Esophageal Cancer Development: Crucial Clues Arising from the Extracellular Matrix

 , ,
, , {kind=link}
{kind=link}
Abstract
1. Introduction
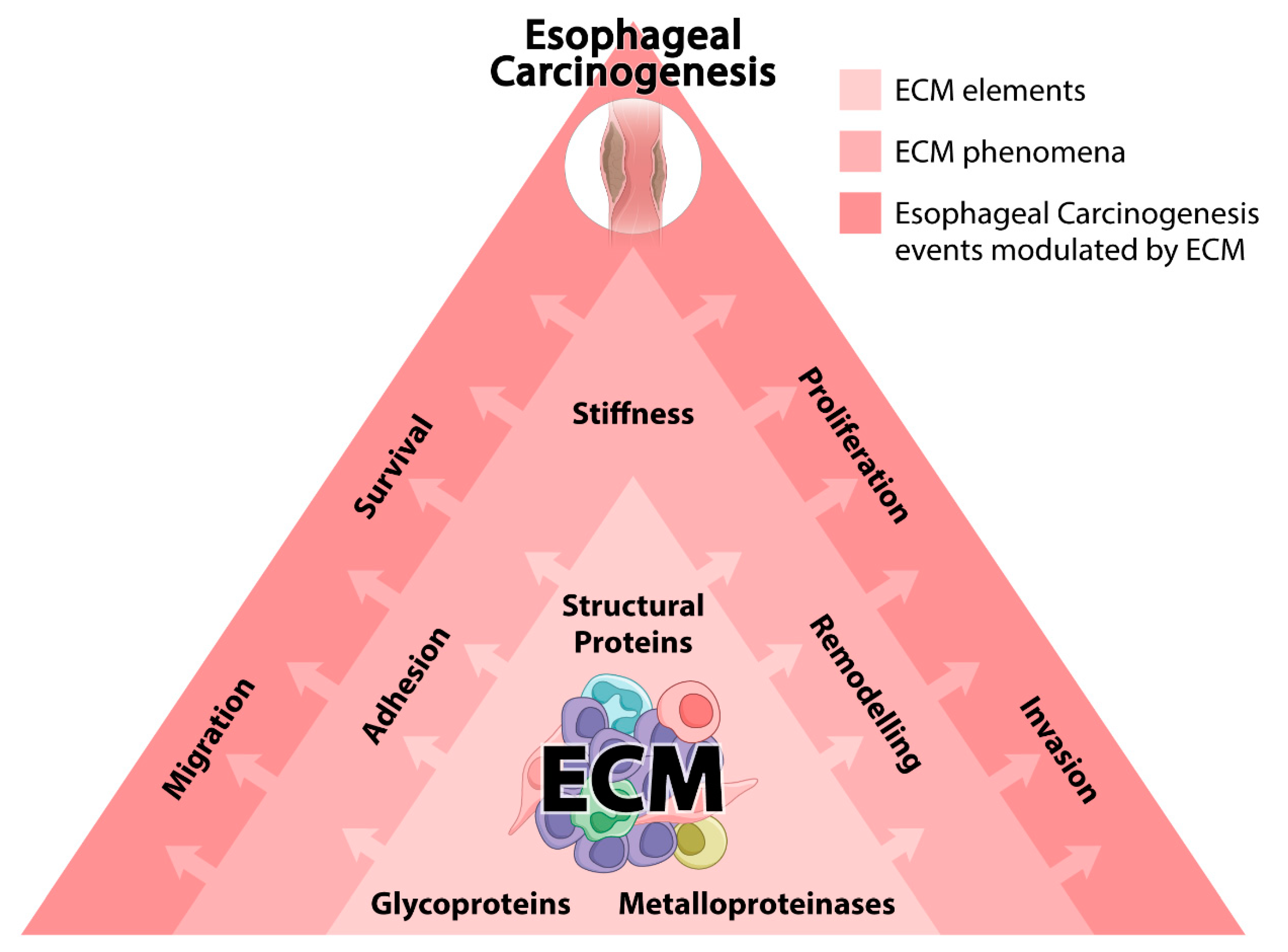
2. EC and ECM
2.1. Structural Proteins and ECM Stiffness
2.2. MMPs and ECM Remodeling
2.3. Glycoproteins and ECM Adhesion and Migration
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| α-SMA | α-smooth muscle actin |
| ABCA12 | 3 ATP Binding Cassette Subfamily A Member 12-3 |
| Akt | protein kinase B |
| AP-1 | activator protein 1 |
| APE1 | apurinic/apyrimidinic endonuclease 1 |
| ARF6 | ADP-ribosylation factor 6 |
| ATF3 | activating transcription factor 3 |
| AURKA | Aurora A kinase |
| BE | Barrett’s esophagus |
| CAF | cancer-associated fibroblasts |
| COX-2 | cyclooxygenase-2 |
| CXCR4 | C-X-C motif chemokine receptor 4 |
| DIRAS1 | Distinct Subgroup of The Ras Family Member 1 |
| DDR | discoidin domain receptor |
| EAC | esophageal adenocarcinoma |
| EC | esophageal cancer |
| ECM | extracellular matrix |
| EGF | epidermal growth factor |
| EGFR | epidermal growth factor receptor |
| EMT | epithelial mesenchymal transition |
| Erk | extracellular-signal regulated kinase |
| ESCC | esophageal squamous cell carcinoma |
| FAK | focal adhesion kinase |
| FAP-α | fibroblast activation protein α |
| GERD | gastroesophageal reflux disease |
| HA | hyaluronic acid |
| HIF-1α | hypoxia-inducible factor 1-α |
| hTERT | human telomerase reverse transcriptase |
| IL-17 | interleukin 17 |
| JNK | c-Jun N-terminal kinase |
| Lam5 (γ2) | chain γ2 of laminin-5 |
| LGALS3BP | galectin-3 binding protein |
| lncRNA | long noncoding RNA |
| LOX | lysyl oxidase |
| LPS | lipopolysaccharide |
| MAPK | mitogen-activated protein kinase |
| miRNA | microRNA |
| MMP | matrix metalloproteinase |
| MT1-MMP | membrane-type 1 matrix metalloproteinase |
| NF-ĸB | nuclear factor kappa B |
| OPN | osteopontin |
| P38 | p38 mitogen-activated protein kinases |
| pErk | phosphorylated extracellular signal-regulated kinases |
| PI3K | phosphoinositide 3-kinase |
| PTK7 | protein tyrosine kinase 7 |
| ROS | reactive oxygen species |
| SOX | sex-determining region Y-box-box transcription factor |
| SPRY4-IT1 | sprouty RTK signaling antagonist 4—Intronic Transcript 1 |
| STAT3 | signal transducer and activator of transcription 3 |
| TAF | tumor associated fibroblasts |
| TLR4 | toll-like receptor 4 |
| VEGF | vascular endothelial growth factor |
References
- Frantz, C.; Stewart, K.M.; Weaver, V.M. The extracellular matrix at a glance. J. Cell Sci. 2010, 123, 4195–4200. [Google Scholar] [CrossRef] [PubMed]
- Huang-Doran, I.; Zhang, C.-Y.; Vidal-Puig, A. Extracellular Vesicles: Novel Mediators of Cell Communication in Metabolic Disease. Trends Endocrinol. Metab. 2017, 28, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Gjorevski, N.; Nelson, C.M. Bidirectional extracellular matrix signaling during tissue morphogenesis. Cytokine Growth Factor Rev. 2009, 20, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Plotnikov, E.Y.; Silachev, D.N.; Popkov, V.A.; Zorova, L.D.; Pevzner, I.B.; Zorov, S.D.; Jankauskas, S.S.; Babenko, V.A.; Sukhikh, G.T.; Zorov, D.B. Intercellular Signalling Cross-Talk: To Kill, To Heal and To Rejuvenate. Heart Lung Circ. 2017, 26, 648–659. [Google Scholar] [CrossRef]
- Bosman, F.T.; Stamenkovic, I. Functional structure and composition of the extracellular matrix. J. Pathol. 2003, 200, 423–428. [Google Scholar] [CrossRef]
- Hynes, R.O. Extracellular matrix: Not just pretty fibrils. Science 2009, 326, 1216–1219. [Google Scholar] [CrossRef]
- Nelson, C.M.; Bissell, M.J. Of extracellular matrix, scaffolds, and signaling: Tissue architecture regulates development, homeostasis, and cancer. Annu. Rev. Cell Dev. Biol. 2006, 22, 287–309. [Google Scholar] [CrossRef]
- Pickup, M.W.; Mouw, J.K.; Weaver, V.M. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep. 2014, 15, 1243–1253. [Google Scholar] [CrossRef]
- Ghajar, C.M.; Bissell, M.J. Extracellular matrix control of mammary gland morphogenesis and tumorigenesis: Insights from imaging. Histochem. Cell Biol. 2008, 130, 1105–1118. [Google Scholar] [CrossRef]
- Pearce, O.M.; Delaine-Smith, R.M.; Maniati, E.; Nichols, S.; Wang, J.; Böhm, S.; Rajeeve, V.; Ullah, D.; Chakravarty, P.; Jones, R.R.; et al. Deconstruction of a Metastatic Tumor Microenvironment Reveals a Common Matrix Response in Human Cancers. Cancer Discov. 2017, 8, 304–319. [Google Scholar] [CrossRef]
- Yuzhalin, A.E.; Urbonas, T.; Silva, M.A.; Muschel, R.J.; Gordon-Weeks, A.N. A core matrisome gene signature predicts cancer outcome. Br. J. Cancer 2018, 118, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Kai, F.; Drain, A.P.; Weaver, V.M. The Extracellular Matrix Modulates the Metastatic Journey. Dev. Cell 2019, 49, 332–346. [Google Scholar] [CrossRef]
- Comoglio, P.M.; Trusolino, L. Cancer: The matrix is now in control. Nat. Med. 2005, 11, 1156–1158. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Weaver, V.M.; Werb, Z. The extracellular matrix: A dynamic niche in cancer progression. J. Cell Biol. 2012, 196, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Lin, E.W.; Karakasheva, T.A.; Hicks, P.D.; Bass, A.J.; Rustgi, A.K. The tumor microenvironment in esophageal cancer. Oncogene 2016, 35, 5337–5349. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Pennathur, A.; Gibson, M.K.; Jobe, B.A.; Luketich, J.D. Oesophageal carcinoma. Lancet 2013, 381, 400–412. [Google Scholar] [CrossRef]
- Lagergren, J.; Lagergren, P. Oesophageal cancer. BMJ 2010, 341, c6280. [Google Scholar] [CrossRef]
- Coleman, H.G.; Xie, S.-H.; Lagergren, J. The Epidemiology of Esophageal Adenocarcinoma. Gastroenterology 2018, 154, 390–405. [Google Scholar] [CrossRef]
- Zhang, Y. Epidemiology of esophageal cancer. World J. Gastroenterol. 2013, 19, 5598–5606. [Google Scholar] [CrossRef]
- Gupta, B.; Kumar, N. Worldwide incidence, mortality and time trends for cancer of the oesophagus. Eur. J. Cancer Prev. 2017, 26, 107–118. [Google Scholar] [CrossRef]
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Morishima, K.; Ui, T.; Matsubara, D.; Tamura, T.; Oguni, S.; Hosoya, Y.; Sata, N.; Lefor, A.T.; Yasuda, Y.; et al. Stromal fibroblasts are predictors of disease-related mortality in esophageal squamous cell carcinoma. Oncol. Rep. 2014, 32, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Kretschmer, I.; Freudenberger, T.; Twarock, S.; Yamaguchi, Y.; Grandoch, M.; Fischer, J.W. Esophageal squamous cell carcinoma cells modulate chemokine expression and hyaluronan synthesis in fibroblasts. J. Biol. Chem. 2016, 291, 4091–4106. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, G.; Wang, J.; Wang, L.; Huang, X.; Cheng, Y. The role of cancer-associated fibroblasts in esophageal cancer. J. Transl. Med. 2016, 14, 30. [Google Scholar] [CrossRef] [PubMed]
- Fan, D.; Creemers, E.E.; Kassiri, Z. Matrix as an Interstitial Transport System. Circ. Res. 2014, 114, 889–902. [Google Scholar] [CrossRef]
- Marks, D.L.; Olson, R.L.; Fernandez-Zapico, E.M. Epigenetic control of the tumor microenvironment. Epigenomics 2016, 8, 1671–1687. [Google Scholar] [CrossRef]
- Fang, M.; Yuan, J.; Peng, C.; Li, Y. Collagen as a double-edged sword in tumor progression. Tumour Biol. 2014, 35, 2871–2882. [Google Scholar] [CrossRef]
- Northcott, J.M.; Dean, I.S.; Mouw, J.K.; Weaver, V.M. Feeling Stress: The Mechanics of Cancer Progression and Aggression. Front. Cell Dev. Biol. 2018, 6, 17. [Google Scholar] [CrossRef]
- Mouw, J.K.; Ou, G.; Weaver, V.M. Extracellular matrix assembly: A multiscale deconstruction. Nat. Rev. Mol. Cell Biol. 2014, 15, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Paszek, M.J.; Zahir, N.; Johnson, K.R.; Lakins, J.N.; Rozenberg, G.I.; Gefen, A.; Reinhart-King, C.A.; Margulies, S.S.; Dembo, M.; Boettiger, D.; et al. Tensional homeostasis and the malignant phenotype. Cancer Cell 2005, 8, 241–254. [Google Scholar] [CrossRef] [PubMed]
- Rudnick, J.A.; Kuperwasser, C. Stromal biomarkers in breast cancer development and progression. Clin. Exp. Metastasis 2012, 29, 663–672. [Google Scholar] [CrossRef]
- Voiles, L.; Lewis, D.E.; Han, L.; Lupov, I.P.; Lin, T.-L.; Robertson, M.J.; Petrache, I.; Chang, H.-C. Overexpression of type VI collagen in neoplastic lung tissues. Oncol. Rep. 2014, 32, 1897–1904. [Google Scholar] [CrossRef] [PubMed]
- Xiong, G.; Deng, L.; Zhu, J.; Rychahou, P.G.; Xu, R. Prolyl-4-hydroxylase α subunit 2 promotes breast cancer progression and metastasis by regulating collagen deposition. BMC Cancer 2014, 14, 1. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Xiong, G.; Trinkle, C.; Xu, R. Integrated extracellular matrix signaling in mammary gland development and breast cancer progression. Histol. Histopathol. 2014, 29, 1083–1092. [Google Scholar]
- Troester, M.A.; Lee, M.H.; Carter, M.; Fan, C.; Cowan, D.W.; Perez, E.R.; Pirone, J.R.; Perou, C.M.; Jerry, D.J.; Schneider, S.S. Activation of Host Wound Responses in Breast Cancer Microenvironment. Clin. Cancer Res. 2009, 15, 7020–7028. [Google Scholar] [CrossRef]
- Senthebane, D.A.; Jonker, T.; Rowe, A.; Thomford, N.E.; Munro, D.; Dandara, C.; Wonkam, A.; Govender, D.; Calder, B.; Soares, N.C.; et al. The Role of Tumor Microenvironment in Chemoresistance: 3D Extracellular Matrices as Accomplices. Int. J. Mol. Sci. 2018, 19, 2861. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Wintzell, M.; Hjerpe, E.; Lundqvist, E.Å.; Shoshan, M. Protein markers of cancer-associated fibroblasts and tumor-initiating cells reveal subpopulations in freshly isolated ovarian cancer ascites. BMC Cancer 2012, 12, 359. [Google Scholar] [CrossRef]
- Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.K.; Zillhardt, M.; Hua, Y.; Tiwari, P.; Murmann, A.E.; Peter, M.E.; Lengyel, E. MicroRNAs reprogram normal fibroblasts into cancer-associated fibroblasts in ovarian cancer. Cancer Discov. 2012, 2, 1100–1108. [Google Scholar] [CrossRef]
- Tanaka, K.; Miyata, H.; Sugimura, K.; Fukuda, S.; Kanemura, T.; Yamashita, K.; Miyazaki, Y.; Takahashi, T.; Kurokawa, Y.; Yamasaki, M.; et al. miR-27 is associated with chemoresistance in esophageal cancer through transformation of normal fibroblasts to cancer-associated fibroblasts. Carcinogenesis 2015, 36, 894–903. [Google Scholar] [CrossRef] [PubMed]
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal Fibroblasts Present in Invasive Human Breast Carcinomas Promote Tumor Growth and Angiogenesis through Elevated SDF-1/CXCL12 Secretion. Cell 2005, 121, 335–348. [Google Scholar] [CrossRef]
- Cat, B.; Stuhlmann, D.; Steinbrenner, H.; Alili, L.; Holtkötter, O.; Sies, H.; Brenneisen, P. Enhancement of tumor invasion depends on transdifferentiation of skin fibroblasts mediated by reactive oxygen species. J. Cell Sci. 2006, 119, 2727–2738. [Google Scholar] [CrossRef]
- Marsh, D.; Suchak, K.; Moutasim, K.A.; Vallath, S.; Hopper, C.; Jerjes, W.; Upile, T.; Kalavrezos, N.; Violette, S.M.; Weinreb, P.H.; et al. Stromal features are predictive of disease mortality in oral cancer patients. J. Pathol. 2011, 223, 470–481. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Torres, S.; Bartolomé, R.A.; Mendes, M.; Barderas, R.; Fernandez-Aceñero, M.J.; Peláez-García, A.; Peña, C.; Lopez-Lucendo, M.; Villar-Vázquez, R.; De Herreros, A.G.; et al. Proteome Profiling of Cancer-Associated Fibroblasts Identifies Novel Proinflammatory Signatures and Prognostic Markers for Colorectal Cancer. Clin. Cancer Res. 2013, 19, 6006–6019. [Google Scholar] [CrossRef]
- Ishibashi, N.; Maebayashi, T.; Aizawa, T.; Sakaguchi, M.; Okada, M. Serum tumor marker levels at the development of intracranial metastasis in patients with lung or breast cancer. J. Thorac. Dis. 2019, 11, 1765–1771. [Google Scholar] [CrossRef]
- Palmieri, D.; Astigiano, S.; Barbieri, O.; Ferrari, N.; Marchisio, S.; Ulivi, V.; Volta, C.; Manduca, P. Procollagen I COOH-terminal fragment induces VEGF-A and CXCR4 expression in breast carcinoma cells. Exp. Cell Res. 2008, 314, 2289–2298. [Google Scholar] [CrossRef]
- Jabłońska-Trypuć, A.; Matejczyk, M.; Rosochacki, S. Matrix metalloproteinases (MMPs), the main extracellular matrix (ECM) enzymes in collagen degradation, as a target for anticancer drugs. J. Enzym. Inhib. Med. Chem. 2016, 31, 177–183. [Google Scholar] [CrossRef]
- Fang, S.; Dai, Y.; Mei, Y.; Yang, M.; Hu, L.; Yang, H.; Guan, X.; Li, J. Clinical significance and biological role of cancer-derived Type I collagen in lung and esophageal cancers. Thorac. Cancer 2019, 10, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, X.; Zheng, K.; Liu, Y.; Li, J.; Wang, S.; Liu, K.; Song, X.; Li, N.; Xie, S.; et al. The clinical significance of collagen family gene expression in esophageal squamous cell carcinoma. PeerJ 2019, 7, e7705. [Google Scholar] [CrossRef] [PubMed]
- Hanley, C.J.; Noble, F.; Ward, M.; Bullock, M.; Drifka, C.; Mellone, M.; Manousopoulou, A.; Johnston, H.E.; Hayden, A.; Thirdborough, S.; et al. A subset of myofibroblastic cancer-associated fibroblasts regulate collagen fiber elongation, which is prognostic in multiple cancers. Oncotarget 2016, 7, 6159–6174. [Google Scholar] [CrossRef] [PubMed]
- Rammal, H.; Saby, C.; Magnien, K.; Van-Gulick, L.; Garnotel, R.; Buache, E.; El Btaouri, H.; Jeannesson, P.; Morjani, H. Discoidin Domain Receptors: Potential actors and targets in cancer. Front. Pharmacol. 2016, 7, 55. [Google Scholar] [CrossRef] [PubMed]
- Croissant, C.; Tuariihionoa, A.; Bacou, M.; Souleyreau, W.; Sala, M.; Henriet, E.; Bikfalvi, A.; Saltel, F.; Auguste, P.; Bikvalvi, A. DDR1 and DDR2 physical interaction leads to signaling interconnection but with possible distinct functions. Cell Adhes. Migr. 2018, 12, 324–334. [Google Scholar] [CrossRef]
- Huang, Y.; Arora, P.; McCulloch, C.A.; Vogel, W.F. The collagen receptor DDR1 regulates cell spreading and motility by associating with myosin IIA. J. Cell Sci. 2009, 122, 1637–1646. [Google Scholar] [CrossRef]
- Ruiz, P.A.; Jarai, G. Collagen I Induces Discoidin Domain Receptor (DDR) 1 Expression through DDR2 and a JAK2-ERK1/2-mediated Mechanism in Primary Human Lung Fibroblasts. J. Biol. Chem. 2011, 286, 12912–12923. [Google Scholar] [CrossRef]
- Fong, S.F.T.; Dietzsch, E.; Fong, K.S.K.; Hollósi, P.; Asuncion, L.; He, Q.; Parker, M.I.; Csiszar, K. Lysyl oxidase-like 2 expression is increased in colon and esophageal tumors and associated with less differentiated colon tumors. Genes Chromosom. Cancer 2007, 46, 644–655. [Google Scholar] [CrossRef]
- Pylayeva, Y.; Gillen, K.M.; Gerald, W.; Beggs, H.E.; Reichardt, L.F.; Giancotti, F.G. Ras- and PI3K-dependent breast tumorigenesis in mice and humans requires focal adhesion kinase signaling. J. Clin. Investig. 2009, 119, 252–266. [Google Scholar] [CrossRef]
- Matte, B.F.; Kumar, A.; Placone, J.K.; Zanella, V.G.; Martins, M.D.; Engler, A.J.; Lamers, M.L. Matrix stiffness mechanically conditions EMT and migratory behavior of oral squamous cell carcinoma. J. Cell Sci. 2019, 132, jcs224360. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.T.; Chen, L.; Wang, H.J.; Tang, X.D.; Fang, D.C.; Yang, S.M. hTERT promotes the invasion of telomerase-negative tumor cells in vitro. Int. J. Oncol. 2009, 35, 329–336. [Google Scholar] [PubMed]
- Pal, J.; Gold, J.S.; Munshi, N.C.; Shammas, M.A. Biology of telomeres: Importance in etiology of esophageal cancer and as therapeutic target. Transl. Res. 2013, 162, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Kunimura, C.; Kikuchi, K.; Ahmed, N.; Shimizu, A.; Yasumoto, S. Telomerase activity in a specific cell subset co-expressing integrin beta1/EGFR but not p75NGFR/bcl2/integrin beta4 in normal human epithelial cells. Oncogene 1998, 17, 187–197. [Google Scholar] [CrossRef]
- Vay, C.; Hosch, S.B.; Stoecklein, N.H.; Klein, C.A.; Vallbohmer, D.; Link, B.-C.; Yekebas, E.F.; Izbicki, J.R.; Knoefel, W.T.; Scheunemann, P. Integrin Expression in Esophageal Squamous Cell Carcinoma: Loss of the Physiological Integrin Expression Pattern Correlates with Disease Progression. PLoS ONE 2014, 9, e109026. [Google Scholar] [CrossRef]
- Kai, F.; Laklai, H.; Weaver, V.M. Force Matters: Biomechanical Regulation of Cell Invasion and Migration in Disease. Trends Cell Biol. 2016, 26, 486–497. [Google Scholar] [CrossRef]
- Wolf, K.; Lindert, M.T.; Krause, M.; Alexander, S.; Riet, J.T.; Willis, A.L.; Hoffman, R.M.; Figdor, C.G.; Weiss, S.J.; Friedl, P. Physical limits of cell migration: Control by ECM space and nuclear deformation and tuning by proteolysis and traction force. J. Cell Biol. 2013, 201, 1069–1084. [Google Scholar] [CrossRef]
- Delcommenne, M.; Streuli, C.H. Control of Integrin Expression by Extracellular Matrix. J. Biol. Chem. 1995, 270, 26794–26801. [Google Scholar] [CrossRef]
- Nukuda, A.; Sasaki, C.; Ishihara, S.; Mizutani, T.; Nakamura, K.; Ayabe, T.; Kawabata, K.; Haga, H. Stiff substrates increase YAP-signaling-mediated matrix metalloproteinase-7 expression. Oncogenesis 2015, 4, e165. [Google Scholar] [CrossRef]
- McGrail, D.J.; Kieu, Q.M.N.; Iandoli, J.A.; Dawson, M.R. Actomyosin tension as a determinant of metastatic cancer mechanical tropism. Phys. Biol. 2015, 12, 026001. [Google Scholar] [CrossRef]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef]
- Lei, Z.; Jian, M.; Li, X.; Wei, J.; Meng, X.; Wang, Z. Biosensors and bioassays for determination of matrix metalloproteinases: State of the art and recent advances. J. Mater. Chem. B 2020. [Google Scholar] [CrossRef] [PubMed]
- Vihinen, P.; Kähäri, V.-M. Matrix metalloproteinases in cancer: Prognostic markers and therapeutic targets. Int. J. Cancer 2002, 99, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Groblewska, M.; Siewko, M.; Mroczko, B.; Szmitkowski, M. The role of matrix metalloproteinases (MMPs) and their inhibitors (TIMPs) in the development of esophageal cancer. Folia Histochem. Cytobiol. 2012, 50, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.-H.; Fu, L.; Chen, L.; Qin, Y.-R.; Liu, H.; Xie, F.; Zeng, T.; Dong, S.-S.; Li, J.; Li, Y.; et al. Downregulation of the Novel Tumor Suppressor DIRAS1 Predicts Poor Prognosis in Esophageal Squamous Cell Carcinoma. Cancer Res. 2013, 73, 2298–2309. [Google Scholar] [CrossRef]
- Zou, S.; Yang, J.; Guo, J.; Su, Y.; He, C.; Wu, J.; Yu, L.; Ding, W.-Q.; Zhou, J. RAD18 promotes the migration and invasion of esophageal squamous cell cancer via the JNK-MMPs pathway. Cancer Lett. 2018, 417, 65–74. [Google Scholar] [CrossRef]
- Zou, S.; Shang, Z.-F.; Liu, B.; Zhang, S.; Wu, J.; Huang, M.; Ding, W.-Q.; Zhou, J. DNA polymerase iota (Pol ι) promotes invasion and metastasis of esophageal squamous cell carcinoma. Oncotarget 2016, 7, 32274–32285. [Google Scholar] [CrossRef]
- Garalla, H.M.; Lertkowit, N.; Tiszlavicz, L.; Reisz, Z.; Holmberg, C.; Beynon, R.; Simpson, D.; Varga, Á.; Kumar, J.D.; Dodd, S.; et al. Matrix metalloproteinase (MMP)-7 in Barrett’s esophagus and esophageal adenocarcinoma: Expression, metabolism, and functional significance. Physiol. Rep. 2018, 6, e13683. [Google Scholar] [CrossRef]
- Adachi, Y.; Itoh, F.; Yamamoto, H.; Matsuno, K.; Arimura, Y.; Kusano, M.; Endoh, T.; Hinoda, Y.; Oohara, M.; Hosokawa, M.; et al. Matrix metalloproteinase matrilysin (MMP-7) participates in the progression of human gastric and esophageal cancers. Int. J. Oncol. 1998, 13, 1031–1036. [Google Scholar] [CrossRef]
- Wang, X.; Li, X.; Li, C.; He, C.; Ren, B.; Deng, Q.; Gao, W.; Wang, B. Aurora-A modulates MMP-2 expression via AKT/NF-κB pathway in esophageal squamous cell carcinoma cells. Acta Biochim. Biophys. Sin. 2016, 48, 520–527. [Google Scholar] [CrossRef]
- Yamashita, K.; Mori, M.; Shiraishi, T.; Shibuta, K.; Sugimachi, K. Clinical significance of matrix metalloproteinase-7 expression in esophageal carcinoma. Clin. Cancer Res. 2000, 6, 1169–1174. [Google Scholar] [PubMed]
- Yoshinaga, K.; Mimori, K.; Inoue, H.; Kamohara, Y.; Yamashita, K.; Tanaka, F.; Mori, M. Activin A enhances MMP-7 activity via the transcription factor AP-1 in an esophageal squamous cell carcinoma cell line. Int. J. Oncol. 2008, 33, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Qi, Y.; Li, X.; He, W.; Fan, Q.-X.; Zong, H. HDAC inhibitor trichostatin A suppresses esophageal squamous cell carcinoma metastasis through HADC2 reduced MMP-2/9. Clin. Investig. Med. 2013, 36, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, J.; Jia, Y.; Wang, Y.; Han, X.; Duan, Y.; Lv, W.; Ma, M.; Liu, L. Methylation decreases the Bin1 tumor suppressor in ESCC and restoration by decitabine inhibits the epithelial mesenchymal transition. Oncotarget 2017, 8, 19661–19673. [Google Scholar] [CrossRef]
- Beales, I.L.; Garcia-Morales, C.; Ogunwobi, O.O.; Mutungi, G. Adiponectin inhibits leptin-induced oncogenic signalling in oesophageal cancer cells by activation of PTP1B. Mol. Cell. Endocrinol. 2014, 382, 150–158. [Google Scholar] [CrossRef]
- Allott, E.H.; Lysaght, J.; Cathcart, M.C.; Donohoe, C.L.; Cummins, R.; McGarrigle, S.A.; Kay, E.; Reynolds, J.V.; Pidgeon, G.P. MMP9 expression in oesophageal adenocarcinoma is upregulated with visceral obesity and is associated with poor tumour differentiation. Mol. Carcinog. 2013, 52, 144–154. [Google Scholar] [CrossRef]
- Fan, F.; Jin, S.; Amundson, A.S.; Tong, T.; Fan, W.; Zhao, H.; Zhu, X.; Mazzacurati, L.; Li, X.; Petrik, K.L.; et al. ATF3 induction following DNA damage is regulated by distinct signaling pathways and over-expression of ATF3 protein suppresses cells growth. Oncogene 2002, 21, 7488–7496. [Google Scholar] [CrossRef]
- Xie, J.-J.; Xie, Y.-M.; Chen, B.; Pan, F.; Guo, J.-C.; Zhao, Q.; Shen, J.-H.; Wu, Z.-Y.; Wu, J.-Y.; Xu, L.-Y.; et al. ATF3 functions as a novel tumor suppressor with prognostic significance in esophageal squamous cell carcinoma. Oncotarget 2014, 5, 8569–8582. [Google Scholar] [CrossRef]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef]
- Da Costa, N.M.; Pinto, L.F.R.; Nasciutti, L.E.; Palumbo, A., Jr. The Prominent Role of HMGA Proteins in the Early Management of Gastrointestinal Cancers. BioMed Res. Int. 2019, 2019, 2059516. [Google Scholar]
- Lu, H.; Bhat, A.A.; Peng, D.; Chen, Z.; Zhu, S.; Hong, J.; Maacha, S.; Yan, J.; Robbins, D.J.; Washington, M.K.; et al. APE1 Upregulates MMP-14 via Redox-Sensitive ARF6-Mediated Recycling to Promote Cell Invasion of Esophageal Adenocarcinoma. Cancer Res. 2019, 79, 4426–4438. [Google Scholar] [CrossRef]
- Da Costa, N.M.; Lima, S.C.S.; Simão, T.D.A.; Pinto, L.F.R. The potential of molecular markers to improve interventions through the natural history of oesophageal squamous cell carcinoma. Biosci. Rep. 2013, 33, 627–636. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; He, W.; Fanghui, P.; Wang, L.; Fan, Q. NF-kBP65 promotes invasion and metastasis of oesophageal squamous cell cancer by regulating matrix metalloproteinase-9 and epithelial-to-mesenchymal transition. Cell Biol. Int. 2013, 37, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Shin, W.-S.; Hong, Y.; Lee, H.W.; Lee, S.-T. Catalytically defective receptor protein tyrosine kinase PTK7 enhances invasive phenotype by inducing MMP-9 through activation of AP-1 and NF-κB in esophageal squamous cell carcinoma cells. Oncotarget 2016, 7, 73242–73256. [Google Scholar] [CrossRef]
- Liu, N.; Zhang, R.; Wu, J.; Pu, Y.; Yin, X.; Cheng, Y.; Wu, J.; Feng, C.; Luo, Y.; Zhang, J. Interleukin-17A promotes esophageal adenocarcinoma cell invasiveness through ROS-dependent, NF-κB-mediated MMP-2/9 activation. Oncol. Rep. 2017, 37, 1779–1785. [Google Scholar] [CrossRef]
- Clemons, N.J.; Shannon, N.B.; Abeyratne, L.R.; Walker, C.E.; Saadi, A.; O’Donovan, M.L.; Lao-Sirieix, P.P.; Fitzgerald, R.C. Nitric oxide-mediated invasion in Barrett’s high-grade dysplasia and adenocarcinoma. Carcinogenesis 2010, 31, 1669–1675. [Google Scholar] [CrossRef]
- Murray, G.I.; Duncan, M.E.; O’Neil, P.; McKay, J.A.; Melvin, W.T.; Fothergill, J.E. Matrix metalloproteinase-1 is associated with poor prognosis in oesophageal cancer. J. Pathol. 1998, 185, 256–261. [Google Scholar] [CrossRef]
- Yamashita, K.; Mori, M.; Kataoka, A.; Inoue, H.; Sugimachi, K. The clinical significance of MMP-1 expression in oesophageal carcinoma. Br. J. Cancer 2001, 84, 276–282. [Google Scholar] [CrossRef][Green Version]
- Cheung, W.Y.; Zhai, R.; Bradbury, P.; Hopkins, J.; Kulke, M.H.; Heist, R.S.; Asomaning, K.; Ma, C.; Xu, W.; Wang, Z.; et al. Single nucleotide polymorphisms in the matrix metalloproteinase gene family and the frequency and duration of gastroesophageal reflux disease influence the risk of esophageal adenocarcinoma. Int. J. Cancer 2012, 131, 2478–2486. [Google Scholar] [CrossRef]
- Bradbury, P.A.; Zhai, R.; Hopkins, J.; Kulke, M.H.; Heist, R.S.; Singh, S.; Zhou, W.; Ma, C.; Xu, W.; Asomaning, K.; et al. Matrix metalloproteinase 1, 3 and 12 polymorphisms and esophageal adenocarcinoma risk and prognosis. Carcinogenesis 2009, 30, 793–798. [Google Scholar] [CrossRef][Green Version]
- Peng, B.; Cao, L.; Ma, X.; Wang, W.; Wang, D.; Yu, L. Meta-analysis of association between matrix metalloproteinases 2,7 and 9 promoter polymorphisms and cancer risk. Mutagenesis 2010, 25, 371–379. [Google Scholar] [CrossRef]
- Petty, R.D.; Dahle-Smith, A.; Stevenson, D.A.; Osborne, A.; Massie, D.; Clark, C.; Murray, G.I.; Dutton, S.J.; Roberts, C.; Chong, I.Y.; et al. Gefitinib and EGFR Gene Copy Number Aberrations in Esophageal Cancer. J. Clin. Oncol. 2017, 35, 2279–2287. [Google Scholar] [CrossRef] [PubMed]
- Shima, I.; Sasaguri, Y.; Arima, N.; Yamana, H.; Fujita, H.; Morimatsu, M.; Nagase, H. Expression of epidermal growth-factor (EGF), matrix metalloproteinase-9 (MMP-9) and proliferating cell nuclear antigen (PCNA) in esophageal cancer. Int. J. Oncol. 1995, 6, 833–839. [Google Scholar] [CrossRef] [PubMed]
- Shima, I.; Sasaguri, Y.; Kusukawa, J.; Nakano, R.; Yamana, H.; Fujita, H.; Kakegawa, T.; Morimatsu, M. Production of matrix metalloproteinase 9 (92-kDa gelatinase) by human oesophageal squamous cell carcinoma in response to epidermal growth factor. Br. J. Cancer 1993, 67, 721–727. [Google Scholar] [CrossRef]
- Okawa, T.; Michaylira, C.Z.; Kalabis, J.; Stairs, U.B.; Nakagawa, H.; Andl, C.; Johnstone, C.N.; Klein-Szanto, A.J.; El-Deiry, W.S.; Cukierman, E.; et al. The functional interplay between EGFR overexpression, hTERT activation, and p53 mutation in esophageal epithelial cells with activation of stromal fibroblasts induces tumor development, invasion, and differentiation. Genome Res. 2007, 21, 2788–2803. [Google Scholar] [CrossRef]
- Ellerbroek, S.M.; Halbleib, J.M.; Benavidez, M.; Warmka, J.K.; Wattenberg, E.V.; Stack, M.S.; Hudson, L.G. Phosphatidylinositol 3-kinase activity in epidermal growth factor-stimulated matrix metalloproteinase-9 production and cell surface association. Cancer Res. 2001, 61, 1855–1861. [Google Scholar]
- Maziveyi, M.; Alahari, S.K. Cell matrix adhesions in cancer: The proteins that form the glue. Oncotarget 2017, 8, 48471–48487. [Google Scholar] [CrossRef]
- Shams, H.; Hoffman, B.D.; Mofrad, M.R.K. The “Stressful” Life of Cell Adhesion Molecules: On the Mechanosensitivity of Integrin Adhesome. J. Biomech. Eng. 2018, 140, 020807. [Google Scholar] [CrossRef]
- Canel, M.; Serrels, A.; Frame, M.C.; Brunton, V.G.; Frisch, S.M.; Schaller, M.; Cieply, B. E-cadherin-integrin crosstalk in cancer invasion and metastasis. J. Cell Sci. 2013, 126, 393–401. [Google Scholar] [CrossRef]
- Liu, B.; Li, X.; Li, C.; Xu, R.; Sun, X. miR-25 mediates metastasis and epithelial–mesenchymal-transition in human esophageal squamous cell carcinoma via regulation of E-cadherin signaling. Bioengineered 2019, 10, 679–688. [Google Scholar] [CrossRef]
- Xu, X.L.; Ling, Z.Q.; Chen, S.Z.; Li, B.; Ji, W.H.; Mao, W.M. The impact of E-cadherin expression on the prognosis of esophageal cancer: A meta-analysis. Dis Esophagus 2014, 27, 79–86. [Google Scholar] [CrossRef]
- Lin, Y.; Shen, L.Y.; Fu, H.; Dong, B.; Yang, H.L.; Yan, W.P.; Kang, X.Z.; Dai, L.; Zhou, H.T.; Yang, Y.B.; et al. P21, COX-2, and E-cadherin are potential prognostic factors for esophageal squamous cell carcinoma. Dis. Esophagus 2017, 30, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Kang, Y.S.; Kim, J.S.; Shin, N.Y.; Hanks, S.K.; Song, W.K. The integrin-coupled signaling adaptor p130Cas suppresses Smad3 function in transforming growth factor-beta signaling. Mol. Biol. Cell 2008, 19, 2135–2146. [Google Scholar] [CrossRef]
- Fu, L.; Qin, Y.R.; Xie, D.; Chow, H.Y.; Ngai, S.M.; Kwong, R.L.W.; Li, Y.; Guan, X.Y. Identification of alpha-actinin 4 and 67 kDa laminin receptor as stage-specific markers in esophageal cancer via proteomic approaches. Cancer 2007, 110, 2672–2681. [Google Scholar] [CrossRef]
- Zhang, J.; Zhi, H.; Zhou, C.; Ding, F.; Luo, A.; Zhang, X.; Sun, Y.; Wang, X.; Wu, M.; Liu, Z. Up-regulation of fibronectin in oesophageal squamous cell carcinoma is associated with activation of the Erk pathway. J. Pathol. 2005, 207, 402–409. [Google Scholar] [CrossRef] [PubMed]
- Durbeej, M. Laminins. Cell Tissue Res. 2010, 339, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Takubo, K.; Tanaka, Y.; Miyama, T.; Fujita, K.; Mafune, K.-I. Basaloid-Squamous Carcinoma of the Esophagus with Marked Deposition of Basement Membrane Substance. Pathol. Int. 1991, 41, 59–64. [Google Scholar] [CrossRef]
- Mori, M.; Shimono, R.; Kido, A.; Kuwano, H.; Akazawa, K.; Sugimachi, K. Distribution of basement membrane antigens in human esophageal lesions: An immunohistochemical study. Int. J. Cancer 1991, 47, 839–842. [Google Scholar] [CrossRef]
- Baba, K.; Kuwano, H.; Kitamura, K.; Sugimachi, K. Carcinomatous invasion and lymphocyte infiltration in early esophageal carcinoma with special regard to the basement membrane. An immunohistochemical study. Hepatogastroenterology 1993, 40, 226–231. [Google Scholar]
- Yamamoto, H.; Itoh, F.; Iku, S.; Hosokawa, M.; Imai, K. Expression of the gamma(2) chain of laminin-5 at the invasive front is associated with recurrence and poor prognosis in human esophageal squamous cell carcinoma. Clin. Cancer Res. 2001, 7, 896–900. [Google Scholar]
- Fukai, Y.; Masuda, N.; Kato, H.; Fukuchi, M.; Miyazaki, T.; Nakajima, M.; Sohda, M.; Kuwano, H.; Nakajima, T. Correlation between laminin-5 gamma2 chain and epidermal growth factor receptor expression in esophageal squamous cell carcinomas. Oncology 2005, 69, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, H.; Miyagi, Y.; Kikkawa, Y.; Yamanaka, N.; Yasumitsu, H.; Misugi, K.; Miyazaki, K. Differential Expression of Laminin-5/Ladsin Subunits in Human Tissues and Cancer Cell Lines and Their Induction by Tumor Promoter and Growth Factors. J. Biochem. 1996, 120, 1196–1202. [Google Scholar] [CrossRef] [PubMed]
- Schenk, S.; Hintermann, E.; Bilban, M.; Koshikawa, N.; Hojilla, C.; Khokha, R.; Quaranta, V. Binding to EGF receptor of a laminin-5 EGF-like fragment liberated during MMP-dependent mammary gland involution. J. Cell Biol. 2003, 161, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.M.; Wu, Y.P.; Feng, Y.B.; Luo, M.L.; Du, X.L.; Zhang, Y.; Cai, Y.; Xu, X.; Han, Y.L.; Zhang, X.; et al. Interaction of MT1-MMP and laminin-5gamma2 chain correlates with metastasis and invasiveness in human esophageal squamous cell carcinoma. Clin. Exp. Metastasis 2007, 24, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Kita, Y.; Mimori, K.; Tanaka, F.; Matsumoto, T.; Haraguchi, N.; Ishikawa, K.; Matsuzaki, S.; Fukuyoshi, Y.; Inoue, H.; Natsugoe, S.; et al. Clinical significance of LAMB3 and COL7A1 mRNA in esophageal squamous cell carcinoma. Eur. J. Surg. Oncol. EJSO 2009, 35, 52–58. [Google Scholar] [CrossRef]
- Baba, Y.; Iyama, K.-I.; Hirashima, K.; Nagai, Y.; Yoshida, N.; Hayashi, N.; Miyanari, N.; Baba, H. Laminin-332 promotes the invasion of oesophageal squamous cell carcinoma via PI3K activation. Br. J. Cancer 2008, 98, 974–980. [Google Scholar] [CrossRef][Green Version]
- Aghcheli, K.; Parsian, H.; Qujeq, D.; Talebi, M.; Mosapour, A.; Khalilipour, E.; Islami, F.; Semnani, S.; Malekzadeh, R. Serum hyaluronic acid and laminin as potential tumor markers for upper gastrointestinal cancers. Eur. J. Intern. Med. 2012, 23, 58–64. [Google Scholar] [CrossRef]
- Lin, J.; Myers, A.L.; Wang, Z.; Nancarrow, D.J.; Ferrer-Torres, D.; Handlogten, A.; Leverenz, K.; Bao, J.; Thomas, D.G.; Wang, T.D.; et al. Osteopontin (OPN/SPP1) isoforms collectively enhance tumor cell invasion and dissemination in esophageal adenocarcinoma. Oncotarget 2015, 6, 22239–22257. [Google Scholar] [CrossRef]
- Meng, X.; Chen, X.; Lu, P.; Ma, W.; Yue, D.; Song, L.; Fan, Q. MicroRNA-202 inhibits tumor progression by targeting LAMA1 in esophageal squamous cell carcinoma. Biochem. Biophys. Res. Commun. 2016, 473, 821–827. [Google Scholar] [CrossRef]
- Yang, Y.; Li, D.; Yang, Y.; Jiang, G. An integrated analysis of the effects of microRNA and mRNA on esophageal squamous cell carcinoma. Mol. Med. Rep. 2015, 12, 945–952. [Google Scholar] [CrossRef]
- Ma, G.; Zhang, F.; Dong, X.; Wang, X.; Ren, Y. Low expression of microRNA-202 is associated with the metastasis of esophageal squamous cell carcinoma. Exp. Ther. Med. 2016, 11, 951–956. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sudo, T.; Iwaya, T.; Nishida, N.; Sawada, G.; Takahashi, Y.; Ishibashi, M.; Shibata, K.; Fujita, H.; Shirouzu, K.; Mori, M.; et al. Expression of mesenchymal markers vimentin and fibronectin: The clinical significance in esophageal squamous cell carcinoma. Ann. Surg. Oncol. 2013, 20, S324–S335. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Yang, W.; Xu, B.; Zhu, H.; Zou, J.; Su, C.; Rong, J.; Wang, T.; Chen, Z. Expression of fibronectin in esophageal squamous cell carcinoma and its role in migration. BMC Cancer 2018, 18, 976. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, M.C.; Hsu, R.Y.; Spicer, J.D.; McDonald, B.; Chan, C.H.; Perera, R.M.; Giannias, B.; Chow, S.C.; Rousseau, S.; Law, S.; et al. Lipopolysaccharide-induced toll-like receptor 4 signaling enhances the migratory ability of human esophageal cancer cells in a selectin-dependent manner. Surgery 2013, 154, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Poehlmann, A.; Kuester, D.; Malfertheiner, P.; Guenther, T.; Roessner, A. Inflammation and Barrett’s carcinogenesis. Pathol. Res. Pract. 2012, 208, 269–280. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Ai, B.; Tian, L. Identification of genes and pathways in esophageal adenocarcinoma using bioinformatics analysis. Biomed. Rep. 2018, 9, 305–312. [Google Scholar] [CrossRef]
- Leppänen, J.; Bogdanoff, S.; Lehenkari, P.P.; Saarnio, J.; Kauppila, J.H.; Karttunen, T.J.; Huhta, H.; Helminen, O. Tenascin-C and fibronectin in normal esophageal mucosa, Barrett’s esophagus, dysplasia and adenocarcinoma. Oncotarget 2017, 8, 66865–66877. [Google Scholar] [CrossRef][Green Version]
- Kuo, I.-Y.; Wu, C.-C.; Chang, J.-M.; Huang, Y.-L.; Lin, C.-H.; Yan, J.-J.; Sheu, B.-S.; Lu, P.-J.; Chang, W.-L.; Lai, W.-W.; et al. Low SOX17 expression is a prognostic factor and drives transcriptional dysregulation and esophageal cancer progression. Int. J. Cancer 2014, 135, 563–573. [Google Scholar] [CrossRef]
- Wei, Q.; Li, X.; Yu, W.; Zhao, K.; Qin, G.; Chen, H.; Gu, Y.; Ding, F.; Zhu, Z.; Fu, X.; et al. microRNA-messenger RNA regulatory network of esophageal squamous cell carcinoma and the identification of miR-1 as a biomarker of patient survival. J. Cell. Biochem. 2019, 120, 12259–12272. [Google Scholar] [CrossRef]
- Ma, J.; Xiao, Y.; Tian, B.; Chen, S.; Zhang, B.; Wu, J.; Wu, Z.; Li, X.; Tang, J.; Yang, D.; et al. Long noncoding RNA lnc-ABCA12-3 promotes cell migration, invasion, and proliferation by regulating fibronectin 1 in esophageal squamous cell carcinoma. J. Cell. Biochem. 2019, 121, 1374–1387. [Google Scholar] [CrossRef]
- Zhang, C.-Y.; Li, R.-K.; Qi, Y.; Yang, Y.; Liu, D.-L.; Zhao, J.; Zhu, D.-Y.; Wu, K.; Zhou, X.-D. Upregulation of long noncoding RNA SPRY4-IT1 promotes metastasis of esophageal squamous cell carcinoma via induction of epithelial–mesenchymal transition. Cell Biol. Toxicol. 2016, 32, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Qiao, B.; Liu, Q.; Zhang, W. Upregulation of extracellular matrix metalloproteinase inducer promotes hypoxia-induced epithelial-mesenchymal transition in esophageal cancer. Mol. Med. Rep. 2015, 12, 7419–7424. [Google Scholar] [CrossRef] [PubMed]
- Broll, R.; Meyer, S.; Neuber, M.; Bruch, H.P. Expression of tenascin in tumors of the esophagus, small intestine and colorectum. An immunohistochemical study. Gen. Diagn. Pathol. 1995, 141, 111–119. [Google Scholar]
- Salmela, M.T.; Karjalainen-Lindsberg, M.L.; Puolakkainen, P.; Saarialho-Kere, U. Upregulation and differential expression of matrilysin (MMP-7) and metalloelastase (MMP-12) and their inhibitors TIMP-1 and TIMP-3 in Barrett’s oesophageal adenocarcinoma. Br. J. Cancer 2001, 85, 383–392. [Google Scholar] [CrossRef]
- Ohtsuka, M.; Yamamoto, H.; Oshiro, R.; Takahashi, H.; Masuzawa, T.; Uemura, M.; Haraguchi, N.; Nishimura, J.; Hata, T.; Yamasaki, M.; et al. Concurrent expression of C4.4A and Tenascin-C in tumor cells relates to poor prognosis of esophageal squamous cell carcinoma. Int. J. Oncol. 2013, 43, 439–446. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yang, Z.T.; Yeo, S.Y.; Yin, Y.X.; Lin, Z.H.; Lee, H.M.; Xuan, Y.H.; Cui, Y.; Kim, S.H. Tenascin-C, a prognostic determinant of esophageal squamous cell carcinoma. PLoS ONE 2016, 11, e0145807. [Google Scholar] [CrossRef]
- Yang, Z.; Zhang, C.; Feng, Y.; Qi, W.; Cui, Y.; Xuan, Y. Tenascin-C is involved in promotion of cancer stemness via the Akt/HIF1α axis in esophageal squamous cell carcinoma. Exp. Mol. Pathol. 2019, 109, 104239. [Google Scholar] [CrossRef]
- Fortuna-Costa, A.; Gomes, A.M.; Kozlowski, E.O.; Stelling, M.P.; Pavão, M.S.G. Extracellular Galectin-3 in Tumor Progression and Metastasis. Front. Oncol. 2014, 4, 138. [Google Scholar] [CrossRef]
- Cardoso, A.C.F.; Andrade, L.N.D.S.; Bustos, S.O.; Chammas, R. Galectin-3 Determines Tumor Cell Adaptive Strategies in Stressed Tumor Microenvironments. Front. Oncol. 2016, 6, 740. [Google Scholar] [CrossRef]
- Kashyap, M.K.; Harsha, H.C.; Renuse, S.; Pawar, H.; Sahasrabuddhe, N.A.; Kim, M.-S.; Marimuthu, A.; Keerthikumar, S.; Muthusamy, B.; Kandasamy, K.; et al. SILAC-based quantitative proteomic approach to identify potential biomarkers from the esophageal squamous cell carcinoma secretome. Cancer Biol. Ther. 2010, 10, 796–810. [Google Scholar] [CrossRef]
- Çobanoğlu, U.; Mergan, D.; Dulger, A.C.; Celik, S.; Kemik, O.; Sayir, F. Are Serum Mac 2-Binding Protein Levels Elevated in Esophageal Cancer? A Control Study of Esophageal Squamous Cell Carcinoma Patients. Dis. Markers 2018, 2018, 3610239. [Google Scholar] [CrossRef] [PubMed]
- Qiao, L.; Liang, N.; Xie, J.; Luo, H.; Zhang, J.; Deng, G.; Li, Y.; Zhang, J. Gene silencing of galectin-3 changes the biological behavior of Eca109 human esophageal cancer cells. Mol. Med. Rep. 2016, 13, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Deng, G.; Qiao, L.; Luo, H.; Liu, O.; Liang, N.; Xie, J.; Zhang, J. Effect of galectin-3 on vasculogenic mimicry in esophageal cancer cells. Oncol. Lett. 2018, 15, 4907–4911. [Google Scholar] [CrossRef] [PubMed]
- Cui, G.; Cui, M.; Li, Y.; Liang, Y.; Li, W.; Guo, H.; Zhao, S. Galectin-3 knockdown increases gefitinib sensitivity to the inhibition of EGFR endocytosis in gefitinib-insensitive esophageal squamous cancer cells. Med. Oncol. 2015, 32, 124. [Google Scholar] [CrossRef]
- Nagahara, K.; Arikawa, T.; Oomizu, S.; Kontani, K.; Nobumoto, A.; Tateno, H.; Watanabe, K.; Niki, T.; Katoh, S.; Miyake, M.; et al. Galectin-9 increases Tim-3+ dendritic cells and CD8+ T cells and enhances antitumor immunity via galectin-9-Tim-3 interactions. J. Immunol. 2008, 181, 7660–7669. [Google Scholar] [CrossRef]
- Chiyo, T.; Fujita, K.; Iwama, H.; Fujihara, S.; Tadokoro, T.; Ohura, K.; Matsui, T.; Goda, Y.; Kobayashi, N.; Nishiyama, N.; et al. Galectin-9 Induces Mitochondria-Mediated Apoptosis of Esophageal Cancer In Vitro and In Vivo in a Xenograft Mouse Model. Int. J. Mol. Sci. 2019, 20, 2634. [Google Scholar] [CrossRef]
- Akashi, E.; Fujihara, S.; Morishita, A.; Tadokoro, T.; Chiyo, T.; Fujikawa, K.; Kobara, H.; Mori, H.; Iwama, H.; Okano, K.; et al. Effects of galectin-9 on apoptosis, cell cycle and autophagy in human esophageal adenocarcinoma cells. Oncol. Rep. 2017, 38, 506–514. [Google Scholar] [CrossRef]
- Zhu, X.; Ding, M.; Yu, M.-L.; Feng, M.-X.; Tan, L.-J.; Zhao, F.-K. Identification of galectin-7 as a potential biomarker for esophageal squamous cell carcinoma by proteomic analysis. BMC Cancer 2010, 10, 290. [Google Scholar] [CrossRef]
- Twarock, S.; Freudenberger, T.; Poscher, E.; Dai, G.; Jannasch, K.; Dullin, C.; Alves, F.; Prenzel, K.; Knoefel, W.T.; Stoecklein, N.H.; et al. Inhibition of Oesophageal Squamous Cell Carcinoma Progression by in vivo Targeting of Hyaluronan Synthesis. Mol. Cancer 2011, 10, 30. [Google Scholar] [CrossRef]
- Thelin, M.A.; Svensson, K.J.; Shi, X.; Bagher, M.; Axelsson, J.; Isinger-Ekstrand, A.; Van Kuppevelt, T.H.; Johansson, J.; Nilbert, M.; Zaia, J.; et al. Dermatan sulfate is involved in the tumorigenic properties of esophagus squamous cell carcinoma. Cancer Res. 2012, 72, 1943–1952. [Google Scholar] [CrossRef]
- Twarock, S.; Tammi, M.I.; Savani, R.C.; Fischer, J.W. Hyaluronan Stabilizes Focal Adhesions, Filopodia, and the Proliferative Phenotype in Esophageal Squamous Carcinoma Cells. J. Biol. Chem. 2010, 285, 23276–23284. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palumbo, A., Jr.; Meireles Da Costa, N.; Pontes, B.; Leite de Oliveira, F.; Lohan Codeço, M.; Ribeiro Pinto, L.F.; Nasciutti, L.E. Esophageal Cancer Development: Crucial Clues Arising from the Extracellular Matrix. Cells 2020, 9, 455. https://doi.org/10.3390/cells9020455
Palumbo A Jr., Meireles Da Costa N, Pontes B, Leite de Oliveira F, Lohan Codeço M, Ribeiro Pinto LF, Nasciutti LE. Esophageal Cancer Development: Crucial Clues Arising from the Extracellular Matrix. Cells. 2020; 9(2):455. https://doi.org/10.3390/cells9020455
Chicago/Turabian StylePalumbo, Antonio, Jr., Nathalia Meireles Da Costa, Bruno Pontes, Felipe Leite de Oliveira, Matheus Lohan Codeço, Luis Felipe Ribeiro Pinto, and Luiz Eurico Nasciutti. 2020. "Esophageal Cancer Development: Crucial Clues Arising from the Extracellular Matrix" Cells 9, no. 2: 455. https://doi.org/10.3390/cells9020455
APA StylePalumbo, A., Jr., Meireles Da Costa, N., Pontes, B., Leite de Oliveira, F., Lohan Codeço, M., Ribeiro Pinto, L. F., & Nasciutti, L. E. (2020). Esophageal Cancer Development: Crucial Clues Arising from the Extracellular Matrix. Cells, 9(2), 455. https://doi.org/10.3390/cells9020455