Telomeres and Telomerase in Heart Ontogenesis, Aging and Regeneration

 ,
, 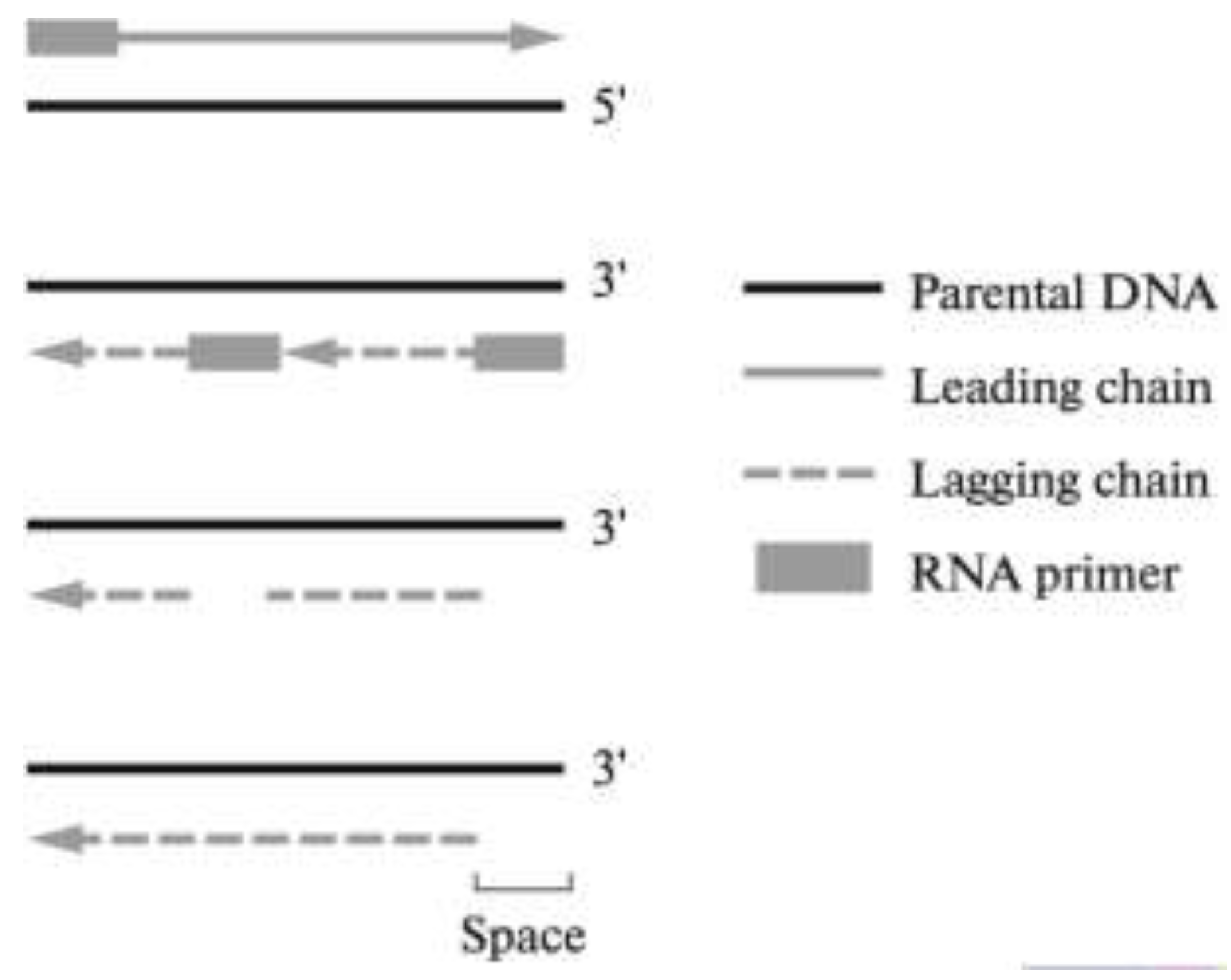{kind=link}
Abstract
1. Introduction
2. Telomeres and Telomere Complex
3. Embryonic Development of the Heart
4. Early Postnatal Heart Development
5. Prepubertal Period
6. Heart of Adult Vertebrates
7. Heart Aging
8. Heart Regeneration
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Experim. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Nalobin, D.S.; Galiakberova, A.A.; Alipkina, S.I.; Glukhov, A.I. Regulation of Telomerase Activity. Biol. Bull. Rev. 2018, 8, 142–154. [Google Scholar] [CrossRef]
- Blackburn, E.H. Switching and signaling at the telomere. Cell 2001, 10, 661–673. [Google Scholar] [CrossRef]
- Palm, W.; de Lange, T. How shelterin protects mammalian telomeres. Annu. Rev. Genet. 2008, 42, 301–334. [Google Scholar] [CrossRef] [PubMed]
- Harley, C.B. Telomere loss: Mitotic clock or genetic time bomb? Mutat. Res. 1991, 256, 271–282. [Google Scholar] [CrossRef]
- Artandi, S.E.; Attardi, L.D. Pathways connecting telomeres and p53 in senescence, apoptosis, and cancer. Biochem. Biophys. Res. Commun. 2005, 331, 881–890. [Google Scholar] [CrossRef] [PubMed]
- Greider, C.W.; Blackburn, E.H. Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell 1985, 43, 405–413. [Google Scholar] [CrossRef]
- Greider, W. Telomerase activity, cell proliferation, and cancer. Proc. Natl. Acad. Sci. USA 1998, 95, 90–92. [Google Scholar] [CrossRef]
- Sun, C.; Kontaridis, M.I. Physiology of cardiac development: From genetics to signaling to therapeutic strategies. Curr. Opin. Physiol. 2018, 1, 123–139. [Google Scholar] [CrossRef]
- Burggren, W.W.; Dubansky, B.; Bautista, N.M. Cardiovascular development in embryonic and larval fishes. Fish. Physiol. 2017, 36, 107–184. [Google Scholar]
- Erokhina, E.L. Proliferation dynamics of cellular elements in the differentiating mouse myocardium. Tsitologiia 1968, 10, 1391–1409. [Google Scholar] [PubMed]
- Nishimura, H. Atlas of Human Prenatal Histology; Igaku-Shoin: Tokyo, Japan, 1983; p. 316. [Google Scholar]
- Aix, E.; Gutierrez-Gutierrez, O.; Sanchez-Ferrer, C.; Aguado, T.; Flores, I. Postnatal telomere dysfunction induces cardiomyocyte cell-cycle arrest through p21 activation. J. Cell Biol. 2016, 13, 571–583. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.; Taffet, G.E.; Youker, K.A.; Entman, M.L.; Overbeek, P.A.; Michael, L.H.; Schneider, M.D. Telomerase reverse transcriptase promotes cardiac muscle cell proliferation, hypertrophy, and survival. Proc. Natl. Acad. Sci. USA 2001, 98, 10308–10313. [Google Scholar] [CrossRef] [PubMed]
- Borges, A.; Liew, C. Telomerase activity during cardiac development. J. Mol. Cell. Cardiol. 1997, 29, 2717–2724. [Google Scholar] [CrossRef]
- Marcela, S.G.; Cristina, R.M.; Angel, P.G.; Manuel, A.M.; Sofía, D.-C.; De La Patricia, R.-S.; Bladimir, R.-R.; Concepción, S.G. Chronological and morphological study of heart development in the rat. Anat. Rec. 2012, 295, 1267–1290. [Google Scholar] [CrossRef]
- Ikenishi, A.; Okayama, H.; Iwamoto, N.; Yoshitome, S.; Tane, S.; Nakamura, K.; Obayashi, T.; Hayashi, T.; Takeuchi, T. Cell cycle regulation in mouse heart during embryonic and postnatal stages. Dev. Growth. Differ. 2012, 54, 731–738. [Google Scholar] [CrossRef]
- Soonpaa, M.H.; Kim, K.K.; Pajak, L.; Franklin, M.; Field, L.J. Cardiomyocyte DNA synthesis and binucleation during murine development. Am. J. Physiol. 1996, 271, 2183–2189. [Google Scholar] [CrossRef]
- Richardson, G.D.; Breault, D.; Horrocks, G.; Cormack, S.; Hole, N.; Owens, W.A. Telomerase expression in the mammalian heart. FASEB J. 2012, 26, 4832–4840. [Google Scholar] [CrossRef]
- Bar, C.; Bernardes de Jesus, B.; Serrano, R.; Tejera, A.; Ayuso, E.; Jimenez, V.; Formentini, I.; Bobadilla, M.; Mizrahi, J.; de Martino, A.; et al. Telomerase expression confers cardioprotection in the adult mouse heart after acute myocardial infarction. Nat. Commun. 2014, 5, 1–14. [Google Scholar] [CrossRef]
- Puente, B.N.; Kimura, W.; Muralidhar, S.A.; Moon, J.; Amatruda, J.F.; Phelps, K.L.; Grinsfelder, D.; Rothermel, B.A.; Chen, R.; Garcia, J.A.; et al. The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response. Cell 2014, 157, 565–579. [Google Scholar] [CrossRef]
- Ghosh, R.; Mitchell, D.L. Effect of oxidative DNA damage in promoter elements on transcription factor binding. Nucleic Acids Res. 1999, 27, 3213–3218. [Google Scholar] [CrossRef] [PubMed]
- Kawanishi, S.; Oikawa, S. Mechanism of telomere shortening by oxidative stress. Ann. NY. Acad. Sci. 2004, 1019, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Fouquerel, E.; Lormand, J.; Bose, A.; Lee, H.T.; Kim, G.S.; Li, J.; Sobol, R.W.; Freudenthal, B.D.; Myong, S.; Opresko, P.L. Oxidative guanine base damage regulates human telomerase activity. Nat. Struct. Mol. Biol. 2016, 23, 1092–1100. [Google Scholar] [CrossRef] [PubMed]
- Pampalona, J.; Frías, C.; Genescà, A.; Tusell, L. Progressive telomere dysfunction causes cytokinesis failure and leads to the accumulation of polyploid cells. PLoS Genet. 2012, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Anchelin, M.; Murcia, L.; Alcaraz-Pérez, F.; García-Navarro, E.M.; Cayuela, M.L. Behavior of telomere and telomerase during aging and regeneration in zebrafish. PLoS ONE 2011, 6, 2. [Google Scholar] [CrossRef] [PubMed]
- Bednarek, D.; González-Rosa, J.M.; Guzmán-Martínez, G.; Gutiérrez-Gutiérrez, Ó.; Aguado, T.; Sánchez-Ferrer, C.; Marques, I.J.; Galardi-Castilla, M.; de Diego, I.; Gómez, M.J.; et al. Telomerase Is Essential for Zebrafish Heart Regeneration. Cell Rep. 2015, 12, 1691–1703. [Google Scholar] [CrossRef]
- Wills, A.A.; Holdway, J.E.; Major, R.J.; Poss, K.D. Regulated addition of new myocardial and epicardial cells fosters homeostatic cardiac growth and maintenance in adult zebrafish. Development 2008, 135, 183–192. [Google Scholar] [CrossRef]
- Gonzalez-Rosa, J.M.; Sharpe, M.; Field, D.; Soonpaa, M.H.; Field, L.J.; Burns, C.E.; Burns, C.G. Myocardial polyploidization creates a barrier to heart regeneration in zebrafish. Dev. Cell. 2018, 44, 433–446. [Google Scholar] [CrossRef]
- Naqvi, N.; Li, M.; Calvert, J.W.; Tejada, T.; Lambert, J.P.; Wu, J.; Kesteven, S.H.; Holman, S.R.; Matsuda, T.; Lovelock, J.D.; et al. A proliferative burst during preadolescence establishes the final cardiomyocyte number. Cell 2014, 157, 795–807. [Google Scholar] [CrossRef]
- Alkass, K.; Panula, J.; Westman, M.; Wu, T.D.; Guerquin-Kern, J.L.; Bergmann, O. No Evidence for cardiomyocyte number expansion in preadolescent mice. Cell 2015, 163, 1026–1036. [Google Scholar] [CrossRef]
- Soonpaa, M.H.; Zebrowski, D.C.; Platt, C.; Rosenzweig, A.; Engel, F.B.; Field, L.J. Cardiomyocyte Cell-Cycle Activity during Preadolescence. Cell 2015, 163, 781–782. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wilson, R.M.; Kubo, H.; Berretta, R.M.; Harris, D.M.; Zhang, X.; Jaleel, N.; MacDonnell, S.M.; Bearzi, C.; Tillmanns, J.; et al. Adolescent feline heart contains a population of small, proliferative ventricular myocytes with immature physiological properties. Circ. Res. 2007, 100, 536–544. [Google Scholar] [CrossRef] [PubMed]
- Mollova, M.; Bersell, K.; Walsh, S.; Savla, J.; Tanmoy Das, L.; Park, S.-Y.; Silberstein, L.E.; dos Remedios, C.G.; Graham, D.; Colan, S.; et al. Cardiomyocyte proliferation contributes to heart growth in young humans. Proc. Natl. Acad. Sci. USA 2013, 110, 1446–1451. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, O.; Bhardwaj, R.D.; Bernard, S.; Zdunek, S.; Barnabé-Heider, F.; Walsh, S.; Zupicich, J.; Alkass, K.; Buchholz, B.A.; Druid, H.; et al. Evidence for cardiomyocyte renewal in humans. Science 2009, 324, 98–102. [Google Scholar] [CrossRef]
- Rota, M.; Hosoda, T.; De Angelis, A.; Arcarese, M.L.; Esposito, G.; Rizzi, R.; Tillmanns, J.; Tugal, D.; Musso, E.; Rimoldi, O.; et al. The young mouse heart is composed of myocytes heterogeneous in age and function. Circ. Res. 2007, 101, 387–399. [Google Scholar] [CrossRef]
- Hara, E.; Smith, R.; Parry, D.; Tahara, H.; Stone, S.; Peters, G. Regulation of p16CDKN2 expression and its implications for cell immortalization and senescence. Mol. Cell Biol. 1996, 16, 859–867. [Google Scholar] [CrossRef]
- Lund, T.C.; Glass, T.J.; Tolar, J.; Blazar, B.R. Expression of telomerase and telomere length are unaffected by either age or limb regeneration in Danio rerio. PLoS ONE 2009, 4, 11. [Google Scholar] [CrossRef]
- Terai, M.; Izumiyama-Shimomura, N.; Aida, J.; Ishikawa, N.; Sawabe, M.; Arai, T.; Fujiwara, M.; Ishii, A.; Nakamura, K.; Takubo, K. Association of telomere shortening in myocardium with heart weight gain and cause of death. Sci. Rep. 2013, 3, 2401. [Google Scholar] [CrossRef]
- Hastings, R.; Li, N.C.; Lacy, P.S.; Patel, H.; Herbert, K.E.; Stanley, A.G.; Williams, B. Rapid telomere attrition in cardiac tissue of the ageing Wistar rat. Exp. Gerontol. 2004, 39, 855–857. [Google Scholar] [CrossRef]
- Ogami, M.; Ikura, Y.; Ohsawa, M.; Matsuo, T.; Kayo, S.; Yoshimi, N.; Hai, E.; Shirai, N.; Ehara, S.; Komatsu, R.; et al. Telomere shortening in human coronary artery diseases. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 546–550. [Google Scholar] [CrossRef]
- Scheller Madrid, A.; Rode, L.; Nordestgaard, B.G.; Bojesen, S.E. Short telomere length and ischemic heart disease: Observational and genetic studies in 290 022 individuals. Clin. Chem. 2016, 62, 1140–1149. [Google Scholar] [CrossRef] [PubMed]
- Lily, L. Pathophysiology of Diseases of the Cardiovascular System, 4th ed.; BINOM: Moscow, Russia, 2016; p. 598. [Google Scholar]
- McNally, E.M.; Mestroni, L. Dilated Cardiomyopathy: Genetic Determinants and Mechanisms. Circ. Res. 2017, 121, 731–748. [Google Scholar] [CrossRef] [PubMed]
- Chimenti, C.; Kajstura, J.; Torella, D.; Urbanek, K.; Heleniak, H.; Colussi, C.; Di Meglio, F.; Nadal-Ginard, B.; Frustaci, A.; Leri, A.; et al. Senescence and death of primitive cells and myocytes lead to premature cardiac aging and heart failure. Circ. Res. 2003, 93, 604–613. [Google Scholar] [CrossRef] [PubMed]
- Boon, R.A.; Iekushi, K.; Lechner, S.; Seeger, T.; Fischer, A.; Heydt, S.; Kaluza, D.; Tréguer, K.; Carmona, G.; Bonauer, A.; et al. MicroRNA-34a regulates cardiac ageing and function. Nature 2013, 495, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Leri, A.; Franco, S.; Zacheo, A.; Barlucchi, L.; Chimenti, S.; Limana, F.; Nadal-Ginard, B.; Kajstura, J.; Anversa, P.; Blasco, M.A. Ablation of telomerase and telomere loss leads to cardiac dilatation and heart failure associated with p53 upregulation. EMBO J. 2003, 22, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Sahin, E.; Colla, S.; Liesa, M.; Moslehi, J.; Müller, F.L.; Guo, M.; Cooper, M.; Kotton, D.; Fabian, A.J.; Walkey, C.; et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature 2011, 470, 359–365. [Google Scholar] [CrossRef]
- Guarente, L. Mitochondria--a nexus for aging, calorie restriction, and sirtuins? Cell 2008, 132, 171–176. [Google Scholar] [CrossRef]
- Ren, J.; Sowers, J.R.; Zhang, Y. Metabolic Stress, Autophagy and Cardiovascular Aging: From Pathophysiology to Therapeutics. Trends Endocrinol. Metab. 2018, 29, 699–711. [Google Scholar] [CrossRef]
- Barton, M.; Husmann, M.; Meyer, M.R. Accelerated Vascular Aging as a Paradigm for Hypertensive Vascular Disease: Prevention and Therapy. Can. J. Cardiol. 2016, 32, 680–686. [Google Scholar] [CrossRef]
- Buford, T.W. Hypertension and aging. Ageing Res. Rev. 2016, 26, 96–111. [Google Scholar] [CrossRef]
- Bertolotti, M.; Lonardo, A.; Mussi, C.; Baldelli, E.; Pellegrini, E.; Ballestri, S.; Romagnoli, D.; Loria, P. Nonalcoholic fatty liver disease and aging: Epidemiology to management. World J. Gastroenterol. 2014, 20, 14185–14204. [Google Scholar] [CrossRef] [PubMed]
- Abel, E.D. Obesity stresses cardiac mitochondria even when you are young. J. Am. Coll Cardiol. 2011, 57, 586–589. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Zhang, Y. Targeting Autophagy in Aging and Aging-Related Cardiovascular Diseases. Trends Pharm. Sci. 2018, 39, 1064–1076. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Yoshimori, T. Autophagy and Longevity. Mol. Cells 2018, 41, 65–72. [Google Scholar] [PubMed]
- Hansen, M.; Rubinsztein, D.C.; Walker, D.W. Autophagy as a promoter of longevity: Insights from model organisms. Nat. Rev. Mol. Cell Biol. 2018, 19, 579–593. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Bergamini, E.; Brunk, U.T.; Dröge, W.; Ffrench, M.; Terman, A. Autophagy and aging: The importance of maintaining “clean” cells. Autophagy 2005, 1, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Schafer, M.J.; Miller, J.D.; LeBrasseur, N.K. Cellular senescence: Implications for metabolic disease. Mol. Cell Endocrinol. 2017, 455, 93–102. [Google Scholar] [CrossRef]
- Alfaras, I.; Di Germanio, C.; Bernier, M.; Csiszar, A.; Ungvari, Z.; Lakatta, E.G.; de Cabo, R. Pharmacological Strategies to Retard Cardiovascular Aging. Circ. Res. 2016, 118, 1626–1642. [Google Scholar] [CrossRef]
- Sonneborn, J.S. Telomerase Biology Associations Offer Keys to Cancer and Aging Therapeutics. Curr. Aging Sci. 2019. [Google Scholar] [CrossRef]
- Haendeler, J.; Hoffmann, J.; Brandes, R.P.; Zeiher, A.M.; Dimmeler, S. Hydrogen peroxide triggers nuclear export of telomerase reverse transcriptase via Src kinase family-dependent phosphorylation of tyrosine 707. Mol. Cell Biol. 2003, 23, 4598–4610. [Google Scholar] [CrossRef]
- Ait-Aissa, K.; Ebben, J.D.; Kadlec, A.O.; Beyer, A.M. Friend or foe? Telomerase as a pharmacological target in cancer and cardiovascular disease. Pharmacol. Res. 2016, 111, 422–433. [Google Scholar] [CrossRef] [PubMed]
- Quryshi, N.; Norwood Toro, L.E.; Ait-Aissa, K.; Kong, A.; Beyer, A.M. Chemotherapeutic-Induced Cardiovascular Dysfunction: Physiological Effects, Early Detection-The Role of Telomerase to Counteract Mitochondrial Defects and Oxidative Stress. Int. J. Mol. Sci 2018, 19, 3. [Google Scholar] [CrossRef] [PubMed]
- Ait-Aissa, K.; Heisner, J.S.; Norwood Toro, L.E.; Bruemmer, D.; Doyon, G.; Harmann, L.; Geurts, A.; Camara, A.K.S.; Beyer, A.M. Telomerase Deficiency Predisposes to Heart Failure and Ischemia-Reperfusion Injury. Front. Cardiovasc. Med. 2019, 6, 31. [Google Scholar] [CrossRef] [PubMed]
- Sharifi-Sanjani, M.; Oyster, N.M.; Tichy, E.D.; Bedi, K.C. Jr.; Harel, O.; Margulies, K.B.; Mourkioti, F. Cardiomyocyte- Specific Telomere Shortening is a Distinct Signature of Heart Failure in Humans. J. Am. Heart Assoc. 2017, 6, 9. [Google Scholar] [CrossRef] [PubMed]
- Porrello, E.R.; Mahmoud, A.I.; Simpson, E.; Johnson, B.A.; Grinsfelder, D.; Canseco, D.; Mammen, P.P. ’ Rothermel, B.A.; Olson, E.N.; Sadek, H.A. Regulation of neonatal and adult mammalian heart regeneration by the miR-15 family. Proc. Natl. Acad. Sci.USA 2013, 110, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Zebrowski, D.C.; Jensen, C.H.; Becker, R.; Ferrazzi, F.; Baun, C.; Hvidsten, S.; Sheikh, S.P.; Polizzotti, B.D.; Andersen, D.C.; Engel, F.B. Cardiac injury of the newborn mammalian heart accelerates cardiomyocyte terminal differentiation. Sci. Rep. 2017, 7, 8362. [Google Scholar] [CrossRef]
- Wang, W.E.; Li, L.; Xia, X.; Fu, W.; Liao, Q.; Lan, C.; Yang, D.; Chen, H.; Yue, R.; Zeng, C.; et al. Dedifferentiation, proliferation, and redifferentiation of adult mammalian cardiomyocytes after ischemic injury. Circulation 2017, 136, 834–848. [Google Scholar] [CrossRef]
- Urbanek, K.; Rota, M.; Cascapera, S.; Bearzi, C.; Nascimbene, A.; De Angelis, A.; Hosoda, T.; Chimenti, S.; Baker, M.; Limana, F.; et al. Cardiac stem cells possess growth factor-receptor systems that after activation regenerate the infarcted myocardium, improving ventricular function and long-term survival. Circ. Res. 2005, 97, 663–673. [Google Scholar] [CrossRef]
- Oh, H.; Wang, S.C.; Prahash, A.; Sano, M.; Moravec, C.S.; Taffet, G.E.; Michael, L.H.; Youker, K.A.; Entman, M.L.; Schneider, M.D. Telomere attrition and Chk2 activation in human heart failure. Proc. Natl. Acad. Sci. USA 2003, 100, 5378–5383. [Google Scholar] [CrossRef]
- Angelos, M.G.; Kutala, V.K.; Torres, C.A.; He, G.; Stoner, J.D.; Mohammad, M.; Kuppusamy, P. Hypoxic reperfusion of the ischemic heart and oxygen radical generation. Am. J. Physiol. Heart. Circ. Physiol. 2006, 290, 341–347. [Google Scholar] [CrossRef]
- Nakada, Y.; Canseco, D.C.; Thet, S.; Abdisalaam, S.; Asaithamby, A.; Santos, C.X.; Shah, A.M.; Zhang, H.; Faber, J.E.; Kinter, M.T.; et al. Hypoxia induces heart regeneration in adult mice. Nature 2017, 541, 222–227. [Google Scholar] [CrossRef] [PubMed]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nalobin, D.; Alipkina, S.; Gaidamaka, A.; Glukhov, A.; Khuchua, Z. Telomeres and Telomerase in Heart Ontogenesis, Aging and Regeneration. Cells 2020, 9, 503. https://doi.org/10.3390/cells9020503
Nalobin D, Alipkina S, Gaidamaka A, Glukhov A, Khuchua Z. Telomeres and Telomerase in Heart Ontogenesis, Aging and Regeneration. Cells. 2020; 9(2):503. https://doi.org/10.3390/cells9020503
Chicago/Turabian StyleNalobin, Denis, Svetlana Alipkina, Anna Gaidamaka, Alexander Glukhov, and Zaza Khuchua. 2020. "Telomeres and Telomerase in Heart Ontogenesis, Aging and Regeneration" Cells 9, no. 2: 503. https://doi.org/10.3390/cells9020503
APA StyleNalobin, D., Alipkina, S., Gaidamaka, A., Glukhov, A., & Khuchua, Z. (2020). Telomeres and Telomerase in Heart Ontogenesis, Aging and Regeneration. Cells, 9(2), 503. https://doi.org/10.3390/cells9020503