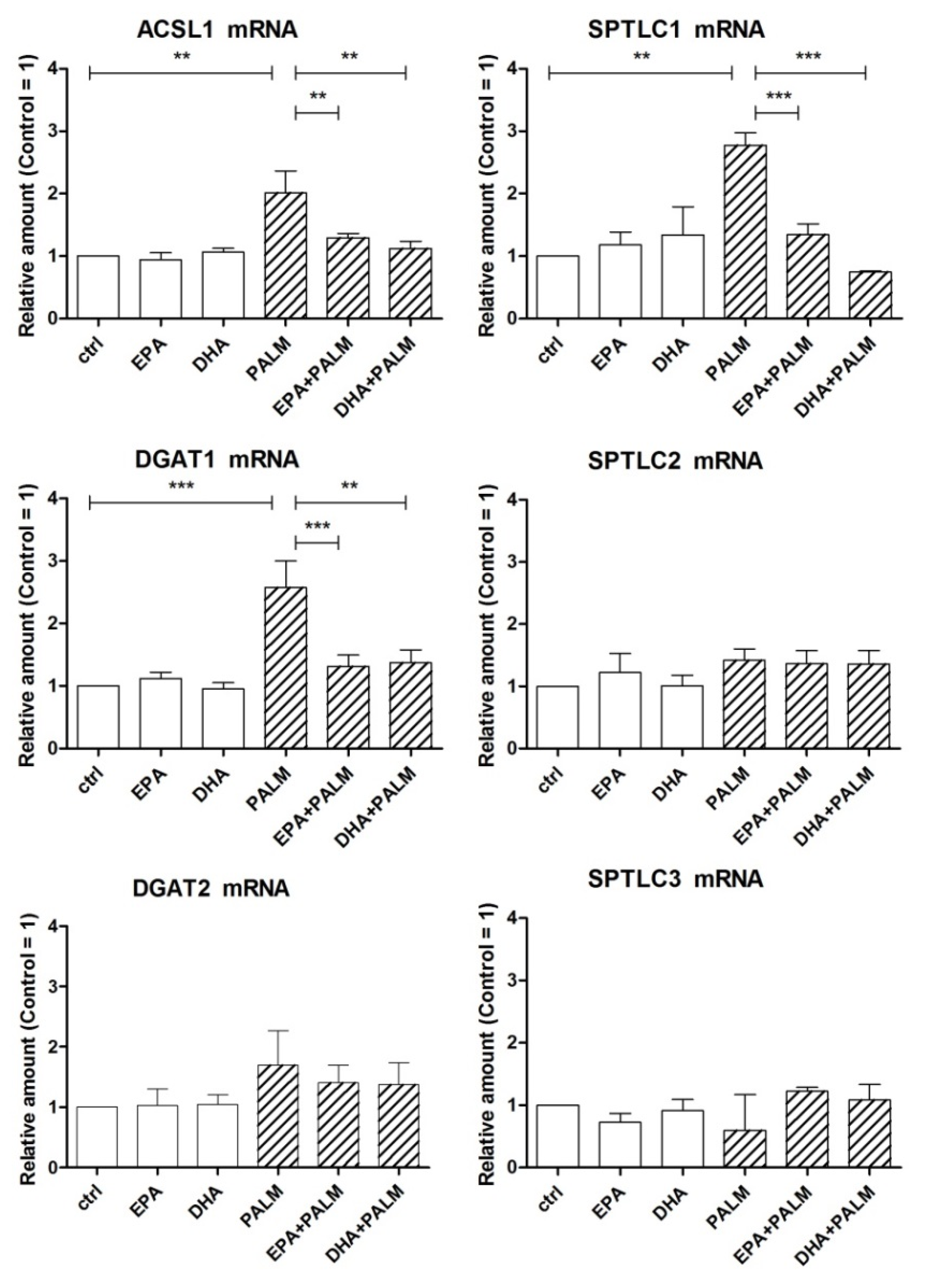
3.2. Effect of Palmitate, EPA, and DHA on the Expression of SREBP Genes and of Genes Encoding Enzymes Driving FA Fate
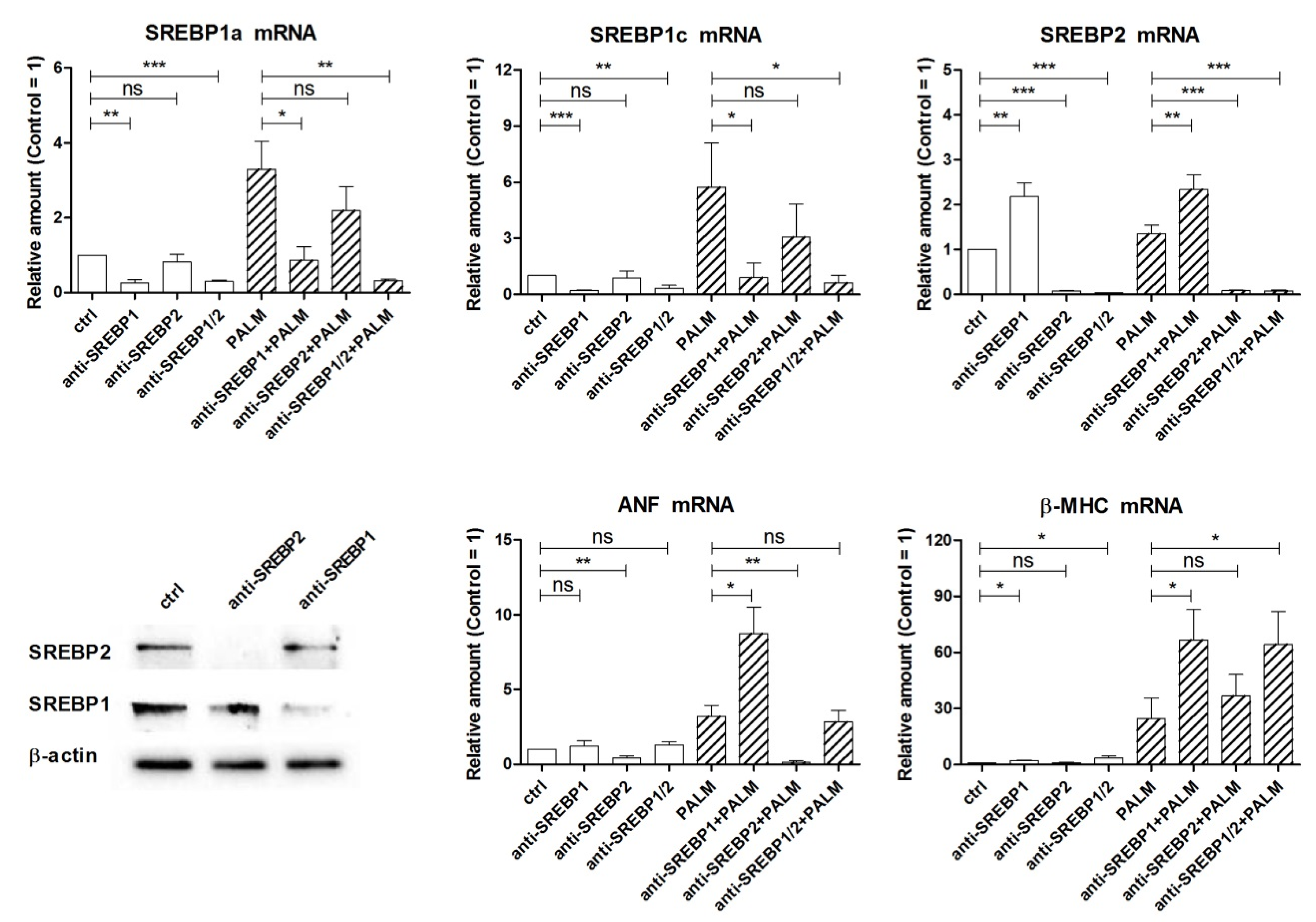
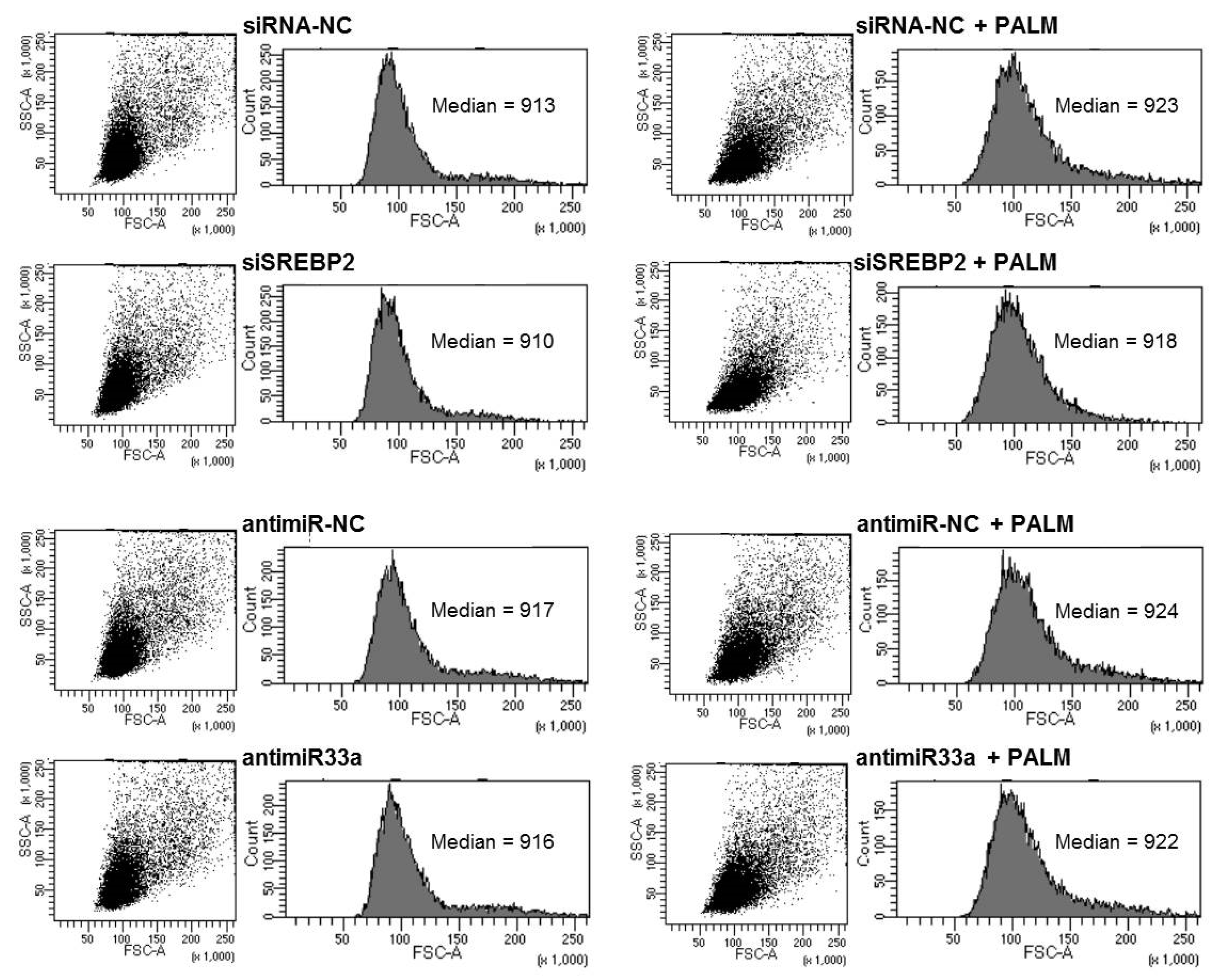
3.3. Effects of Manipulating the Expression of miR-33a and SREBPs on Cell Death and Hypertrophy
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hooper, L.; Summerbell, C.D.; Thompson, R.; Sills, D.; Roberts, F.G.; Moore, H.; Davey Smith, G. Reduced or modified dietary fat for preventing cardiovascular disease. Cochrane Database Syst. Rev. 2011. [Google Scholar] [CrossRef]
- Chang, W.; Zhang, M.; Meng, Z.; Yu, Y.; Yao, F.; Hatch, G.M.; Chen, L. Berberine treatment prevents cardiac dysfunction and remodeling through activation of 5’-adenosine monophosphate-activated protein kinase in type 2 diabetic rats and in palmitate-induced hypertrophic h9c2 cells. Eur. J. Pharm. 2015, 769, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Kuwabara, Y.; Horie, T.; Baba, O.; Watanabe, S.; Nishiga, M.; Usami, S.; Izuhara, M.; Nakao, T.; Nishino, T.; Otsu, K.; et al. Microrna-451 exacerbates lipotoxicity in cardiac myocytes and high-fat diet-induced cardiac hypertrophy in mice through suppression of the lkb1/ampk pathway. Circ. Res. 2015, 116, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Nobuhara, M.; Saotome, M.; Watanabe, T.; Urushida, T.; Katoh, H.; Satoh, H.; Funaki, M.; Hayashi, H. Mitochondrial dysfunction caused by saturated fatty acid loading induces myocardial insulin-resistance in differentiated h9c2 myocytes: A novel ex vivo myocardial insulin-resistance model. Exp. Cell Res. 2013, 319, 955–966. [Google Scholar] [CrossRef] [PubMed]
- Park, T.S.; Hu, Y.; Noh, H.L.; Drosatos, K.; Okajima, K.; Buchanan, J.; Tuinei, J.; Homma, S.; Jiang, X.C.; Abel, E.D.; et al. Ceramide is a cardiotoxin in lipotoxic cardiomyopathy. J. Lipid Res. 2008, 49, 2101–2112. [Google Scholar] [CrossRef] [PubMed]
- Haffar, T.; Akoumi, A.; Bousette, N. Lipotoxic palmitate impairs the rate of beta-oxidation and citric acid cycle flux in rat neonatal cardiomyocytes. Cell Physiol. Biochem. 2016, 40, 969–981. [Google Scholar] [CrossRef] [PubMed]
- van de Weijer, T.; Schrauwen-Hinderling, V.B.; Schrauwen, P. Lipotoxicity in type 2 diabetic cardiomyopathy. Cardiovasc. Res. 2011, 92, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Shi, X.; Bharadwaj, K.G.; Ikeda, S.; Yamashita, H.; Yagyu, H.; Schaffer, J.E.; Yu, Y.H.; Goldberg, I.J. Dgat1 expression increases heart triglyceride content but ameliorates lipotoxicity. J. Biol. Chem. 2009, 284, 36312–36323. [Google Scholar] [CrossRef] [PubMed]
- Eberle, D.; Hegarty, B.; Bossard, P.; Ferre, P.; Foufelle, F. Srebp transcription factors: Master regulators of lipid homeostasis. Biochimie 2004, 86, 839–848. [Google Scholar] [CrossRef]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. Srebps: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Invest. 2002, 109, 1125–1131. [Google Scholar] [CrossRef]
- Shimano, H. Srebps: Physiology and pathophysiology of the srebp family. Febs. J. 2009, 276, 616–621. [Google Scholar] [CrossRef] [PubMed]
- Danesi, F.; Govoni, M.; D’Antuono, L.F.; Bordoni, A. The molecular mechanism of the cholesterol-lowering effect of dill and kale: The influence of the food matrix components. Electrophoresis 2016, 37, 1805–1813. [Google Scholar] [CrossRef] [PubMed]
- Najafi-Shoushtari, S.H.; Kristo, F.; Li, Y.; Shioda, T.; Cohen, D.E.; Gerszten, R.E.; Naar, A.M. Microrna-33 and the srebp host genes cooperate to control cholesterol homeostasis. Science 2010, 328, 1566–1569. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Young, M.E.; Chatham, J.C.; Crossman, D.K.; Dell’Italia, L.J.; Shalev, A. Txnip regulates myocardial fatty acid oxidation via mir-33a signaling. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H64–H75. [Google Scholar] [CrossRef]
- Davalos, A.; Goedeke, L.; Smibert, P.; Ramirez, C.M.; Warrier, N.P.; Andreo, U.; Cirera-Salinas, D.; Rayner, K.; Suresh, U.; Pastor-Pareja, J.C.; et al. Mir-33a/b contribute to the regulation of fatty acid metabolism and insulin signaling. Proc. Natl. Acad. Sci. USA 2011, 108, 9232–9237. [Google Scholar] [CrossRef]
- Kalupahana, N.S.; Claycombe, K.J.; Moustaid-Moussa, N. (n-3) fatty acids alleviate adipose tissue inflammation and insulin resistance: Mechanistic insights. Adv. Nutr. 2011, 2, 304–316. [Google Scholar] [CrossRef]
- Mozaffarian, D.; Wu, J.H. Omega-3 fatty acids and cardiovascular disease: Effects on risk factors, molecular pathways, and clinical events. J. Am. Coll. Cardiol. 2011, 58, 2047–2067. [Google Scholar] [CrossRef]
- Davidson, M.H. Omega-3 fatty acids: New insights into the pharmacology and biology of docosahexaenoic acid, docosapentaenoic acid, and eicosapentaenoic acid. Curr. Opin. Lipidol. 2013, 24, 467–474. [Google Scholar] [CrossRef]
- Drouin, G.; Rioux, V.; Legrand, P. The n-3 docosapentaenoic acid (dpa): A new player in the n-3 long chain polyunsaturated fatty acid family. Biochimie 2019, 159, 36–48. [Google Scholar] [CrossRef]
- Cetrullo, S.; Tantini, B.; Flamigni, F.; Pazzini, C.; Facchini, A.; Stefanelli, C.; Caldarera, C.M.; Pignatti, C. Antiapoptotic and antiautophagic effects of eicosapentaenoic acid in cardiac myoblasts exposed to palmitic acid. Nutrients 2012, 4, 78–90. [Google Scholar] [CrossRef]
- Cetrullo, S.; D’Adamo, S.; Guidotti, S.; Borzi, R.M.; Flamigni, F. Hydroxytyrosol prevents chondrocyte death under oxidative stress by inducing autophagy through sirtuin 1-dependent and -independent mechanisms. Biochim. Biophys. Acta 2016, 1860, 1181–1191. [Google Scholar] [CrossRef] [PubMed]
- Stefanelli, C.; Pignatti, C.; Tantini, B.; Fattori, M.; Stanic, I.; Mackintosh, C.A.; Flamigni, F.; Guarnieri, C.; Caldarera, C.M.; Pegg, A.E. Effect of polyamine depletion on caspase activation: A study with spermine synthase-deficient cells. Biochem. J. 2001, 355, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Greenspan, P.; Mayer, E.P.; Fowler, S.D. Nile red: A selective fluorescent stain for intracellular lipid droplets. J. Cell. Biol. 1985, 100, 965–973. [Google Scholar] [CrossRef] [PubMed]
- D’Adamo, S.; Cetrullo, S.; Guidotti, S.; Borzi, R.M.; Flamigni, F. Hydroxytyrosol modulates the levels of microrna-9 and its target sirtuin-1 thereby counteracting oxidative stress-induced chondrocyte death. Osteoarthr. Cartil. 2017, 25, 600–610. [Google Scholar] [CrossRef]
- Chiu, H.C.; Kovacs, A.; Ford, D.A.; Hsu, F.F.; Garcia, R.; Herrero, P.; Saffitz, J.E.; Schaffer, J.E. A novel mouse model of lipotoxic cardiomyopathy. J. Clin. Invest. 2001, 107, 813–822. [Google Scholar] [CrossRef]
- Goldenberg, J.R.; Wang, X.; Lewandowski, E.D. Acyl coa synthetase-1 links facilitated long chain fatty acid uptake to intracellular metabolic trafficking differently in hearts of male versus female mice. J. Mol. Cell Cardiol. 2016, 94, 1–9. [Google Scholar] [CrossRef]
- Nakakuki, M.; Kawano, H.; Notsu, T.; Imada, K. Eicosapentaenoic acid suppresses palmitate-induced cytokine production by modulating long-chain acyl-coa synthetase 1 expression in human thp-1 macrophages. Atherosclerosis 2013, 227, 289–296. [Google Scholar] [CrossRef]
- Yen, C.L.E.; Monetti, M.; Burri, B.J.; Farese, R.V. The triacylglycerol synthesis enzyme dgat1 also catalyzes the synthesis of diacylglycerols, waxes, and retinyl esters. J. Lipid Res. 2005, 46, 1502–1511. [Google Scholar] [CrossRef]
- Stone, S.J.; Myers, H.M.; Watkins, S.M.; Brown, B.E.; Feingold, K.R.; Elias, P.M.; Farese, R.V. Lipopenia and skin barrier abnormalities in dgat2-deficient mice. J. Biol. Chem. 2004, 279, 11767–11776. [Google Scholar] [CrossRef]
- Ruangsiriluk, W.; Grosskurth, S.E.; Ziemek, D.; Kuhn, M.; des Etages, S.G.; Francone, O.L. Silencing of enzymes involved in ceramide biosynthesis causes distinct global alterations of lipid homeostasis and gene expression. J. Lipid Res. 2012, 53, 1459–1471. [Google Scholar] [CrossRef]
- Sakamoto, A.; Saotome, M.; Hasan, P.; Satoh, T.; Ohtani, H.; Urushida, T.; Katoh, H.; Satoh, H.; Hayashi, H. Eicosapentaenoic acid ameliorates palmitate-induced lipotoxicity via the amp kinase/dynamin-related protein-1 signaling pathway in differentiated h9c2 myocytes. Exp. Cell Res. 2017, 351, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Shimojo, N.; Jesmin, S.; Sakai, S.; Maeda, S.; Miyauchi, T.; Mizutani, T.; Aonuma, K.; Kawano, S. Fish oil constituent eicosapentaenoic acid inhibits endothelin-induced cardiomyocyte hypertrophy via ppar-alpha. Life Sci. 2014, 118, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Folino, A.; Sprio, A.E.; Di Scipio, F.; Berta, G.N.; Rastaldo, R. Alpha-linolenic acid protects against cardiac injury and remodelling induced by beta-adrenergic overstimulation. Food Funct. 2015, 6, 2231–2239. [Google Scholar] [CrossRef] [PubMed]
- Cheema, S.K.; Tappia, P.S.; Dhalla, N.S. Modification of gene expression in rat cardiomyocytes by linoleic and docosahexaenoic acids (1). Can J. Physiol. Pharm. 2019, 97, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Akoumi, A.; Haffar, T.; Mousterji, M.; Kiss, R.S.; Bousette, N. Palmitate mediated diacylglycerol accumulation causes endoplasmic reticulum stress, plin2 degradation, and cell death in h9c2 cardiomyoblasts. Exp. Cell Res. 2017, 354, 85–94. [Google Scholar] [CrossRef]
- Bosma, M.; Dapito, D.H.; Drosatos-Tampakaki, Z.; Huiping-Son, N.; Huang, L.S.; Kersten, S.; Drosatos, K.; Goldberg, I.J. Sequestration of fatty acids in triglycerides prevents endoplasmic reticulum stress in an in vitro model of cardiomyocyte lipotoxicity. Biochim Biophys. Acta 2014, 1841, 1648–1655. [Google Scholar] [CrossRef]
- Righi, V.; Di Nunzio, M.; Danesi, F.; Schenetti, L.; Mucci, A.; Boschetti, E.; Biagi, P.; Bonora, S.; Tugnoli, V.; Bordoni, A. Epa or dha supplementation increases triacylglycerol, but not phospholipid, levels in isolated rat cardiomyocytes. Lipids 2011, 46, 627–636. [Google Scholar] [CrossRef]
- He, L.; Kim, T.; Long, Q.; Liu, J.; Wang, P.; Zhou, Y.; Ding, Y.; Prasain, J.; Wood, P.A.; Yang, Q. Carnitine palmitoyltransferase-1b deficiency aggravates pressure overload-induced cardiac hypertrophy caused by lipotoxicity. Circulation 2012, 126, 1705–1716. [Google Scholar] [CrossRef]
- Henique, C.; Mansouri, A.; Fumey, G.; Lenoir, V.; Girard, J.; Bouillaud, F.; Prip-Buus, C.; Cohen, I. Increased mitochondrial fatty acid oxidation is sufficient to protect skeletal muscle cells from palmitate-induced apoptosis. J. Biol. Chem. 2010, 285, 36818–36827. [Google Scholar] [CrossRef]
- Haffar, T.; Berube-Simard, F.; Bousette, N. Impaired fatty acid oxidation as a cause for lipotoxicity in cardiomyocytes. Biochem Biophys. Res. Commun. 2015, 468, 73–78. [Google Scholar] [CrossRef]
- Sun, Y.; Zhang, D.; Liu, X.; Li, X.; Liu, F.; Yu, Y.; Jia, S.; Zhou, Y.; Zhao, Y. Endoplasmic reticulum stress affects lipid metabolism in atherosclerosis via chop activation and over-expression of mir-33. Cell Physiol. Biochem. 2018, 48, 1995–2010. [Google Scholar] [CrossRef] [PubMed]
- Rotllan, N.; Price, N.; Pati, P.; Goedeke, L.; Fernandez-Hernando, C. Micrornas in lipoprotein metabolism and cardiometabolic disorders. Atherosclerosis 2016, 246, 352–360. [Google Scholar] [CrossRef] [PubMed]
- Nishiga, M.; Horie, T.; Kuwabara, Y.; Nagao, K.; Baba, O.; Nakao, T.; Nishino, T.; Hakuno, D.; Nakashima, Y.; Nishi, H.; et al. Microrna-33 controls adaptive fibrotic response in the remodeling heart by preserving lipid raft cholesterol. Circ. Res. 2017, 120, 835–847. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Ma, P. Mir-33 may be a biological marker for coronary heart disease. Clin. Lab. 2018, 64, 1755–1760. [Google Scholar] [CrossRef]
- Shao, S.; Liu, Z.; Yang, Y.; Zhang, M.; Yu, X. Srebp-1c, pdx-1, and glp-1r involved in palmitate-epa regulated glucose-stimulated insulin secretion in ins-1 cells. J. Cell Biochem. 2010, 111, 634–642. [Google Scholar] [CrossRef]
- Tang, S.; Wu, W.; Tang, W.; Ge, Z.; Wang, H.; Hong, T.; Zhu, D.; Bi, Y. Suppression of rho-kinase 1 is responsible for insulin regulation of the ampk/srebp-1c pathway in skeletal muscle cells exposed to palmitate. Acta Diabetol. 2017, 54, 635–644. [Google Scholar] [CrossRef]
- Hong, S.W.; Lee, J.; Park, S.E.; Rhee, E.J.; Park, C.Y.; Oh, K.W.; Park, S.W.; Lee, W.Y. Repression of sterol regulatory element-binding protein 1-c is involved in the protective effects of exendin-4 in pancreatic beta-cell line. Mol. Cell Endocrinol. 2012, 362, 242–252. [Google Scholar] [CrossRef]
- Natalicchio, A.; Biondi, G.; Marrano, N.; Labarbuta, R.; Tortosa, F.; Spagnuolo, R.; D’Oria, R.; Carchia, E.; Leonardini, A.; Cignarelli, A.; et al. Long-term exposure of pancreatic beta-cells to palmitate results in srebp-1c-dependent decreases in glp-1 receptor signaling via creb and akt and insulin secretory response. Endocrinology 2016, 157, 2243–2258. [Google Scholar] [CrossRef]
- Chen, H.; Huang, X.N.; Stewart, A.F.; Sepulveda, J.L. Gene expression changes associated with fibronectin-induced cardiac myocyte hypertrophy. Physiol. Genom. 2004, 18, 273–283. [Google Scholar] [CrossRef]
- Marfella, R.; Di Filippo, C.; Portoghese, M.; Barbieri, M.; Ferraraccio, F.; Siniscalchi, M.; Cacciapuoti, F.; Rossi, F.; D’Amico, M.; Paolisso, G. Myocardial lipid accumulation in patients with pressure-overloaded heart and metabolic syndrome. J. Lipid Res. 2009, 50, 2314–2323. [Google Scholar] [CrossRef]
- Harmancey, R.; Haight, D.L.; Watts, K.A.; Taegtmeyer, H. Chronic hyperinsulinemia causes selective insulin resistance and down-regulates uncoupling protein 3 (ucp3) through the activation of sterol regulatory element-binding protein (srebp)-1 transcription factor in the mouse heart. J. Biol. Chem. 2015, 290, 30947–30961. [Google Scholar] [CrossRef] [PubMed]
- Perez-Belmonte, L.M.; Moreno-Santos, I.; Cabrera-Bueno, F.; Sanchez-Espin, G.; Castellano, D.; Such, M.; Crespo-Leiro, M.G.; Carrasco-Chinchilla, F.; Alonso-Pulpon, L.; Lopez-Garrido, M.; et al. Expression of sterol regulatory element-binding proteins in epicardial adipose tissue in patients with coronary artery disease and diabetes mellitus: Preliminary study. Int. J. Med. Sci. 2017, 14, 268–274. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward | Reverse |
|---|---|---|
| GAPDH | GACCTCAACTACATGGTCTACA | ACTCCACGACATACTCAGCAC |
| SREBF1A | CCATGGACGAGCTACCCTTC | GGCACTGGCTCCTCTTTGAT |
| SREBF1C | GATTGCACATTTGAAGACATGC | GCACGGACGGGTACATCTTTA |
| SREBF2 | AGCACACTTGTCGAGATCCA | CCTTGGCTGCTGACTTGATC |
| ANF | TGGGCTCCTTCTCCATCACC | GCCAAAAGGCCAGGAAGAGG |
| β-MHC | GCCTACCTCATGGGACTGAA | ACATTCTGCCCTTTGGTGAC |
| DGAT1 | CCCATACCCGGGACAAAGAC | AGAGTCTTGCAGACGATGGC |
| DGAT2 | CGTGAGGCGGCTTCCTG | GAGGATGCTGGAGCCAGTG |
| SPTLC1 | GAGGGTACGGGGATGAGTCT | TGAGCAAGCGGCTATCCAAA |
| SPTLC2 | CTCTACATGCCGGCCAAAAT | TCAAAGCCGTGTCAAGTATTTCT |
| SPTLC3 | CCAAGGCATCCGAGAGTTGT | AGCACTATGCGACTGAACCC |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cetrullo, S.; D’Adamo, S.; Panichi, V.; Borzì, R.M.; Pignatti, C.; Flamigni, F. Modulation of Fatty Acid-Related Genes in the Response of H9c2 Cardiac Cells to Palmitate and n-3 Polyunsaturated Fatty Acids. Cells 2020, 9, 537. https://doi.org/10.3390/cells9030537
Cetrullo S, D’Adamo S, Panichi V, Borzì RM, Pignatti C, Flamigni F. Modulation of Fatty Acid-Related Genes in the Response of H9c2 Cardiac Cells to Palmitate and n-3 Polyunsaturated Fatty Acids. Cells. 2020; 9(3):537. https://doi.org/10.3390/cells9030537
Chicago/Turabian StyleCetrullo, Silvia, Stefania D’Adamo, Veronica Panichi, Rosa Maria Borzì, Carla Pignatti, and Flavio Flamigni. 2020. "Modulation of Fatty Acid-Related Genes in the Response of H9c2 Cardiac Cells to Palmitate and n-3 Polyunsaturated Fatty Acids" Cells 9, no. 3: 537. https://doi.org/10.3390/cells9030537
APA StyleCetrullo, S., D’Adamo, S., Panichi, V., Borzì, R. M., Pignatti, C., & Flamigni, F. (2020). Modulation of Fatty Acid-Related Genes in the Response of H9c2 Cardiac Cells to Palmitate and n-3 Polyunsaturated Fatty Acids. Cells, 9(3), 537. https://doi.org/10.3390/cells9030537