Human Plasmacytoid Dendritic Cells and Cutaneous Melanoma

 ,
,
Abstract
1. Introduction
2. Human Plasmacytoid Dendritic Cells: Biology and Functions
2.1. Development, Phenotype and Trafficking of Plasmacytoid Dendritic Cells
2.2. The Type I and Type III Interferons Production by Plasmacytoid Dendritic Cells
2.3. The Multifaceted Function of Plasmacytoid Dendritic Cells: Not Only Interferon Producing Cells
3. Plasmacytoid Dendritic Cells and Cancer
3.1. Murine Pre-Clinical Models
3.2. Clinical Significance of the pDCs Compartment in Human Cancer Patients
3.3. The Dual Role of pDCs in Human Cancer
4. Human Cutaneous Melanoma: From Immunogenicity to Therapy
4.1. Melanoma as a Model of Hypermutated Hot Tumor
4.2. Novel Systemic Treatments Partially Restrain Melanoma Dissemination
4.3. The Immuno-Mediated Component of the Clinical Response to Systemic Treatments
5. Plasmacytoid Dendritic Cells and Melanoma
5.1. Clinical Relevance of Melanoma-Associated pDCs
5.2. pDC Function in Human Cutaneous Melanoma
5.3. Therapeutic Implications of Human pDCs in Melanoma
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Li, S.; Wu, J.; Zhu, S.; Liu, Y.J.; Chen, J. Disease-Associated Plasmacytoid Dendritic Cells. Front. Immunol. 2017, 8, 1268. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Koebel, C.M.; Schreiber, R.D. Interferons, immunity and cancer immunoediting. Nat. Rev. Immunol. 2006, 6, 836–848. [Google Scholar] [CrossRef] [PubMed]
- Swiecki, M.; Colonna, M. Unraveling the functions of plasmacytoid dendritic cells during viral infections, autoimmunity, and tolerance. Immunol. Rev. 2010, 234, 142–162. [Google Scholar] [CrossRef] [PubMed]
- Swiecki, M.; Colonna, M. The multifaceted biology of plasmacytoid dendritic cells. Nat. Rev. Immunol. 2015, 15, 471–485. [Google Scholar] [CrossRef] [PubMed]
- Karrich, J.J.; Jachimowski, L.C.; Uittenbogaart, C.H.; Blom, B. The plasmacytoid dendritic cell as the Swiss army knife of the immune system: Molecular regulation of its multifaceted functions. J. Immunol. 2014, 193, 5772–5778. [Google Scholar] [CrossRef]
- Bao, M.; Liu, Y.J. Regulation of TLR7/9 signaling in plasmacytoid dendritic cells. Protein Cell 2013, 4, 40–52. [Google Scholar] [CrossRef]
- Villadangos, J.A.; Young, L. Antigen-presentation properties of plasmacytoid dendritic cells. Immunity 2008, 29, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Musumeci, A.; Lutz, K.; Winheim, E.; Krug, A.B. What Makes a pDC: Recent Advances in Understanding Plasmacytoid DC Development and Heterogeneity. Front. Immunol. 2019, 10, 1222. [Google Scholar] [CrossRef]
- Sozzani, S.; Vermi, W.; Del Prete, A.; Facchetti, F. Trafficking properties of plasmacytoid dendritic cells in health and disease. Trends Immunol. 2010, 31, 270–277. [Google Scholar] [CrossRef]
- Vermi, W.; Soncini, M.; Melocchi, L.; Sozzani, S.; Facchetti, F. Plasmacytoid dendritic cells and cancer. J. Leukoc. Biol. 2011, 90, 681–690. [Google Scholar] [CrossRef]
- Koucky, V.; Boucek, J.; Fialova, A. Immunology of Plasmacytoid Dendritic Cells in Solid Tumors: A Brief Review. Cancers (Basel) 2019, 11, 470. [Google Scholar] [CrossRef] [PubMed]
- Lennert, K.; Remmele, W. Karyometric research on lymph node cells in man. I. Germinoblasts, lymphoblasts & lymphocytes. Acta Haematol. 1958, 19, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Facchetti, F.; de Wolf-Peeters, C.; Mason, D.Y.; Pulford, K.; van den Oord, J.J.; Desmet, V.J. Plasmacytoid T cells. Immunohistochemical evidence for their monocyte/macrophage origin. Am. J. Pathol. 1988, 133, 15–21. [Google Scholar]
- Facchetti, F.; Vermi, W.; Mason, D.; Colonna, M. The plasmacytoid monocyte/interferon producing cells. Virchows Arch. 2003, 443, 703–717. [Google Scholar] [CrossRef] [PubMed]
- Vermi, W.; Bonecchi, R.; Facchetti, F.; Bianchi, D.; Sozzani, S.; Festa, S.; Berenzi, A.; Cella, M.; Colonna, M. Recruitment of immature plasmacytoid dendritic cells (plasmacytoid monocytes) and myeloid dendritic cells in primary cutaneous melanomas. J. Pathol. 2003, 200, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Siegal, F.P.; Kadowaki, N.; Shodell, M.; Fitzgerald-Bocarsly, P.A.; Shah, K.; Ho, S.; Antonenko, S.; Liu, Y.J. The nature of the principal type 1 interferon-producing cells in human blood. Science 1999, 284, 1835–1837. [Google Scholar] [CrossRef] [PubMed]
- Cella, M.; Jarrossay, D.; Facchetti, F.; Alebardi, O.; Nakajima, H.; Lanzavecchia, A.; Colonna, M. Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amounts of type I interferon. Nat. Med. 1999, 5, 919–923. [Google Scholar] [CrossRef]
- Dzionek, A.; Inagaki, Y.; Okawa, K.; Nagafune, J.; Rock, J.; Sohma, Y.; Winkels, G.; Zysk, M.; Yamaguchi, Y.; Schmitz, J. Plasmacytoid dendritic cells: From specific surface markers to specific cellular functions. Hum. Immunol. 2002, 63, 1133–1148. [Google Scholar] [CrossRef]
- Facchetti, F.; Candiago, E.; Vermi, W. Plasmacytoid monocytes express IL3-receptor alpha and differentiate into dendritic cells. Histopathology 1999, 35, 88–89. [Google Scholar] [CrossRef]
- Demoulin, S.; Roncarati, P.; Delvenne, P.; Hubert, P. Production of large numbers of plasmacytoid dendritic cells with functional activities from CD34(+) hematopoietic progenitor cells: Use of interleukin-3. Exp. Hematol. 2012, 40, 268–278. [Google Scholar] [CrossRef]
- Dzionek, A.; Fuchs, A.; Schmidt, P.; Cremer, S.; Zysk, M.; Miltenyi, S.; Buck, D.W.; Schmitz, J. BDCA-2, BDCA-3, and BDCA-4: Three markers for distinct subsets of dendritic cells in human peripheral blood. J. Immunol. 2000, 165, 6037–6046. [Google Scholar] [CrossRef] [PubMed]
- Matsui, T.; Connolly, J.E.; Michnevitz, M.; Chaussabel, D.; Yu, C.I.; Glaser, C.; Tindle, S.; Pypaert, M.; Freitas, H.; Piqueras, B.; et al. CD2 distinguishes two subsets of human plasmacytoid dendritic cells with distinct phenotype and functions. J. Immunol. 2009, 182, 6815–6823. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Gregorio, J.D.; Iwahori, T.; Zhang, X.; Choi, O.; Tolentino, L.L.; Prestwood, T.; Carmi, Y.; Engleman, E.G. A distinct subset of plasmacytoid dendritic cells induces activation and differentiation of B and T lymphocytes. Proc. Natl. Acad. Sci. USA 2017, 114, 1988–1993. [Google Scholar] [CrossRef] [PubMed]
- Alculumbre, S.G.; Saint-Andre, V.; Di Domizio, J.; Vargas, P.; Sirven, P.; Bost, P.; Maurin, M.; Maiuri, P.; Wery, M.; Roman, M.S.; et al. Diversification of human plasmacytoid predendritic cells in response to a single stimulus. Nat. Immunol. 2018, 19, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Wimmers, F.; Subedi, N.; van Buuringen, N.; Heister, D.; Vivie, J.; Beeren-Reinieren, I.; Woestenenk, R.; Dolstra, H.; Piruska, A.; Jacobs, J.F.M.; et al. Single-cell analysis reveals that stochasticity and paracrine signaling control interferon-alpha production by plasmacytoid dendritic cells. Nat. Commun. 2018, 9, 3317. [Google Scholar] [CrossRef]
- Marsman, C.; Lafouresse, F.; Liao, Y.; Baldwin, T.M.; Mielke, L.A.; Hu, Y.; Mack, M.; Hertzog, P.J.; de Graaf, C.A.; Shi, W.; et al. Plasmacytoid dendritic cell heterogeneity is defined by CXCL10 expression following TLR7 stimulation. Immunol. Cell Biol. 2018, 96, 1083–1094. [Google Scholar] [CrossRef]
- Maraskovsky, E.; Daro, E.; Roux, E.; Teepe, M.; Maliszewski, C.R.; Hoek, J.; Caron, D.; Lebsack, M.E.; McKenna, H.J. In vivo generation of human dendritic cell subsets by Flt3 ligand. Blood 2000, 96, 878–884. [Google Scholar] [CrossRef]
- Karsunky, H.; Merad, M.; Cozzio, A.; Weissman, I.L.; Manz, M.G. Flt3 ligand regulates dendritic cell development from Flt3+ lymphoid and myeloid-committed progenitors to Flt3+ dendritic cells in vivo. J. Exp. Med. 2003, 198, 305–313. [Google Scholar] [CrossRef]
- D’Amico, A.; Wu, L. The early progenitors of mouse dendritic cells and plasmacytoid predendritic cells are within the bone marrow hemopoietic precursors expressing Flt3. J. Exp. Med. 2003, 198, 293–303. [Google Scholar] [CrossRef]
- Lee, J.; Breton, G.; Oliveira, T.Y.; Zhou, Y.J.; Aljoufi, A.; Puhr, S.; Cameron, M.J.; Sekaly, R.P.; Nussenzweig, M.C.; Liu, K. Restricted dendritic cell and monocyte progenitors in human cord blood and bone marrow. J. Exp. Med. 2015, 212, 385–399. [Google Scholar] [CrossRef]
- Helft, J.; Anjos-Afonso, F.; van der Veen, A.G.; Chakravarty, P.; Bonnet, D.; Reis e Sousa, C. Dendritic Cell Lineage Potential in Human Early Hematopoietic Progenitors. Cell Rep. 2017, 20, 529–537. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.X.; Lian, Z.X.; Kikuchi, K.; Moritoki, Y.; Ansari, A.A.; Liu, Y.J.; Ikehara, S.; Gershwin, M.E. Plasmacytoid dendritic cells of different origins have distinct characteristics and function: Studies of lymphoid progenitors versus myeloid progenitors. J. Immunol. 2005, 175, 7281–7287. [Google Scholar] [CrossRef] [PubMed]
- Cisse, B.; Caton, M.L.; Lehner, M.; Maeda, T.; Scheu, S.; Locksley, R.; Holmberg, D.; Zweier, C.; den Hollander, N.S.; Kant, S.G.; et al. Transcription factor E2-2 is an essential and specific regulator of plasmacytoid dendritic cell development. Cell 2008, 135, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Carotta, S.; Dakic, A.; D’Amico, A.; Pang, S.H.; Greig, K.T.; Nutt, S.L.; Wu, L. The transcription factor PU.1 controls dendritic cell development and Flt3 cytokine receptor expression in a dose-dependent manner. Immunity 2010, 32, 628–641. [Google Scholar] [CrossRef]
- Schiavoni, G.; Mattei, F.; Sestili, P.; Borghi, P.; Venditti, M.; Morse, H.C., 3rd; Belardelli, F.; Gabriele, L. ICSBP is essential for the development of mouse type I interferon-producing cells and for the generation and activation of CD8alpha(+) dendritic cells. J. Exp. Med. 2002, 196, 1415–1425. [Google Scholar] [CrossRef]
- Schotte, R.; Nagasawa, M.; Weijer, K.; Spits, H.; Blom, B. The ETS transcription factor Spi-B is required for human plasmacytoid dendritic cell development. J. Exp. Med. 2004, 200, 1503–1509. [Google Scholar] [CrossRef]
- Chen, Y.L.; Chen, T.T.; Pai, L.M.; Wesoly, J.; Bluyssen, H.A.; Lee, C.K. A type I IFN-Flt3 ligand axis augments plasmacytoid dendritic cell development from common lymphoid progenitors. J. Exp. Med. 2013, 210, 2515–2522. [Google Scholar] [CrossRef]
- Ghosh, H.S.; Cisse, B.; Bunin, A.; Lewis, K.L.; Reizis, B. Continuous expression of the transcription factor e2-2 maintains the cell fate of mature plasmacytoid dendritic cells. Immunity 2010, 33, 905–916. [Google Scholar] [CrossRef]
- Karrich, J.J.; Balzarolo, M.; Schmidlin, H.; Libouban, M.; Nagasawa, M.; Gentek, R.; Kamihira, S.; Maeda, T.; Amsen, D.; Wolkers, M.C.; et al. The transcription factor Spi-B regulates human plasmacytoid dendritic cell survival through direct induction of the antiapoptotic gene BCL2-A1. Blood 2012, 119, 5191–5200. [Google Scholar] [CrossRef]
- Laouar, Y.; Welte, T.; Fu, X.Y.; Flavell, R.A. STAT3 is required for Flt3L-dependent dendritic cell differentiation. Immunity 2003, 19, 903–912. [Google Scholar] [CrossRef]
- Esashi, E.; Wang, Y.H.; Perng, O.; Qin, X.F.; Liu, Y.J.; Watowich, S.S. The signal transducer STAT5 inhibits plasmacytoid dendritic cell development by suppressing transcription factor IRF8. Immunity 2008, 28, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Li, H.S.; Yang, C.Y.; Nallaparaju, K.C.; Zhang, H.; Liu, Y.J.; Goldrath, A.W.; Watowich, S.S. The signal transducers STAT5 and STAT3 control expression of Id2 and E2-2 during dendritic cell development. Blood 2012, 120, 4363–4373. [Google Scholar] [CrossRef] [PubMed]
- Naik, S.H.; Perie, L.; Swart, E.; Gerlach, C.; van Rooij, N.; de Boer, R.J.; Schumacher, T.N. Diverse and heritable lineage imprinting of early haematopoietic progenitors. Nature 2013, 496, 229–232. [Google Scholar] [CrossRef]
- Vermi, W.; Riboldi, E.; Wittamer, V.; Gentili, F.; Luini, W.; Marrelli, S.; Vecchi, A.; Franssen, J.D.; Communi, D.; Massardi, L.; et al. Role of ChemR23 in directing the migration of myeloid and plasmacytoid dendritic cells to lymphoid organs and inflamed skin. J. Exp. Med. 2005, 201, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Krug, A.; Uppaluri, R.; Facchetti, F.; Dorner, B.G.; Sheehan, K.C.; Schreiber, R.D.; Cella, M.; Colonna, M. IFN-producing cells respond to CXCR3 ligands in the presence of CXCL12 and secrete inflammatory chemokines upon activation. J. Immunol. 2002, 169, 6079–6083. [Google Scholar] [CrossRef] [PubMed]
- Seth, S.; Oberdorfer, L.; Hyde, R.; Hoff, K.; Thies, V.; Worbs, T.; Schmitz, S.; Forster, R. CCR7 essentially contributes to the homing of plasmacytoid dendritic cells to lymph nodes under steady-state as well as inflammatory conditions. J. Immunol. 2011, 186, 3364–3372. [Google Scholar] [CrossRef]
- Umemoto, E.; Otani, K.; Ikeno, T.; Verjan Garcia, N.; Hayasaka, H.; Bai, Z.; Jang, M.H.; Tanaka, T.; Nagasawa, T.; Ueda, K.; et al. Constitutive plasmacytoid dendritic cell migration to the splenic white pulp is cooperatively regulated by CCR7- and CXCR4-mediated signaling. J. Immunol. 2012, 189, 191–199. [Google Scholar] [CrossRef]
- Kadowaki, N.; Antonenko, S.; Lau, J.Y.; Liu, Y.J. Natural interferon alpha/beta-producing cells link innate and adaptive immunity. J. Exp. Med. 2000, 192, 219–226. [Google Scholar] [CrossRef]
- Feldman, S.B.; Ferraro, M.; Zheng, H.M.; Patel, N.; Gould-Fogerite, S.; Fitzgerald-Bocarsly, P. Viral induction of low frequency interferon-alpha producing cells. Virology 1994, 204, 1–7. [Google Scholar] [CrossRef]
- Ito, T.; Kanzler, H.; Duramad, O.; Cao, W.; Liu, Y.J. Specialization, kinetics, and repertoire of type 1 interferon responses by human plasmacytoid predendritic cells. Blood 2006, 107, 2423–2431. [Google Scholar] [CrossRef]
- Fitzgerald-Bocarsly, P. Natural interferon-alpha producing cells: The plasmacytoid dendritic cells. Biotechniques 2002, 33, S16–S20. [Google Scholar] [CrossRef]
- Tailor, P.; Tamura, T.; Kong, H.J.; Kubota, T.; Kubota, M.; Borghi, P.; Gabriele, L.; Ozato, K. The feedback phase of type I interferon induction in dendritic cells requires interferon regulatory factor 8. Immunity 2007, 27, 228–239. [Google Scholar] [CrossRef] [PubMed]
- Swiecki, M.; Wang, Y.; Vermi, W.; Gilfillan, S.; Schreiber, R.D.; Colonna, M. Type I interferon negatively controls plasmacytoid dendritic cell numbers in vivo. J. Exp. Med. 2011, 208, 2367–2374. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.K.; Lund, J.M.; Ramanathan, B.; Mizushima, N.; Iwasaki, A. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science 2007, 315, 1398–1401. [Google Scholar] [CrossRef] [PubMed]
- Bave, U.; Magnusson, M.; Eloranta, M.L.; Perers, A.; Alm, G.V.; Ronnblom, L. Fc gamma RIIa is expressed on natural IFN-alpha-producing cells (plasmacytoid dendritic cells) and is required for the IFN-alpha production induced by apoptotic cells combined with lupus IgG. J. Immunol. 2003, 171, 3296–3302. [Google Scholar] [CrossRef]
- Wang, J.P.; Asher, D.R.; Chan, M.; Kurt-Jones, E.A.; Finberg, R.W. Cutting Edge: Antibody-mediated TLR7-dependent recognition of viral RNA. J. Immunol. 2007, 178, 3363–3367. [Google Scholar] [CrossRef]
- Honda, K.; Yanai, H.; Mizutani, T.; Negishi, H.; Shimada, N.; Suzuki, N.; Ohba, Y.; Takaoka, A.; Yeh, W.C.; Taniguchi, T. Role of a transductional-transcriptional processor complex involving MyD88 and IRF-7 in Toll-like receptor signaling. Proc. Natl. Acad. Sci. USA 2004, 101, 15416–15421. [Google Scholar] [CrossRef]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef]
- Honda, K.; Ohba, Y.; Yanai, H.; Negishi, H.; Mizutani, T.; Takaoka, A.; Taya, C.; Taniguchi, T. Spatiotemporal regulation of MyD88-IRF-7 signalling for robust type-I interferon induction. Nature 2005, 434, 1035–1040. [Google Scholar] [CrossRef]
- Guiducci, C.; Ott, G.; Chan, J.H.; Damon, E.; Calacsan, C.; Matray, T.; Lee, K.D.; Coffman, R.L.; Barrat, F.J. Properties regulating the nature of the plasmacytoid dendritic cell response to Toll-like receptor 9 activation. J. Exp. Med. 2006, 203, 1999–2008. [Google Scholar] [CrossRef]
- Majer, O.; Liu, B.; Woo, B.J.; Kreuk, L.S.M.; Van Dis, E.; Barton, G.M. Release from UNC93B1 reinforces the compartmentalized activation of select TLRs. Nature 2019, 575, 371–374. [Google Scholar] [CrossRef] [PubMed]
- Majer, O.; Liu, B.; Kreuk, L.S.M.; Krogan, N.; Barton, G.M. UNC93B1 recruits syntenin-1 to dampen TLR7 signalling and prevent autoimmunity. Nature 2019, 575, 366–370. [Google Scholar] [CrossRef] [PubMed]
- Dzionek, A.; Sohma, Y.; Nagafune, J.; Cella, M.; Colonna, M.; Facchetti, F.; Gunther, G.; Johnston, I.; Lanzavecchia, A.; Nagasaka, T.; et al. BDCA-2, a novel plasmacytoid dendritic cell-specific type II C-type lectin, mediates antigen capture and is a potent inhibitor of interferon alpha/beta induction. J. Exp. Med. 2001, 194, 1823–1834. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Zhang, L.; Rosen, D.B.; Bover, L.; Watanabe, G.; Bao, M.; Lanier, L.L.; Liu, Y.J. BDCA2/Fc epsilon RI gamma complex signals through a novel BCR-like pathway in human plasmacytoid dendritic cells. PLoS Biol. 2007, 5, e248. [Google Scholar] [CrossRef]
- Pellerin, A.; Otero, K.; Czerkowicz, J.M.; Kerns, H.M.; Shapiro, R.I.; Ranger, A.M.; Otipoby, K.L.; Taylor, F.R.; Cameron, T.O.; Viney, J.L.; et al. Anti-BDCA2 monoclonal antibody inhibits plasmacytoid dendritic cell activation through Fc-dependent and Fc-independent mechanisms. EMBO Mol. Med. 2015, 7, 464–476. [Google Scholar] [CrossRef]
- Riboldi, E.; Daniele, R.; Parola, C.; Inforzato, A.; Arnold, P.L.; Bosisio, D.; Fremont, D.H.; Bastone, A.; Colonna, M.; Sozzani, S. Human C-type lectin domain family 4, member C (CLEC4C/BDCA-2/CD303) is a receptor for asialo-galactosyl-oligosaccharides. J. Biol Chem. 2011, 286, 35329–35333. [Google Scholar] [CrossRef]
- Cao, W.; Rosen, D.B.; Ito, T.; Bover, L.; Bao, M.; Watanabe, G.; Yao, Z.; Zhang, L.; Lanier, L.L.; Liu, Y.J. Plasmacytoid dendritic cell-specific receptor ILT7-Fc epsilonRI gamma inhibits Toll-like receptor-induced interferon production. J. Exp. Med. 2006, 203, 1399–1405. [Google Scholar] [CrossRef]
- Cao, W.; Bover, L.; Cho, M.; Wen, X.; Hanabuchi, S.; Bao, M.; Rosen, D.B.; Wang, Y.H.; Shaw, J.L.; Du, Q.; et al. Regulation of TLR7/9 responses in plasmacytoid dendritic cells by BST2 and ILT7 receptor interaction. J. Exp. Med. 2009, 206, 1603–1614. [Google Scholar] [CrossRef]
- Fuchs, A.; Cella, M.; Kondo, T.; Colonna, M. Paradoxic inhibition of human natural interferon-producing cells by the activating receptor NKp44. Blood 2005, 106, 2076–2082. [Google Scholar] [CrossRef]
- Rosental, B.; Brusilovsky, M.; Hadad, U.; Oz, D.; Appel, M.Y.; Afergan, F.; Yossef, R.; Rosenberg, L.A.; Aharoni, A.; Cerwenka, A.; et al. Proliferating cell nuclear antigen is a novel inhibitory ligand for the natural cytotoxicity receptor NKp44. J. Immunol. 2011, 187, 5693–5702. [Google Scholar] [CrossRef]
- Bloem, K.; Vuist, I.M.; van den Berk, M.; Klaver, E.J.; van Die, I.; Knippels, L.M.; Garssen, J.; Garcia-Vallejo, J.J.; van Vliet, S.J.; van Kooyk, Y. DCIR interacts with ligands from both endogenous and pathogenic origin. Immunol. Lett. 2014, 158, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Meyer-Wentrup, F.; Benitez-Ribas, D.; Tacken, P.J.; Punt, C.J.; Figdor, C.G.; de Vries, I.J.; Adema, G.J. Targeting DCIR on human plasmacytoid dendritic cells results in antigen presentation and inhibits IFN-alpha production. Blood 2008, 111, 4245–4253. [Google Scholar] [CrossRef] [PubMed]
- Szabo, A.; Magyarics, Z.; Pazmandi, K.; Gopcsa, L.; Rajnavolgyi, E.; Bacsi, A. TLR ligands upregulate RIG-I expression in human plasmacytoid dendritic cells in a type I IFN-independent manner. Immunol. Cell Biol. 2014, 92, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Loo, Y.M.; Gale, M., Jr. Immune signaling by RIG-I-like receptors. Immunity 2011, 34, 680–692. [Google Scholar] [CrossRef]
- Fekete, T.; Bencze, D.; Szabo, A.; Csoma, E.; Biro, T.; Bacsi, A.; Pazmandi, K. Regulatory NLRs Control the RLR-Mediated Type I Interferon and Inflammatory Responses in Human Dendritic Cells. Front. Immunol. 2018, 9, 2314. [Google Scholar] [CrossRef]
- Bode, C.; Fox, M.; Tewary, P.; Steinhagen, A.; Ellerkmann, R.K.; Klinman, D.; Baumgarten, G.; Hornung, V.; Steinhagen, F. Human plasmacytoid dentritic cells elicit a Type I Interferon response by sensing DNA via the cGAS-STING signaling pathway. Eur. J. Immunol. 2016, 46, 1615–1621. [Google Scholar] [CrossRef]
- Ishikawa, H.; Ma, Z.; Barber, G.N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 2009, 461, 788–792. [Google Scholar] [CrossRef]
- Cai, X.; Chiu, Y.H.; Chen, Z.J. The cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling. Mol. Cell 2014, 54, 289–296. [Google Scholar] [CrossRef]
- Yu, X.; Cai, B.; Wang, M.; Tan, P.; Ding, X.; Wu, J.; Li, J.; Li, Q.; Liu, P.; Xing, C.; et al. Cross-Regulation of Two Type I Interferon Signaling Pathways in Plasmacytoid Dendritic Cells Controls Anti-malaria Immunity and Host Mortality. Immunity 2016, 45, 1093–1107. [Google Scholar] [CrossRef]
- Kotenko, S.V.; Gallagher, G.; Baurin, V.V.; Lewis-Antes, A.; Shen, M.; Shah, N.K.; Langer, J.A.; Sheikh, F.; Dickensheets, H.; Donnelly, R.P. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat. Immunol. 2003, 4, 69–77. [Google Scholar] [CrossRef]
- Sheppard, P.; Kindsvogel, W.; Xu, W.; Henderson, K.; Schlutsmeyer, S.; Whitmore, T.E.; Kuestner, R.; Garrigues, U.; Birks, C.; Roraback, J.; et al. IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat. Immunol. 2003, 4, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Prokunina-Olsson, L.; Muchmore, B.; Tang, W.; Pfeiffer, R.M.; Park, H.; Dickensheets, H.; Hergott, D.; Porter-Gill, P.; Mumy, A.; Kohaar, I.; et al. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat. Genet. 2013, 45, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Schnepf, D.; Staeheli, P. Interferon-lambda orchestrates innate and adaptive mucosal immune responses. Nat. Rev. Immunol. 2019, 19, 614–625. [Google Scholar] [CrossRef] [PubMed]
- Lasfar, A.; Gogas, H.; Zloza, A.; Kaufman, H.L.; Kirkwood, J.M. IFN-lambda cancer immunotherapy: New kid on the block. Immunotherapy 2016, 8, 877–888. [Google Scholar] [CrossRef]
- Sato, A.; Ohtsuki, M.; Hata, M.; Kobayashi, E.; Murakami, T. Antitumor activity of IFN-lambda in murine tumor models. J. Immunol. 2006, 176, 7686–7694. [Google Scholar] [CrossRef]
- Stiff, A.; Carson, W., III. Investigations of interferon-lambda for the treatment of cancer. J. Innate Immun 2015, 7, 243–250. [Google Scholar] [CrossRef]
- Coccia, E.M.; Severa, M.; Giacomini, E.; Monneron, D.; Remoli, M.E.; Julkunen, I.; Cella, M.; Lande, R.; Uze, G. Viral infection and Toll-like receptor agonists induce a differential expression of type I and lambda interferons in human plasmacytoid and monocyte-derived dendritic cells. Eur. J. Immunol. 2004, 34, 796–805. [Google Scholar] [CrossRef]
- Yin, Z.; Dai, J.; Deng, J.; Sheikh, F.; Natalia, M.; Shih, T.; Lewis-Antes, A.; Amrute, S.B.; Garrigues, U.; Doyle, S.; et al. Type III IFNs are produced by and stimulate human plasmacytoid dendritic cells. J. Immunol. 2012, 189, 2735–2745. [Google Scholar] [CrossRef]
- Finotti, G.; Tamassia, N.; Calzetti, F.; Fattovich, G.; Cassatella, M.A. Endogenously produced TNF-alpha contributes to the expression of CXCL10/IP-10 in IFN-lambda3-activated plasmacytoid dendritic cells. J. Leukoc. Biol. 2016, 99, 107–119. [Google Scholar] [CrossRef]
- Megjugorac, N.J.; Gallagher, G.E.; Gallagher, G. Modulation of human plasmacytoid DC function by IFN-lambda1 (IL-29). J. Leukoc. Biol. 2009, 86, 1359–1363. [Google Scholar] [CrossRef]
- Finotti, G.; Tamassia, N.; Cassatella, M.A. Interferon-lambdas and Plasmacytoid Dendritic Cells: A Close Relationship. Front. Immunol. 2017, 8, 1015. [Google Scholar] [CrossRef] [PubMed]
- Chaperot, L.; Blum, A.; Manches, O.; Lui, G.; Angel, J.; Molens, J.P.; Plumas, J. Virus or TLR agonists induce TRAIL-mediated cytotoxic activity of plasmacytoid dendritic cells. J. Immunol. 2006, 176, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Blum, A.; Chaperot, L.; Molens, J.P.; Foissaud, V.; Plantaz, D.; Plumas, J. Mechanisms of TRAIL-induced apoptosis in leukemic plasmacytoid dendritic cells. Exp. Hematol. 2006, 34, 1655–1662. [Google Scholar] [CrossRef] [PubMed]
- Balzarolo, M.; Karrich, J.J.; Engels, S.; Blom, B.; Medema, J.P.; Wolkers, M.C. The transcriptional regulator NAB2 reveals a two-step induction of TRAIL in activated plasmacytoid DCs. Eur. J. Immunol. 2012, 42, 3019–3027. [Google Scholar] [CrossRef] [PubMed]
- Drobits, B.; Holcmann, M.; Amberg, N.; Swiecki, M.; Grundtner, R.; Hammer, M.; Colonna, M.; Sibilia, M. Imiquimod clears tumors in mice independent of adaptive immunity by converting pDCs into tumor-killing effector cells. J. Clin. Investig. 2012, 122, 575–585. [Google Scholar] [CrossRef]
- Kalb, M.L.; Glaser, A.; Stary, G.; Koszik, F.; Stingl, G. TRAIL(+) human plasmacytoid dendritic cells kill tumor cells in vitro: Mechanisms of imiquimod- and IFN-alpha-mediated antitumor reactivity. J. Immunol. 2012, 188, 1583–1591. [Google Scholar] [CrossRef]
- Rissoan, M.C.; Duhen, T.; Bridon, J.M.; Bendriss-Vermare, N.; Peronne, C.; de Saint Vis, B.; Briere, F.; Bates, E.E. Subtractive hybridization reveals the expression of immunoglobulin-like transcript 7, Eph-B1, granzyme B, and 3 novel transcripts in human plasmacytoid dendritic cells. Blood 2002, 100, 3295–3303. [Google Scholar] [CrossRef]
- Salvi, V.; Vermi, W.; Cavani, A.; Lonardi, S.; Carbone, T.; Facchetti, F.; Bosisio, D.; Sozzani, S. IL-21 May Promote Granzyme B-Dependent NK/Plasmacytoid Dendritic Cell Functional Interaction in Cutaneous Lupus Erythematosus. J. Investig. Dermatol. 2017, 137, 1493–1500. [Google Scholar] [CrossRef]
- Facchetti, F.; Vermi, W.; Santoro, A.; Vergoni, F.; Chilosi, M.; Doglioni, C. Neoplasms derived from plasmacytoid monocytes/interferon-producing cells: Variability of CD56 and granzyme B expression. Am. J. Surg. Pathol. 2003, 27, 1489–1492. [Google Scholar] [CrossRef]
- Yu, H.; Zhang, P.; Yin, X.; Yin, Z.; Shi, Q.; Cui, Y.; Liu, G.; Wang, S.; Piccaluga, P.P.; Jiang, T.; et al. Human BDCA2+CD123+CD56+ dendritic cells (DCs) related to blastic plasmacytoid dendritic cell neoplasm represent a unique myeloid DC subset. Protein Cell 2015, 6, 297–306. [Google Scholar] [CrossRef]
- Steinman, R.M. The dendritic cell system and its role in immunogenicity. Annu. Rev. Immunol. 1991, 9, 271–296. [Google Scholar] [CrossRef] [PubMed]
- Young, L.J.; Wilson, N.S.; Schnorrer, P.; Proietto, A.; ten Broeke, T.; Matsuki, Y.; Mount, A.M.; Belz, G.T.; O’Keeffe, M.; Ohmura-Hoshino, M.; et al. Differential MHC class II synthesis and ubiquitination confers distinct antigen-presenting properties on conventional and plasmacytoid dendritic cells. Nat. Immunol. 2008, 9, 1244–1252. [Google Scholar] [CrossRef] [PubMed]
- Hoeffel, G.; Ripoche, A.C.; Matheoud, D.; Nascimbeni, M.; Escriou, N.; Lebon, P.; Heshmati, F.; Guillet, J.G.; Gannage, M.; Caillat-Zucman, S.; et al. Antigen crosspresentation by human plasmacytoid dendritic cells. Immunity 2007, 27, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Di Pucchio, T.; Chatterjee, B.; Smed-Sorensen, A.; Clayton, S.; Palazzo, A.; Montes, M.; Xue, Y.; Mellman, I.; Banchereau, J.; Connolly, J.E. Direct proteasome-independent cross-presentation of viral antigen by plasmacytoid dendritic cells on major histocompatibility complex class I. Nat. Immunol. 2008, 9, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Tel, J.; Schreibelt, G.; Sittig, S.P.; Mathan, T.S.; Buschow, S.I.; Cruz, L.J.; Lambeck, A.J.; Figdor, C.G.; de Vries, I.J. Human plasmacytoid dendritic cells efficiently cross-present exogenous Ags to CD8+ T cells despite lower Ag uptake than myeloid dendritic cell subsets. Blood 2013, 121, 459–467. [Google Scholar] [CrossRef]
- Soumelis, V.; Liu, Y.J. From plasmacytoid to dendritic cell: Morphological and functional switches during plasmacytoid pre-dendritic cell differentiation. Eur. J. Immunol. 2006, 36, 2286–2292. [Google Scholar] [CrossRef]
- Bjorck, P.; Leong, H.X.; Engleman, E.G. Plasmacytoid dendritic cell dichotomy: Identification of IFN-alpha producing cells as a phenotypically and functionally distinct subset. J. Immunol. 2011, 186, 1477–1485. [Google Scholar] [CrossRef]
- Benitez-Ribas, D.; Tacken, P.; Punt, C.J.; de Vries, I.J.; Figdor, C.G. Activation of human plasmacytoid dendritic cells by TLR9 impairs Fc gammaRII-mediated uptake of immune complexes and presentation by MHC class II. J. Immunol. 2008, 181, 5219–5224. [Google Scholar] [CrossRef]
- Ito, T.; Yang, M.; Wang, Y.H.; Lande, R.; Gregorio, J.; Perng, O.A.; Qin, X.F.; Liu, Y.J.; Gilliet, M. Plasmacytoid dendritic cells prime IL-10-producing T regulatory cells by inducible costimulator ligand. J. Exp. Med. 2007, 204, 105–115. [Google Scholar] [CrossRef]
- Gilliet, M.; Liu, Y.J. Generation of human CD8 T regulatory cells by CD40 ligand-activated plasmacytoid dendritic cells. J. Exp. Med. 2002, 195, 695–704. [Google Scholar] [CrossRef]
- Ochando, J.C.; Homma, C.; Yang, Y.; Hidalgo, A.; Garin, A.; Tacke, F.; Angeli, V.; Li, Y.; Boros, P.; Ding, Y.; et al. Alloantigen-presenting plasmacytoid dendritic cells mediate tolerance to vascularized grafts. Nat. Immunol. 2006, 7, 652–662. [Google Scholar] [CrossRef] [PubMed]
- Guery, L.; Hugues, S. Tolerogenic and activatory plasmacytoid dendritic cells in autoimmunity. Front. Immunol. 2013, 4, 59. [Google Scholar] [CrossRef] [PubMed]
- Munn, D.H.; Sharma, M.D.; Hou, D.; Baban, B.; Lee, J.R.; Antonia, S.J.; Messina, J.L.; Chandler, P.; Koni, P.A.; Mellor, A.L. Expression of indoleamine 2,3-dioxygenase by plasmacytoid dendritic cells in tumor-draining lymph nodes. J. Clin. Investig. 2004, 114, 280–290. [Google Scholar] [CrossRef] [PubMed]
- Araujo, E.F.; Medeiros, D.H.; Galdino, N.A.; Condino-Neto, A.; Calich, V.L.; Loures, F.V. Tolerogenic Plasmacytoid Dendritic Cells Control Paracoccidioides brasiliensis Infection by Inducting Regulatory T Cells in an IDO-Dependent Manner. PLoS Pathog. 2016, 12, e1006115. [Google Scholar] [CrossRef]
- Sharma, M.D.; Baban, B.; Chandler, P.; Hou, D.Y.; Singh, N.; Yagita, H.; Azuma, M.; Blazar, B.R.; Mellor, A.L.; Munn, D.H. Plasmacytoid dendritic cells from mouse tumor-draining lymph nodes directly activate mature Tregs via indoleamine 2,3-dioxygenase. J. Clin. Investig. 2007, 117, 2570–2582. [Google Scholar] [CrossRef]
- Diana, J.; Griseri, T.; Lagaye, S.; Beaudoin, L.; Autrusseau, E.; Gautron, A.S.; Tomkiewicz, C.; Herbelin, A.; Barouki, R.; von Herrath, M.; et al. NKT cell-plasmacytoid dendritic cell cooperation via OX40 controls viral infection in a tissue-specific manner. Immunity 2009, 30, 289–299. [Google Scholar] [CrossRef]
- Ito, T.; Amakawa, R.; Inaba, M.; Hori, T.; Ota, M.; Nakamura, K.; Takebayashi, M.; Miyaji, M.; Yoshimura, T.; Inaba, K.; et al. Plasmacytoid dendritic cells regulate Th cell responses through OX40 ligand and type I IFNs. J. Immunol. 2004, 172, 4253–4259. [Google Scholar] [CrossRef]
- Diana, J.; Brezar, V.; Beaudoin, L.; Dalod, M.; Mellor, A.; Tafuri, A.; von Herrath, M.; Boitard, C.; Mallone, R.; Lehuen, A. Viral infection prevents diabetes by inducing regulatory T cells through NKT cell-plasmacytoid dendritic cell interplay. J. Exp. Med. 2011, 208, 729–745. [Google Scholar] [CrossRef]
- Jahrsdorfer, B.; Vollmer, A.; Blackwell, S.E.; Maier, J.; Sontheimer, K.; Beyer, T.; Mandel, B.; Lunov, O.; Tron, K.; Nienhaus, G.U.; et al. Granzyme B produced by human plasmacytoid dendritic cells suppresses T-cell expansion. Blood 2010, 115, 1156–1165. [Google Scholar] [CrossRef]
- Dunn, G.P.; Bruce, A.T.; Sheehan, K.C.; Shankaran, V.; Uppaluri, R.; Bui, J.D.; Diamond, M.S.; Koebel, C.M.; Arthur, C.; White, J.M.; et al. A critical function for type I interferons in cancer immunoediting. Nat. Immunol. 2005, 6, 722–729. [Google Scholar] [CrossRef]
- Liu, C.; Lou, Y.; Lizee, G.; Qin, H.; Liu, S.; Rabinovich, B.; Kim, G.J.; Wang, Y.H.; Ye, Y.; Sikora, A.G.; et al. Plasmacytoid dendritic cells induce NK cell-dependent, tumor antigen-specific T cell cross-priming and tumor regression in mice. J. Clin. Investig. 2008, 118, 1165–1175. [Google Scholar] [CrossRef] [PubMed]
- Blasius, A.L.; Giurisato, E.; Cella, M.; Schreiber, R.D.; Shaw, A.S.; Colonna, M. Bone marrow stromal cell antigen 2 is a specific marker of type I IFN-producing cells in the naive mouse, but a promiscuous cell surface antigen following IFN stimulation. J. Immunol. 2006, 177, 3260–3265. [Google Scholar] [CrossRef] [PubMed]
- Wrammert, J.; Kallberg, E.; Agace, W.W.; Leanderson, T. Ly6C expression differentiates plasma cells from other B cell subsets in mice. Eur J. Immunol. 2002, 32, 97–103. [Google Scholar] [CrossRef]
- Yang, L.L.; Mao, L.; Wu, H.; Chen, L.; Deng, W.W.; Xiao, Y.; Li, H.; Zhang, L.; Sun, Z.J. pDC depletion induced by CD317 blockade drives the antitumor immune response in head and neck squamous cell carcinoma. Oral Oncol. 2019, 96, 131–139. [Google Scholar] [CrossRef]
- Jung, S.; Unutmaz, D.; Wong, P.; Sano, G.; De los Santos, K.; Sparwasser, T.; Wu, S.; Vuthoori, S.; Ko, K.; Zavala, F.; et al. In vivo depletion of CD11c+ dendritic cells abrogates priming of CD8+ T cells by exogenous cell-associated antigens. Immunity 2002, 17, 211–220. [Google Scholar] [CrossRef]
- Swiecki, M.; Gilfillan, S.; Vermi, W.; Wang, Y.; Colonna, M. Plasmacytoid dendritic cell ablation impacts early interferon responses and antiviral NK and CD8(+) T cell accrual. Immunity 2010, 33, 955–966. [Google Scholar] [CrossRef]
- Mandl, M.; Drechsler, M.; Jansen, Y.; Neideck, C.; Noels, H.; Faussner, A.; Soehnlein, O.; Weber, C.; Doring, Y. Evaluation of the BDCA2-DTR Transgenic Mouse Model in Chronic and Acute Inflammation. PLoS ONE 2015, 10, e0134176. [Google Scholar] [CrossRef]
- Lippitsch, A.; Baal, N.; Chukovetskyi, Y.; Cunningham, S.; Michel, G.; Dietert, K.; Gurtner, C.; Gruber, A.D.; Bein, G.; Hackstein, H. Plasmacytoid dendritic cell depletion modifies FoxP3+ T cell homeostasis and the clinical course of bacterial pneumonia in mice. J. Leukocyte Biol. 2019, 106, 977–985. [Google Scholar] [CrossRef]
- Rowland, S.L.; Riggs, J.M.; Gilfillan, S.; Bugatti, M.; Vermi, W.; Kolbeck, R.; Unanue, E.R.; Sanjuan, M.A.; Colonna, M. Early, transient depletion of plasmacytoid dendritic cells ameliorates autoimmunity in a lupus model. J. Exp. Med. 2014, 211, 1977–1991. [Google Scholar] [CrossRef]
- Zou, W.; Machelon, V.; Coulomb-L’Hermin, A.; Borvak, J.; Nome, F.; Isaeva, T.; Wei, S.; Krzysiek, R.; Durand-Gasselin, I.; Gordon, A.; et al. Stromal-derived factor-1 in human tumors recruits and alters the function of plasmacytoid precursor dendritic cells. Nat. Med. 2001, 7, 1339–1346. [Google Scholar] [CrossRef]
- Thiel, A.; Pries, R.; Jeske, S.; Trenkle, T.; Wollenberg, B. Effect of head and neck cancer supernatant and CpG-oligonucleotides on migration and IFN-alpha production of plasmacytoid dendritic cells. Anticancer Res. 2009, 29, 3019–3025. [Google Scholar] [PubMed]
- Penna, G.; Sozzani, S.; Adorini, L. Cutting edge: Selective usage of chemokine receptors by plasmacytoid dendritic cells. J. Immunol. 2001, 167, 1862–1866. [Google Scholar] [CrossRef] [PubMed]
- Vanbervliet, B.; Bendriss-Vermare, N.; Massacrier, C.; Homey, B.; de Bouteiller, O.; Briere, F.; Trinchieri, G.; Caux, C. The inducible CXCR3 ligands control plasmacytoid dendritic cell responsiveness to the constitutive chemokine stromal cell-derived factor 1 (SDF-1)/CXCL12. J. Exp. Med. 2003, 198, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Treilleux, I.; Blay, J.Y.; Bendriss-Vermare, N.; Ray-Coquard, I.; Bachelot, T.; Guastalla, J.P.; Bremond, A.; Goddard, S.; Pin, J.J.; Barthelemy-Dubois, C.; et al. Dendritic cell infiltration and prognosis of early stage breast cancer. Clin. Cancer Res. 2004, 10, 7466–7474. [Google Scholar] [CrossRef]
- Gadalla, R.; Hassan, H.; Ibrahim, S.A.; Abdullah, M.S.; Gaballah, A.; Greve, B.; El-Deeb, S.; El-Shinawi, M.; Mohamed, M.M. Tumor microenvironmental plasmacytoid dendritic cells contribute to breast cancer lymph node metastasis via CXCR4/SDF-1 axis. Breast Cancer Res. Treat. 2019, 174, 679–691. [Google Scholar] [CrossRef]
- Mansfield, A.S.; Heikkila, P.; von Smitten, K.; Vakkila, J.; Leidenius, M. Metastasis to sentinel lymph nodes in breast cancer is associated with maturation arrest of dendritic cells and poor co-localization of dendritic cells and CD8+ T cells. Virchows Arch. 2011, 459, 391–398. [Google Scholar] [CrossRef]
- Labidi-Galy, S.I.; Sisirak, V.; Meeus, P.; Gobert, M.; Treilleux, I.; Bajard, A.; Combes, J.D.; Faget, J.; Mithieux, F.; Cassignol, A.; et al. Quantitative and functional alterations of plasmacytoid dendritic cells contribute to immune tolerance in ovarian cancer. Cancer Res. 2011, 71, 5423–5434. [Google Scholar] [CrossRef]
- Conrad, C.; Gregorio, J.; Wang, Y.H.; Ito, T.; Meller, S.; Hanabuchi, S.; Anderson, S.; Atkinson, N.; Ramirez, P.T.; Liu, Y.J.; et al. Plasmacytoid dendritic cells promote immunosuppression in ovarian cancer via ICOS costimulation of Foxp3(+) T-regulatory cells. Cancer Res. 2012, 72, 5240–5249. [Google Scholar] [CrossRef]
- Curiel, T.J.; Cheng, P.; Mottram, P.; Alvarez, X.; Moons, L.; Evdemon-Hogan, M.; Wei, S.; Zou, L.; Kryczek, I.; Hoyle, G.; et al. Dendritic cell subsets differentially regulate angiogenesis in human ovarian cancer. Cancer Res. 2004, 64, 5535–5538. [Google Scholar] [CrossRef]
- Hartmann, E.; Wollenberg, B.; Rothenfusser, S.; Wagner, M.; Wellisch, D.; Mack, B.; Giese, T.; Gires, O.; Endres, S.; Hartmann, G. Identification and functional analysis of tumor-infiltrating plasmacytoid dendritic cells in head and neck cancer. Cancer Res. 2003, 63, 6478–6487. [Google Scholar]
- Han, N.; Zhang, Z.; Liu, S.; Ow, A.; Ruan, M.; Yang, W.; Zhang, C. Increased tumor-infiltrating plasmacytoid dendritic cells predicts poor prognosis in oral squamous cell carcinoma. Arch. Oral Biol. 2017, 78, 129–134. [Google Scholar] [CrossRef]
- Han, N.; Zhang, Z.; Jv, H.; Hu, J.; Ruan, M.; Zhang, C. Culture supernatants of oral cancer cells induce impaired IFN-alpha production of pDCs partly through the down-regulation of TLR-9 expression. Arch. Oral Biol. 2018, 93, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Gai, X.D.; Song, Y.; Li, C.; Lei, Y.M.; Yang, B. Potential role of plasmacytoid dendritic cells for FOXP3+ regulatory T cell development in human colorectal cancer and tumor draining lymph node. Pathol. Res. Pract. 2013, 209, 774–778. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Li, X.; Porter, J.L.; Ostrodi, D.H.; Yang, B.; Li, J.; Wang, Y.; Zhang, J.; Bai, L.; Jiao, S. Level of plasmacytoid dendritic cells is increased in non-small cell lung carcinoma. Tumour Biol. 2014, 35, 2247–2252. [Google Scholar] [CrossRef]
- Riemann, D.; Cwikowski, M.; Turzer, S.; Giese, T.; Grallert, M.; Schutte, W.; Seliger, B. Blood immune cell biomarkers in lung cancer. Clin. Exp. Immunol. 2019, 195, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Rossi, R.; Lichtner, M.; Iori, F.; Ermocida, A.; Mascia, C.; Mengoni, F.; Sauzullo, I.; Dini, D.; Mastroianni, C.M.; Vullo, V. Dendritic cells in blood and urine samples from bladder cancer patients undergoing BCG immunotherapy. Arch. Ital. Urol. Androl. 2013, 85, 157–163. [Google Scholar] [CrossRef]
- Orsini, G.; Legitimo, A.; Failli, A.; Ferrari, P.; Nicolini, A.; Spisni, R.; Miccoli, P.; Consolini, R. Quantification of blood dendritic cells in colorectal cancer patients during the course of disease. Pathol. Oncol. Res. 2014, 20, 267–276. [Google Scholar] [CrossRef]
- Kini Bailur, J.; Gueckel, B.; Pawelec, G. Prognostic impact of high levels of circulating plasmacytoid dendritic cells in breast cancer. J. Transl. Med. 2016, 14, 151. [Google Scholar] [CrossRef]
- Sakakura, K.; Chikamatsu, K.; Takahashi, K.; Whiteside, T.L.; Furuya, N. Maturation of circulating dendritic cells and imbalance of T-cell subsets in patients with squamous cell carcinoma of the head and neck. Cancer Immunol. Immunother. 2006, 55, 151–159. [Google Scholar] [CrossRef]
- Faget, J.; Bendriss-Vermare, N.; Gobert, M.; Durand, I.; Olive, D.; Biota, C.; Bachelot, T.; Treilleux, I.; Goddard-Leon, S.; Lavergne, E.; et al. ICOS-ligand expression on plasmacytoid dendritic cells supports breast cancer progression by promoting the accumulation of immunosuppressive CD4+ T cells. Cancer Res. 2012, 72, 6130–6141. [Google Scholar] [CrossRef]
- Labidi-Galy, S.I.; Treilleux, I.; Goddard-Leon, S.; Combes, J.D.; Blay, J.Y.; Ray-Coquard, I.; Caux, C.; Bendriss-Vermare, N. Plasmacytoid dendritic cells infiltrating ovarian cancer are associated with poor prognosis. Oncoimmunology 2012, 1, 380–382. [Google Scholar] [CrossRef] [PubMed]
- Faget, J.; Sisirak, V.; Blay, J.Y.; Caux, C.; Bendriss-Vermare, N.; Menetrier-Caux, C. ICOS is associated with poor prognosis in breast cancer as it promotes the amplification of immunosuppressive CD4(+) T cells by plasmacytoid dendritic cells. Oncoimmunology 2013, 2, e23185. [Google Scholar] [CrossRef] [PubMed]
- Sawant, A.; Ponnazhagan, S. Role of plasmacytoid dendritic cells in breast cancer bone dissemination. Oncoimmunology 2013, 2, e22983. [Google Scholar] [CrossRef] [PubMed]
- Sisirak, V.; Faget, J.; Gobert, M.; Goutagny, N.; Vey, N.; Treilleux, I.; Renaudineau, S.; Poyet, G.; Labidi-Galy, S.I.; Goddard-Leon, S.; et al. Impaired IFN-alpha production by plasmacytoid dendritic cells favors regulatory T-cell expansion that may contribute to breast cancer progression. Cancer Res. 2012, 72, 5188–5197. [Google Scholar] [CrossRef]
- Tjomsland, V.; Sandstrom, P.; Spangeus, A.; Messmer, D.; Emilsson, J.; Falkmer, U.; Falkmer, S.; Magnusson, K.E.; Borch, K.; Larsson, M. Pancreatic adenocarcinoma exerts systemic effects on the peripheral blood myeloid and plasmacytoid dendritic cells: An indicator of disease severity? BMC Cancer 2010, 10, 87. [Google Scholar] [CrossRef]
- Pedroza-Gonzalez, A.; Zhou, G.; Vargas-Mendez, E.; Boor, P.P.; Mancham, S.; Verhoef, C.; Polak, W.G.; Grunhagen, D.; Pan, Q.; Janssen, H.; et al. Tumor-infiltrating plasmacytoid dendritic cells promote immunosuppression by Tr1 cells in human liver tumors. Oncoimmunology 2015, 4, e1008355. [Google Scholar] [CrossRef]
- Aspord, C.; Leccia, M.T.; Charles, J.; Plumas, J. Plasmacytoid dendritic cells support melanoma progression by promoting Th2 and regulatory immunity through OX40L and ICOSL. Cancer Immunol. Res. 2013, 1, 402–415. [Google Scholar] [CrossRef]
- Perrot, I.; Blanchard, D.; Freymond, N.; Isaac, S.; Guibert, B.; Pacheco, Y.; Lebecque, S. Dendritic cells infiltrating human non-small cell lung cancer are blocked at immature stage. J. Immunol. 2007, 178, 2763–2769. [Google Scholar] [CrossRef]
- Beckebaum, S.; Zhang, X.; Chen, X.; Yu, Z.; Frilling, A.; Dworacki, G.; Grosse-Wilde, H.; Broelsch, C.E.; Gerken, G.; Cicinnati, V.R. Increased levels of interleukin-10 in serum from patients with hepatocellular carcinoma correlate with profound numerical deficiencies and immature phenotype of circulating dendritic cell subsets. Clin. Cancer Res. 2004, 10, 7260–7269. [Google Scholar] [CrossRef]
- Fabricius, D.; Neubauer, M.; Mandel, B.; Schutz, C.; Viardot, A.; Vollmer, A.; Jahrsdorfer, B.; Debatin, K.M. Prostaglandin E2 inhibits IFN-alpha secretion and Th1 costimulation by human plasmacytoid dendritic cells via E-prostanoid 2 and E-prostanoid 4 receptor engagement. J. Immunol. 2010, 184, 677–684. [Google Scholar] [CrossRef]
- Bekeredjian-Ding, I.; Schafer, M.; Hartmann, E.; Pries, R.; Parcina, M.; Schneider, P.; Giese, T.; Endres, S.; Wollenberg, B.; Hartmann, G. Tumour-derived prostaglandin E and transforming growth factor-beta synergize to inhibit plasmacytoid dendritic cell-derived interferon-alpha. Immunology 2009, 128, 439–450. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, J.T.; Bieneman, A.P.; Xiao, H.; Chichester, K.L.; Vasagar, K.; Saini, S.; Liu, M.C. TLR9- and FcepsilonRI-mediated responses oppose one another in plasmacytoid dendritic cells by down-regulating receptor expression. J. Immunol. 2005, 175, 5724–5731. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, D.; Singh, A.V.; Brahmandam, M.; Carrasco, R.; Bandi, M.; Hideshima, T.; Bianchi, G.; Podar, K.; Tai, Y.T.; Mitsiades, C.; et al. Functional interaction of plasmacytoid dendritic cells with multiple myeloma cells: A therapeutic target. Cancer Cell 2009, 16, 309–323. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Chung, H.Y.; Ehrlich, L.A.; Jelinek, D.F.; Callander, N.S.; Roodman, G.D.; Choi, S.J. IL-3 expression by myeloma cells increases both osteoclast formation and growth of myeloma cells. Blood 2004, 103, 2308–2315. [Google Scholar] [CrossRef]
- Fabricius, D.; O’Dorisio, M.S.; Blackwell, S.; Jahrsdorfer, B. Human plasmacytoid dendritic cell function: Inhibition of IFN-alpha secretion and modulation of immune phenotype by vasoactive intestinal peptide. J. Immunol. 2006, 177, 5920–5927. [Google Scholar] [CrossRef]
- Dissanayake, S.K.; Wade, M.; Johnson, C.E.; O’Connell, M.P.; Leotlela, P.D.; French, A.D.; Shah, K.V.; Hewitt, K.J.; Rosenthal, D.T.; Indig, F.E.; et al. The Wnt5A/protein kinase C pathway mediates motility in melanoma cells via the inhibition of metastasis suppressors and initiation of an epithelial to mesenchymal transition. J. Biol. Chem. 2007, 282, 17259–17271. [Google Scholar] [CrossRef]
- Hack, K.; Reilly, L.; Proby, C.; Fleming, C.; Leigh, I.; Foerster, J. Wnt5a inhibits the CpG oligodeoxynucleotide-triggered activation of human plasmacytoid dendritic cells. Clin. Exp. Dermatol. 2012, 37, 557–561. [Google Scholar] [CrossRef]
- Demoulin, S.; Herfs, M.; Somja, J.; Roncarati, P.; Delvenne, P.; Hubert, P. HMGB1 secretion during cervical carcinogenesis promotes the acquisition of a tolerogenic functionality by plasmacytoid dendritic cells. Int. J. Cancer 2015, 137, 345–358. [Google Scholar] [CrossRef]
- Cao, W.; Bover, L. Signaling and ligand interaction of ILT7: Receptor-mediated regulatory mechanisms for plasmacytoid dendritic cells. Immunol. Rev. 2010, 234, 163–176. [Google Scholar] [CrossRef]
- Tsukamoto, N.; Okada, S.; Onami, Y.; Sasaki, Y.; Umezawa, K.; Kawakami, Y. Impairment of plasmacytoid dendritic cells for IFN production by the ligand for immunoglobulin-like transcript 7 expressed on human cancer cells. Clin. Cancer Res. 2009, 15, 5733–5743. [Google Scholar] [CrossRef]
- Saadeh, D.; Kurban, M.; Abbas, O. Plasmacytoid dendritic cell role in cutaneous malignancies. J. Dermatol Sci. 2016, 83, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Cao, J.; Li, Z.; Zheng, X.; Yao, Y.; Li, W.; Yuan, Z. Up-regulation of bone marrow stromal protein 2 (BST2) in breast cancer with bone metastasis. BMC Cancer 2009, 9, 102. [Google Scholar] [CrossRef] [PubMed]
- Bruchhage, K.L.; Heinrichs, S.; Wollenberg, B.; Pries, R. IL-10 in the microenvironment of HNSCC inhibits the CpG ODN induced IFN-alpha secretion of pDCs. Oncol. Lett. 2018, 15, 3985–3990. [Google Scholar] [CrossRef] [PubMed]
- Sisirak, V.; Vey, N.; Goutagny, N.; Renaudineau, S.; Malfroy, M.; Thys, S.; Treilleux, I.; Labidi-Galy, S.I.; Bachelot, T.; Dezutter-Dambuyant, C.; et al. Breast cancer-derived transforming growth factor-beta and tumor necrosis factor-alpha compromise interferon-alpha production by tumor-associated plasmacytoid dendritic cells. Int. J. Cancer 2013, 133, 771–778. [Google Scholar] [CrossRef] [PubMed]
- Fallarino, F.; Gizzi, S.; Mosci, P.; Grohmann, U.; Puccetti, P. Tryptophan catabolism in IDO+ plasmacytoid dendritic cells. Curr. Drug Metab. 2007, 8, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Sisirak, V.; Faget, J.; Vey, N.; Blay, J.Y.; Menetrier-Caux, C.; Caux, C.; Bendriss-Vermare, N. Plasmacytoid dendritic cells deficient in IFNalpha production promote the amplification of FOXP3(+) regulatory T cells and are associated with poor prognosis in breast cancer patients. Oncoimmunology 2013, 2, e22338. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Kryczek, I.; Zou, L.; Daniel, B.; Cheng, P.; Mottram, P.; Curiel, T.; Lange, A.; Zou, W. Plasmacytoid dendritic cells induce CD8+ regulatory T cells in human ovarian carcinoma. Cancer Res. 2005, 65, 5020–5026. [Google Scholar] [CrossRef]
- Munn, D.H.; Mellor, A.L. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J. Clin. Investig. 2007, 117, 1147–1154. [Google Scholar] [CrossRef]
- Boasso, A.; Herbeuval, J.P.; Hardy, A.W.; Anderson, S.A.; Dolan, M.J.; Fuchs, D.; Shearer, G.M. HIV inhibits CD4+ T-cell proliferation by inducing indoleamine 2,3-dioxygenase in plasmacytoid dendritic cells. Blood 2007, 109, 3351–3359. [Google Scholar] [CrossRef]
- Gerlini, G.; Di Gennaro, P.; Mariotti, G.; Urso, C.; Chiarugi, A.; Pimpinelli, N.; Borgognoni, L. Indoleamine 2,3-dioxygenase+ cells correspond to the BDCA2+ plasmacytoid dendritic cells in human melanoma sentinel nodes. J. Investig. Dermatol. 2010, 130, 898–901. [Google Scholar] [CrossRef]
- Chen, W.; Liang, X.; Peterson, A.J.; Munn, D.H.; Blazar, B.R. The indoleamine 2,3-dioxygenase pathway is essential for human plasmacytoid dendritic cell-induced adaptive T regulatory cell generation. J. Immunol. 2008, 181, 5396–5404. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, R.; Terlizzi, M.; Di Crescenzo, V.G.; Popolo, A.; Pecoraro, M.; Perillo, G.; Galderisi, A.; Pinto, A. Human lung cancer-derived immunosuppressive plasmacytoid dendritic cells release IL-1alpha in an AIM2 inflammasome-dependent manner. Am. J. Pathol. 2015, 185, 3115–3124. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.R.; Zhu, Z.; Hansen, D.M.; Bai, Q.; Fang, Y. The role of IL-21 in immunity and cancer. Cancer Lett. 2015, 358, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Le Mercier, I.; Poujol, D.; Sanlaville, A.; Sisirak, V.; Gobert, M.; Durand, I.; Dubois, B.; Treilleux, I.; Marvel, J.; Vlach, J.; et al. Tumor promotion by intratumoral plasmacytoid dendritic cells is reversed by TLR7 ligand treatment. Cancer Res. 2013, 73, 4629–4640. [Google Scholar] [CrossRef]
- Lou, Y.; Liu, C.; Kim, G.J.; Liu, Y.J.; Hwu, P.; Wang, G. Plasmacytoid dendritic cells synergize with myeloid dendritic cells in the induction of antigen-specific antitumor immune responses. J. Immunol. 2007, 178, 1534–1541. [Google Scholar] [CrossRef]
- Borden, E.C. Interferons alpha and beta in cancer: Therapeutic opportunities from new insights. Nat. Rev. Drug Discovery 2019, 18, 219–234. [Google Scholar] [CrossRef]
- Wu, J.; Li, S.; Yang, Y.; Zhu, S.; Zhang, M.; Qiao, Y.; Liu, Y.J.; Chen, J. TLR-activated plasmacytoid dendritic cells inhibit breast cancer cell growth in vitro and in vivo. Oncotarget 2017, 8, 11708–11718. [Google Scholar] [CrossRef]
- Stary, G.; Bangert, C.; Tauber, M.; Strohal, R.; Kopp, T.; Stingl, G. Tumoricidal activity of TLR7/8-activated inflammatory dendritic cells. J. Exp. Med. 2007, 204, 1441–1451. [Google Scholar] [CrossRef]
- Shain, A.H.; Bastian, B.C. From melanocytes to melanomas. Nat. Rev. Cancer 2016, 16, 345–358. [Google Scholar] [CrossRef]
- Palmieri, G.; Capone, M.; Ascierto, M.L.; Gentilcore, G.; Stroncek, D.F.; Casula, M.; Sini, M.C.; Palla, M.; Mozzillo, N.; Ascierto, P.A. Main roads to melanoma. J. Transl. Med. 2009, 7, 86. [Google Scholar] [CrossRef]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Curtin, J.A.; Fridlyand, J.; Kageshita, T.; Patel, H.N.; Busam, K.J.; Kutzner, H.; Cho, K.H.; Aiba, S.; Brocker, E.B.; LeBoit, P.E.; et al. Distinct sets of genetic alterations in melanoma. N. Engl. J. Med. 2005, 353, 2135–2147. [Google Scholar] [CrossRef] [PubMed]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A landscape of driver mutations in melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef]
- Pfeifer, G.P. Environmental exposures and mutational patterns of cancer genomes. Genome Med. 2010, 2, 54. [Google Scholar] [CrossRef]
- Turajlic, S.; Litchfield, K.; Xu, H.; Rosenthal, R.; McGranahan, N.; Reading, J.L.; Wong, Y.N.S.; Rowan, A.; Kanu, N.; Al Bakir, M.; et al. Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: A pan-cancer analysis. Lancet Oncol. 2017, 18, 1009–1021. [Google Scholar] [CrossRef]
- Gubin, M.M.; Zhang, X.; Schuster, H.; Caron, E.; Ward, J.P.; Noguchi, T.; Ivanova, Y.; Hundal, J.; Arthur, C.D.; Krebber, W.J.; et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 2014, 515, 577–581. [Google Scholar] [CrossRef]
- Van Allen, E.M.; Miao, D.; Schilling, B.; Shukla, S.A.; Blank, C.; Zimmer, L.; Sucker, A.; Hillen, U.; Foppen, M.H.G.; Goldinger, S.M.; et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015, 350, 207–211. [Google Scholar] [CrossRef]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef]
- Linnebacher, M.; Gebert, J.; Rudy, W.; Woerner, S.; Yuan, Y.P.; Bork, P.; von Knebel Doeberitz, M. Frameshift peptide-derived T-cell epitopes: A source of novel tumor-specific antigens. Int. J. Cancer 2001, 93, 6–11. [Google Scholar] [CrossRef]
- Saleh, F.; Renno, W.; Klepacek, I.; Ibrahim, G.; Dashti, H.; Asfar, S.; Behbehani, A.; Al-Sayer, H.; Dashti, A. Direct evidence on the immune-mediated spontaneous regression of human cancer: An incentive for pharmaceutical companies to develop a novel anti-cancer vaccine. Curr. Pharm. Des. 2005, 11, 3531–3543. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Gupta, R.; Filipp, F.V. Cancer systems biology of TCGA SKCM: Efficient detection of genomic drivers in melanoma. Sci. Rep. 2015, 5, 7857. [Google Scholar] [CrossRef] [PubMed]
- Savage, P.A.; Leventhal, D.S.; Malchow, S. Shaping the repertoire of tumor-infiltrating effector and regulatory T cells. Immunol. Rev. 2014, 259, 245–258. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S. Mechanisms of tumor escape in the context of the T-cell-inflamed and the non-T-cell-inflamed tumor microenvironment. Int. Immunol. 2016, 28, 383–391. [Google Scholar] [CrossRef]
- Robbins, P.F.; Lu, Y.C.; El-Gamil, M.; Li, Y.F.; Gross, C.; Gartner, J.; Lin, J.C.; Teer, J.K.; Cliften, P.; Tycksen, E.; et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat. Med. 2013, 19, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Akbani, R.; Akdemir, K.C.; Aksoy, B.A.; Albert, M.; Ally, A.; Amin, S.B. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef]
- Spranger, S.; Luke, J.J.; Bao, R.; Zha, Y.; Hernandez, K.M.; Li, Y.; Gajewski, A.P.; Andrade, J.; Gajewski, T.F. Density of immunogenic antigens does not explain the presence or absence of the T-cell-inflamed tumor microenvironment in melanoma. Proc. Natl. Acad. Sci. USA 2016, 113, E7759–E7768. [Google Scholar] [CrossRef]
- Dabrosin, N.; Sloth Juul, K.; Baehr Georgsen, J.; Andrup, S.; Schmidt, H.; Steiniche, T.; Heide Ollegaard, T.; Bonnelykke Behrndtz, L. Innate immune cell infiltration in melanoma metastases affects survival and is associated with BRAFV600E mutation status. Melanoma Res. 2019, 29, 30–37. [Google Scholar] [CrossRef]
- Vescovi, R.; Monti, M.; Moratto, D.; Paolini, L.; Consoli, F.; Benerini, L.; Melocchi, L.; Calza, S.; Chiudinelli, M.; Rossi, G.; et al. Collapse of the Plasmacytoid Dendritic Cell Compartment in Advanced Cutaneous Melanomas by Components of the Tumor Cell Secretome. Cancer Immunol. Res. 2019, 7, 12–28. [Google Scholar] [CrossRef]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef]
- Gentles, A.J.; Newman, A.M.; Liu, C.L.; Bratman, S.V.; Feng, W.; Kim, D.; Nair, V.S.; Xu, Y.; Khuong, A.; Hoang, C.D.; et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med. 2015, 21, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Steen, C.B.; Liu, C.L.; Gentles, A.J.; Chaudhuri, A.A.; Scherer, F.; Khodadoust, M.S.; Esfahani, M.S.; Luca, B.A.; Steiner, D.; et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 2019, 37, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Korn, E.L.; Liu, P.Y.; Lee, S.J.; Chapman, J.A.; Niedzwiecki, D.; Suman, V.J.; Moon, J.; Sondak, V.K.; Atkins, M.B.; Eisenhauer, E.A.; et al. Meta-analysis of phase II cooperative group trials in metastatic stage IV melanoma to determine progression-free and overall survival benchmarks for future phase II trials. J. Clin. Oncol. 2008, 26, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Ugurel, S.; Rohmel, J.; Ascierto, P.A.; Flaherty, K.T.; Grob, J.J.; Hauschild, A.; Larkin, J.; Long, G.V.; Lorigan, P.; McArthur, G.A.; et al. Survival of patients with advanced metastatic melanoma: The impact of novel therapies-update 2017. Eur. J. Cancer 2017, 83, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Silva, I.P.; Long, G.V. Systemic therapy in advanced melanoma: Integrating targeted therapy and immunotherapy into clinical practice. Curr. Opin. Oncol 2017, 29, 484–492. [Google Scholar] [CrossRef]
- Luke, J.J.; Flaherty, K.T.; Ribas, A.; Long, G.V. Targeted agents and immunotherapies: Optimizing outcomes in melanoma. Nat. Rev. Clin. Oncol. 2017, 14, 463–482. [Google Scholar] [CrossRef]
- Garbe, C.; Amaral, T.; Peris, K.; Hauschild, A.; Arenberger, P.; Bastholt, L.; Bataille, V.; Del Marmol, V.; Dreno, B.; Fargnoli, M.C.; et al. European consensus-based interdisciplinary guideline for melanoma. Part 2: Treatment - Update 2019. Eur. J. Cancer 2019. [Google Scholar] [CrossRef]
- Dankner, M.; Rose, A.A.N.; Rajkumar, S.; Siegel, P.M.; Watson, I.R. Classifying BRAF alterations in cancer: New rational therapeutic strategies for actionable mutations. Oncogene 2018, 37, 3183–3199. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Puzanov, I.; Kim, K.B.; Ribas, A.; McArthur, G.A.; Sosman, J.A.; O’Dwyer, P.J.; Lee, R.J.; Grippo, J.F.; Nolop, K.; et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N. Engl. J. Med. 2010, 363, 809–819. [Google Scholar] [CrossRef]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef]
- Hauschild, A.; Grob, J.J.; Demidov, L.V.; Jouary, T.; Gutzmer, R.; Millward, M.; Rutkowski, P.; Blank, C.U.; Miller, W.H., Jr.; Kaempgen, E.; et al. Dabrafenib in BRAF-mutated metastatic melanoma: A multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012, 380, 358–365. [Google Scholar] [CrossRef]
- Robert, C.; Karaszewska, B.; Schachter, J.; Rutkowski, P.; Mackiewicz, A.; Stroiakovski, D.; Lichinitser, M.; Dummer, R.; Grange, F.; Mortier, L.; et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N. Engl. J. Med. 2015, 372, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N. Engl. J. Med. 2014, 371, 1877–1888. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; McArthur, G.A.; Dreno, B.; Atkinson, V.; Liszkay, G.; Di Giacomo, A.M.; Mandala, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Cobimetinib combined with vemurafenib in advanced BRAF(V600)-mutant melanoma (coBRIM): Updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol. 2016, 17, 1248–1260. [Google Scholar] [CrossRef]
- Dummer, R.; Ascierto, P.A.; Gogas, H.J.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; Gutzmer, R.; et al. Overall survival in patients with BRAF-mutant melanoma receiving encorafenib plus binimetinib versus vemurafenib or encorafenib (COLUMBUS): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2018, 19, 1315–1327. [Google Scholar] [CrossRef]
- Long, G.V.; Grob, J.J.; Nathan, P.; Ribas, A.; Robert, C.; Schadendorf, D.; Lane, S.R.; Mak, C.; Legenne, P.; Flaherty, K.T.; et al. Factors predictive of response, disease progression, and overall survival after dabrafenib and trametinib combination treatment: A pooled analysis of individual patient data from randomised trials. Lancet Oncol. 2016, 17, 1743–1754. [Google Scholar] [CrossRef]
- Robert, C.; Grob, J.J.; Stroyakovskiy, D.; Karaszewska, B.; Hauschild, A.; Levchenko, E.; Chiarion Sileni, V.; Schachter, J.; Garbe, C.; Bondarenko, I.; et al. Five-Year Outcomes with Dabrafenib plus Trametinib in Metastatic Melanoma. N. Engl. J. Med. 2019, 381, 626–636. [Google Scholar] [CrossRef]
- Yan, Y.; Wongchenko, M.J.; Robert, C.; Larkin, J.; Ascierto, P.A.; Dreno, B.; Maio, M.; Garbe, C.; Chapman, P.B.; Sosman, J.A.; et al. Genomic Features of Exceptional Response in Vemurafenib +/- Cobimetinib-treated Patients with BRAF (V600)-mutated Metastatic Melanoma. Clin. Cancer Res. 2019, 25, 3239–3246. [Google Scholar] [CrossRef]
- Freeman, G.J. Structures of PD-1 with its ligands: Sideways and dancing cheek to cheek. Proc. Natl. Acad. Sci. USA 2008, 105, 10275–10276. [Google Scholar] [CrossRef]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef]
- Schadendorf, D.; Hodi, F.S.; Robert, C.; Weber, J.S.; Margolin, K.; Hamid, O.; Patt, D.; Chen, T.T.; Berman, D.M.; Wolchok, J.D. Pooled Analysis of Long-Term Survival Data From Phase II and Phase III Trials of Ipilimumab in Unresectable or Metastatic Melanoma. J. Clin. Oncol. 2015, 33, 1889–1894. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Ribas, A.; Schachter, J.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.M.; Lotem, M.; et al. Pembrolizumab versus ipilimumab in advanced melanoma (KEYNOTE-006): Post-hoc 5-year results from an open-label, multicentre, randomised, controlled, phase 3 study. Lancet Oncol. 2019, 20, 1239–1251. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef]
- Eggermont, A.M.M.; Robert, C.; Ribas, A. The new era of adjuvant therapies for melanoma. Nat. Rev. Clin. Oncol. 2018, 15, 535–536. [Google Scholar] [CrossRef]
- Eggermont, A.M.M.; Blank, C.U.; Mandala, M.; Long, G.V.; Atkinson, V.; Dalle, S.; Haydon, A.; Lichinitser, M.; Khattak, A.; Carlino, M.S.; et al. Adjuvant Pembrolizumab versus Placebo in Resected Stage III Melanoma. N. Engl. J. Med. 2018, 378, 1789–1801. [Google Scholar] [CrossRef]
- Weber, J.; Mandala, M.; Del Vecchio, M.; Gogas, H.J.; Arance, A.M.; Cowey, C.L.; Dalle, S.; Schenker, M.; Chiarion-Sileni, V.; Marquez-Rodas, I.; et al. Adjuvant Nivolumab versus Ipilimumab in Resected Stage III or IV Melanoma. N. Engl. J. Med. 2017, 377, 1824–1835. [Google Scholar] [CrossRef]
- Long, G.V.; Hauschild, A.; Santinami, M.; Atkinson, V.; Mandala, M.; Chiarion-Sileni, V.; Larkin, J.; Nyakas, M.; Dutriaux, C.; Haydon, A.; et al. Adjuvant Dabrafenib plus Trametinib in Stage III BRAF-Mutated Melanoma. N. Engl. J. Med. 2017, 377, 1813–1823. [Google Scholar] [CrossRef]
- Robert, C. Is earlier better for melanoma checkpoint blockade? Nat. Med. 2018, 24, 1645–1648. [Google Scholar] [CrossRef] [PubMed]
- Blank, C.U.; Rozeman, E.A.; Fanchi, L.F.; Sikorska, K.; van de Wiel, B.; Kvistborg, P.; Krijgsman, O.; van den Braber, M.; Philips, D.; Broeks, A.; et al. Neoadjuvant versus adjuvant ipilimumab plus nivolumab in macroscopic stage III melanoma. Nat. Med. 2018, 24, 1655–1661. [Google Scholar] [CrossRef] [PubMed]
- Amaria, R.N.; Reddy, S.M.; Tawbi, H.A.; Davies, M.A.; Ross, M.I.; Glitza, I.C.; Cormier, J.N.; Lewis, C.; Hwu, W.J.; Hanna, E.; et al. Neoadjuvant immune checkpoint blockade in high-risk resectable melanoma. Nat. Med. 2018, 24, 1649–1654. [Google Scholar] [CrossRef] [PubMed]
- Amaria, R.N.; Prieto, P.A.; Tetzlaff, M.T.; Reuben, A.; Andrews, M.C.; Ross, M.I.; Glitza, I.C.; Cormier, J.; Hwu, W.J.; Tawbi, H.A.; et al. Neoadjuvant plus adjuvant dabrafenib and trametinib versus standard of care in patients with high-risk, surgically resectable melanoma: A single-centre, open-label, randomised, phase 2 trial. Lancet Oncol. 2018, 19, 181–193. [Google Scholar] [CrossRef]
- Mandala, M.; De Logu, F.; Merelli, B.; Nassini, R.; Massi, D. Immunomodulating property of MAPK inhibitors: From translational knowledge to clinical implementation. Lab. Investig. 2017, 97, 166–175. [Google Scholar] [CrossRef]
- Sumimoto, H.; Imabayashi, F.; Iwata, T.; Kawakami, Y. The BRAF-MAPK signaling pathway is essential for cancer-immune evasion in human melanoma cells. J. Exp. Med. 2006, 203, 1651–1656. [Google Scholar] [CrossRef]
- Ott, P.A.; Henry, T.; Baranda, S.J.; Frleta, D.; Manches, O.; Bogunovic, D.; Bhardwaj, N. Inhibition of both BRAF and MEK in BRAF(V600E) mutant melanoma restores compromised dendritic cell (DC) function while having differential direct effects on DC properties. Cancer Immunol. Immunother. 2013, 62, 811–822. [Google Scholar] [CrossRef]
- Khalili, J.S.; Liu, S.; Rodriguez-Cruz, T.G.; Whittington, M.; Wardell, S.; Liu, C.; Zhang, M.; Cooper, Z.A.; Frederick, D.T.; Li, Y.; et al. Oncogenic BRAF(V600E) promotes stromal cell-mediated immunosuppression via induction of interleukin-1 in melanoma. Clin. Cancer Res. 2012, 18, 5329–5340. [Google Scholar] [CrossRef]
- Bradley, S.D.; Chen, Z.; Melendez, B.; Talukder, A.; Khalili, J.S.; Rodriguez-Cruz, T.; Liu, S.; Whittington, M.; Deng, W.; Li, F.; et al. BRAFV600E Co-opts a Conserved MHC Class I Internalization Pathway to Diminish Antigen Presentation and CD8+ T-cell Recognition of Melanoma. Cancer Immunol. Res. 2015, 3, 602–609. [Google Scholar] [CrossRef]
- Frederick, D.T.; Piris, A.; Cogdill, A.P.; Cooper, Z.A.; Lezcano, C.; Ferrone, C.R.; Mitra, D.; Boni, A.; Newton, L.P.; Liu, C.; et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin. Cancer Res. 2013, 19, 1225–1231. [Google Scholar] [CrossRef]
- Wilmott, J.S.; Long, G.V.; Howle, J.R.; Haydu, L.E.; Sharma, R.N.; Thompson, J.F.; Kefford, R.F.; Hersey, P.; Scolyer, R.A. Selective BRAF inhibitors induce marked T-cell infiltration into human metastatic melanoma. Clin. Cancer Res. 2012, 18, 1386–1394. [Google Scholar] [CrossRef] [PubMed]
- Spagnolo, F.; Ghiorzo, P.; Orgiano, L.; Pastorino, L.; Picasso, V.; Tornari, E.; Ottaviano, V.; Queirolo, P. BRAF-mutant melanoma: Treatment approaches, resistance mechanisms, and diagnostic strategies. Onco. Targets Ther. 2015, 8, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Young, H.L.; Rowling, E.J.; Bugatti, M.; Giurisato, E.; Luheshi, N.; Arozarena, I.; Acosta, J.C.; Kamarashev, J.; Frederick, D.T.; Cooper, Z.A.; et al. An adaptive signaling network in melanoma inflammatory niches confers tolerance to MAPK signaling inhibition. J. Exp. Med. 2017, 214, 1691–1710. [Google Scholar] [CrossRef]
- Cooper, Z.A.; Juneja, V.R.; Sage, P.T.; Frederick, D.T.; Piris, A.; Mitra, D.; Lo, J.A.; Hodi, F.S.; Freeman, G.J.; Bosenberg, M.W.; et al. Response to BRAF inhibition in melanoma is enhanced when combined with immune checkpoint blockade. Cancer Immunol. Res. 2014, 2, 643–654. [Google Scholar] [CrossRef] [PubMed]
- Hu-Lieskovan, S.; Mok, S.; Homet Moreno, B.; Tsoi, J.; Robert, L.; Goedert, L.; Pinheiro, E.M.; Koya, R.C.; Graeber, T.G.; Comin-Anduix, B.; et al. Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in BRAF(V600E) melanoma. Sci. Transl. Med. 2015, 7, 279ra241. [Google Scholar] [CrossRef] [PubMed]
- Kono, M.; Dunn, I.S.; Durda, P.J.; Butera, D.; Rose, L.B.; Haggerty, T.J.; Benson, E.M.; Kurnick, J.T. Role of the mitogen-activated protein kinase signaling pathway in the regulation of human melanocytic antigen expression. Mol. Cancer Res. 2006, 4, 779–792. [Google Scholar] [CrossRef]
- Boni, A.; Cogdill, A.P.; Dang, P.; Udayakumar, D.; Njauw, C.N.; Sloss, C.M.; Ferrone, C.R.; Flaherty, K.T.; Lawrence, D.P.; Fisher, D.E.; et al. Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer Res. 2010, 70, 5213–5219. [Google Scholar] [CrossRef]
- Vella, L.J.; Pasam, A.; Dimopoulos, N.; Andrews, M.; Knights, A.; Puaux, A.L.; Louahed, J.; Chen, W.; Woods, K.; Cebon, J.S. MEK inhibition, alone or in combination with BRAF inhibition, affects multiple functions of isolated normal human lymphocytes and dendritic cells. Cancer Immunol. Res. 2014, 2, 351–360. [Google Scholar] [CrossRef]
- Ebert, P.J.R.; Cheung, J.; Yang, Y.; McNamara, E.; Hong, R.; Moskalenko, M.; Gould, S.E.; Maecker, H.; Irving, B.A.; Kim, J.M.; et al. MAP Kinase Inhibition Promotes T Cell and Anti-tumor Activity in Combination with PD-L1 Checkpoint Blockade. Immunity 2016, 44, 609–621. [Google Scholar] [CrossRef]
- Cooper, Z.A.; Reuben, A.; Austin-Breneman, J.; Wargo, J.A. Does It MEK a Difference? Understanding Immune Effects of Targeted Therapy. Clin. Cancer Res. 2015, 21, 3102–3104. [Google Scholar] [CrossRef]
- Salio, M.; Cella, M.; Vermi, W.; Facchetti, F.; Palmowski, M.J.; Smith, C.L.; Shepherd, D.; Colonna, M.; Cerundolo, V. Plasmacytoid dendritic cells prime IFN-gamma-secreting melanoma-specific CD8 lymphocytes and are found in primary melanoma lesions. Eur. J. Immunol. 2003, 33, 1052–1062. [Google Scholar] [CrossRef] [PubMed]
- Charles, J.; Di Domizio, J.; Salameire, D.; Bendriss-Vermare, N.; Aspord, C.; Muhammad, R.; Lefebvre, C.; Plumas, J.; Leccia, M.T.; Chaperot, L. Characterization of circulating dendritic cells in melanoma: Role of CCR6 in plasmacytoid dendritic cell recruitment to the tumor. J. Investig. Dermatol. 2010, 130, 1646–1656. [Google Scholar] [CrossRef] [PubMed]
- Gerlini, G.; Urso, C.; Mariotti, G.; Di Gennaro, P.; Palli, D.; Brandani, P.; Salvadori, A.; Pimpinelli, N.; Reali, U.M.; Borgognoni, L. Plasmacytoid dendritic cells represent a major dendritic cell subset in sentinel lymph nodes of melanoma patients and accumulate in metastatic nodes. Clin. Immunol. 2007, 125, 184–193. [Google Scholar] [CrossRef] [PubMed]
- van den Hout, M.; Koster, B.D.; Sluijter, B.J.R.; Molenkamp, B.G.; van de Ven, R.; van den Eertwegh, A.J.M.; Scheper, R.J.; van Leeuwen, P.A.M.; van den Tol, M.P.; de Gruijl, T.D. Melanoma Sequentially Suppresses Different DC Subsets in the Sentinel Lymph Node, Affecting Disease Spread and Recurrence. Cancer Immunol. Res. 2017, 5, 969–977. [Google Scholar] [CrossRef]
- Jensen, T.O.; Schmidt, H.; Moller, H.J.; Donskov, F.; Hoyer, M.; Sjoegren, P.; Christensen, I.J.; Steiniche, T. Intratumoral neutrophils and plasmacytoid dendritic cells indicate poor prognosis and are associated with pSTAT3 expression in AJCC stage I/II melanoma. Cancer 2012, 118, 2476–2485. [Google Scholar] [CrossRef]
- Chevolet, I.; Speeckaert, R.; Schreuer, M.; Neyns, B.; Krysko, O.; Bachert, C.; Van Gele, M.; van Geel, N.; Brochez, L. Clinical significance of plasmacytoid dendritic cells and myeloid-derived suppressor cells in melanoma. J. Transl. Med. 2015, 13, 9. [Google Scholar] [CrossRef]
- Failli, A.; Legitimo, A.; Orsini, G.; Romanini, A.; Consolini, R. Numerical defect of circulating dendritic cell subsets and defective dendritic cell generation from monocytes of patients with advanced melanoma. Cancer Lett. 2013, 337, 184–192. [Google Scholar] [CrossRef]
- Wenzel, J.; Bekisch, B.; Uerlich, M.; Haller, O.; Bieber, T.; Tuting, T. Type I interferon-associated recruitment of cytotoxic lymphocytes: A common mechanism in regressive melanocytic lesions. Am. J. Clin. Pathol. 2005, 124, 37–48. [Google Scholar] [CrossRef]
- McCarter, M.D.; Baumgartner, J.; Escobar, G.A.; Richter, D.; Lewis, K.; Robinson, W.; Wilson, C.; Palmer, B.E.; Gonzalez, R. Immunosuppressive dendritic and regulatory T cells are upregulated in melanoma patients. Ann. Surg. Oncol. 2007, 14, 2854–2860. [Google Scholar] [CrossRef]
- Boiocchi, L.; Lonardi, S.; Vermi, W.; Fisogni, S.; Facchetti, F. BDCA-2 (CD303): A highly specific marker for normal and neoplastic plasmacytoid dendritic cells. Blood 2013, 122, 296–297. [Google Scholar] [CrossRef]
- Camisaschi, C.; De Filippo, A.; Beretta, V.; Vergani, B.; Villa, A.; Vergani, E.; Santinami, M.; Cabras, A.D.; Arienti, F.; Triebel, F.; et al. Alternative activation of human plasmacytoid DCs in vitro and in melanoma lesions: Involvement of LAG-3. J. Investig. Dermatol. 2014, 134, 1893–1902. [Google Scholar] [CrossRef]
- Zitvogel, L.; Galluzzi, L.; Kepp, O.; Smyth, M.J.; Kroemer, G. Type I interferons in anticancer immunity. Nat. Rev. Immunol. 2015, 15, 405–414. [Google Scholar] [CrossRef]
- Asmana Ningrum, R. Human interferon alpha-2b: A therapeutic protein for cancer treatment. Scientifica (Cairo) 2014, 2014, 970315. [Google Scholar] [CrossRef] [PubMed]
- Atkins, M.B.; Hodi, F.S.; Thompson, J.A.; McDermott, D.F.; Hwu, W.J.; Lawrence, D.P.; Dawson, N.A.; Wong, D.J.; Bhatia, S.; James, M.; et al. Pembrolizumab Plus Pegylated Interferon alfa-2b or Ipilimumab for Advanced Melanoma or Renal Cell Carcinoma: Dose-Finding Results from the Phase Ib KEYNOTE-029 Study. Clin. Cancer Res. 2018, 24, 1805–1815. [Google Scholar] [CrossRef] [PubMed]
- Chevolet, I.; Schreuer, M.; Speeckaert, R.; Neyns, B.; Hoorens, I.; van Geel, N.; Kruse, V.; Hennart, B.; Allorge, D.; Van Gele, M.; et al. Systemic immune changes associated with adjuvant interferon-alpha2b-therapy in stage III melanoma patients: Failure at the effector phase? Melanoma Res. 2015, 25, 357–361. [Google Scholar] [CrossRef]
- Lou, Y.; Liu, C.; Lizee, G.; Peng, W.; Xu, C.; Ye, Y.; Rabinovich, B.A.; Hailemichael, Y.; Gelbard, A.; Zhou, D.; et al. Antitumor activity mediated by CpG: The route of administration is critical. J. Immunother. 2011, 34, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Gungor, B.; Yagci, F.C.; Tincer, G.; Bayyurt, B.; Alpdundar, E.; Yildiz, S.; Ozcan, M.; Gursel, I.; Gursel, M. CpG ODN nanorings induce IFNalpha from plasmacytoid dendritic cells and demonstrate potent vaccine adjuvant activity. Sci. Transl. Med. 2014, 6, 235ra261. [Google Scholar] [CrossRef]
- Kim, Y.H.; Gratzinger, D.; Harrison, C.; Brody, J.D.; Czerwinski, D.K.; Ai, W.Z.; Morales, A.; Abdulla, F.; Xing, L.; Navi, D.; et al. In situ vaccination against mycosis fungoides by intratumoral injection of a TLR9 agonist combined with radiation: A phase 1/2 study. Blood 2012, 119, 355–363. [Google Scholar] [CrossRef]
- Kneme, S. Warming “Cold” Melanoma with TLR9 Agonists. Cancer Discov. 2018, 8, 670. [Google Scholar]
- Krieg, A.M. Therapeutic potential of Toll-like receptor 9 activation. Nat. Rev. Drug Discov. 2006, 5, 471–484. [Google Scholar] [CrossRef]
- Pashenkov, M.; Goess, G.; Wagner, C.; Hormann, M.; Jandl, T.; Moser, A.; Britten, C.M.; Smolle, J.; Koller, S.; Mauch, C.; et al. Phase II trial of a toll-like receptor 9-activating oligonucleotide in patients with metastatic melanoma. J. Clin. Oncol. 2006, 24, 5716–5724. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, M.A.; Kors, C.; Audring, H.; Walden, P.; Sterry, W.; Trefzer, U. Phase 1 evaluation of intralesionally injected TLR9-agonist PF-3512676 in patients with basal cell carcinoma or metastatic melanoma. J. Immunother. 2008, 31, 520–527. [Google Scholar] [CrossRef] [PubMed]
- Aspord, C.; Tramcourt, L.; Leloup, C.; Molens, J.P.; Leccia, M.T.; Charles, J.; Plumas, J. Imiquimod inhibits melanoma development by promoting pDC cytotoxic functions and impeding tumor vascularization. J. Investig. Dermatol. 2014, 134, 2551–2561. [Google Scholar] [CrossRef] [PubMed]
- Teulings, H.E.; Tjin, E.P.M.; Willemsen, K.J.; van der Kleij, S.; Ter Meulen, S.; Kemp, E.H.; Krebbers, G.; van Noesel, C.J.M.; Franken, C.; Drijfhout, J.W.; et al. Anti-Melanoma immunity and local regression of cutaneous metastases in melanoma patients treated with monobenzone and imiquimod; a phase 2 a trial. Oncoimmunology 2018, 7, e1419113. [Google Scholar] [CrossRef] [PubMed]
- Shackleton, M.; Davis, I.D.; Hopkins, W.; Jackson, H.; Dimopoulos, N.; Tai, T.; Chen, Q.; Parente, P.; Jefford, M.; Masterman, K.A.; et al. The impact of imiquimod, a Toll-like receptor-7 ligand (TLR7L), on the immunogenicity of melanoma peptide vaccination with adjuvant Flt3 ligand. Cancer Immun. 2004, 4, 9. [Google Scholar]
- Adams, S.; O’Neill, D.W.; Nonaka, D.; Hardin, E.; Chiriboga, L.; Siu, K.; Cruz, C.M.; Angiulli, A.; Angiulli, F.; Ritter, E.; et al. Immunization of malignant melanoma patients with full-length NY-ESO-1 protein using TLR7 agonist imiquimod as vaccine adjuvant. J. Immunol. 2008, 181, 776–784. [Google Scholar] [CrossRef]
- Tel, J.; Aarntzen, E.H.; Baba, T.; Schreibelt, G.; Schulte, B.M.; Benitez-Ribas, D.; Boerman, O.C.; Croockewit, S.; Oyen, W.J.; van Rossum, M.; et al. Natural human plasmacytoid dendritic cells induce antigen-specific T-cell responses in melanoma patients. Cancer Res. 2013, 73, 1063–1075. [Google Scholar] [CrossRef] [PubMed]
- Aspord, C.; Charles, J.; Leccia, M.T.; Laurin, D.; Richard, M.J.; Chaperot, L.; Plumas, J. A novel cancer vaccine strategy based on HLA-A*0201 matched allogeneic plasmacytoid dendritic cells. PLoS ONE 2010, 5, e10458. [Google Scholar] [CrossRef]
- Aspord, C.; Leccia, M.T.; Salameire, D.; Laurin, D.; Chaperot, L.; Charles, J.; Plumas, J. HLA-A(*)0201(+) plasmacytoid dendritic cells provide a cell-based immunotherapy for melanoma patients. J. Investig. Dermatol. 2012, 132, 2395–2406. [Google Scholar] [CrossRef]
- Long, G.V.; Atkinson, V.; Cebon, J.S.; Jameson, M.B.; Fitzharris, B.M.; McNeil, C.M.; Hill, A.G.; Ribas, A.; Atkins, M.B.; Thompson, J.A.; et al. Standard-dose pembrolizumab in combination with reduced-dose ipilimumab for patients with advanced melanoma (KEYNOTE-029): An open-label, phase 1b trial. Lancet Oncol. 2017, 18, 1202–1210. [Google Scholar] [CrossRef]






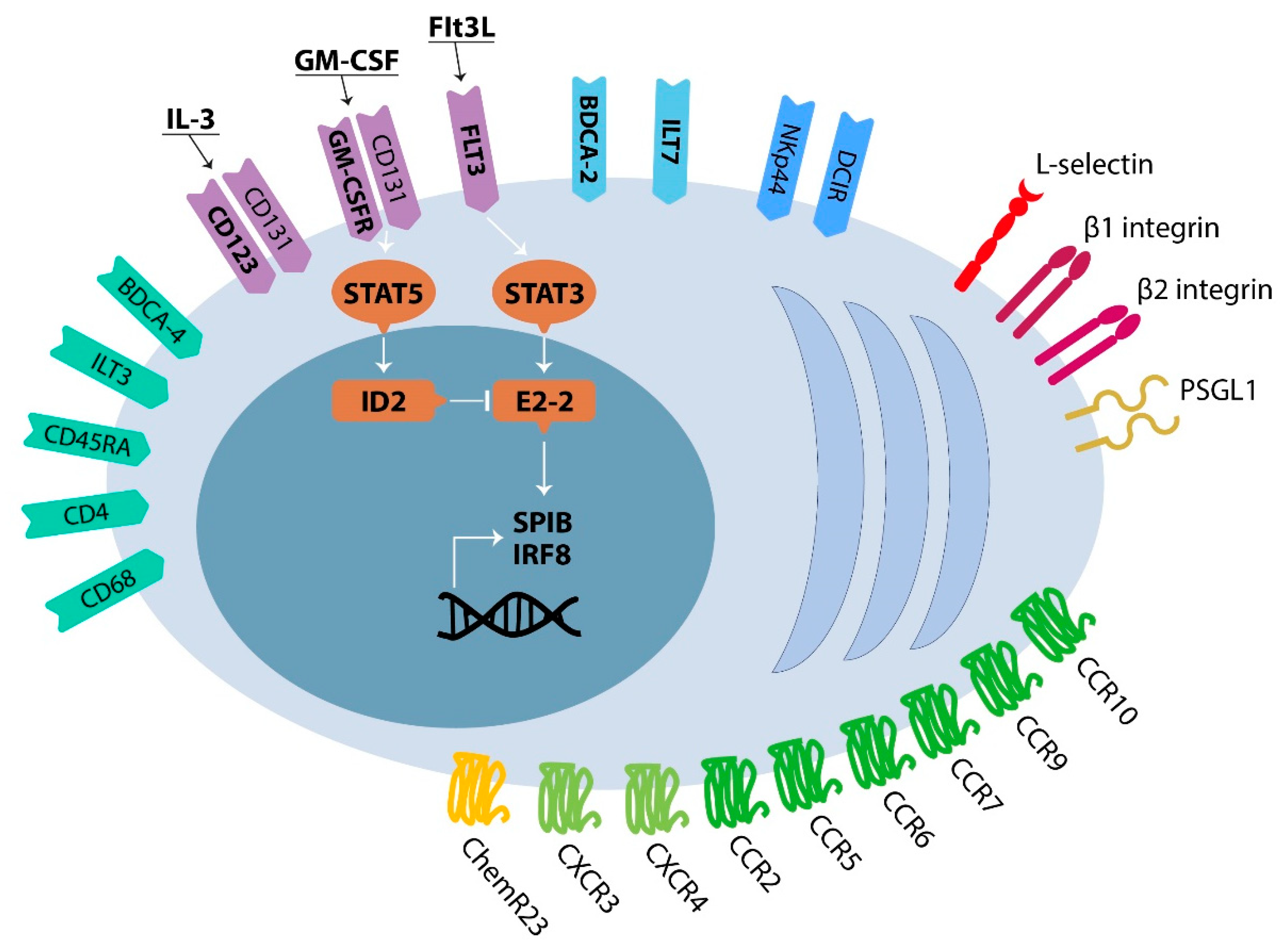{kind=link}
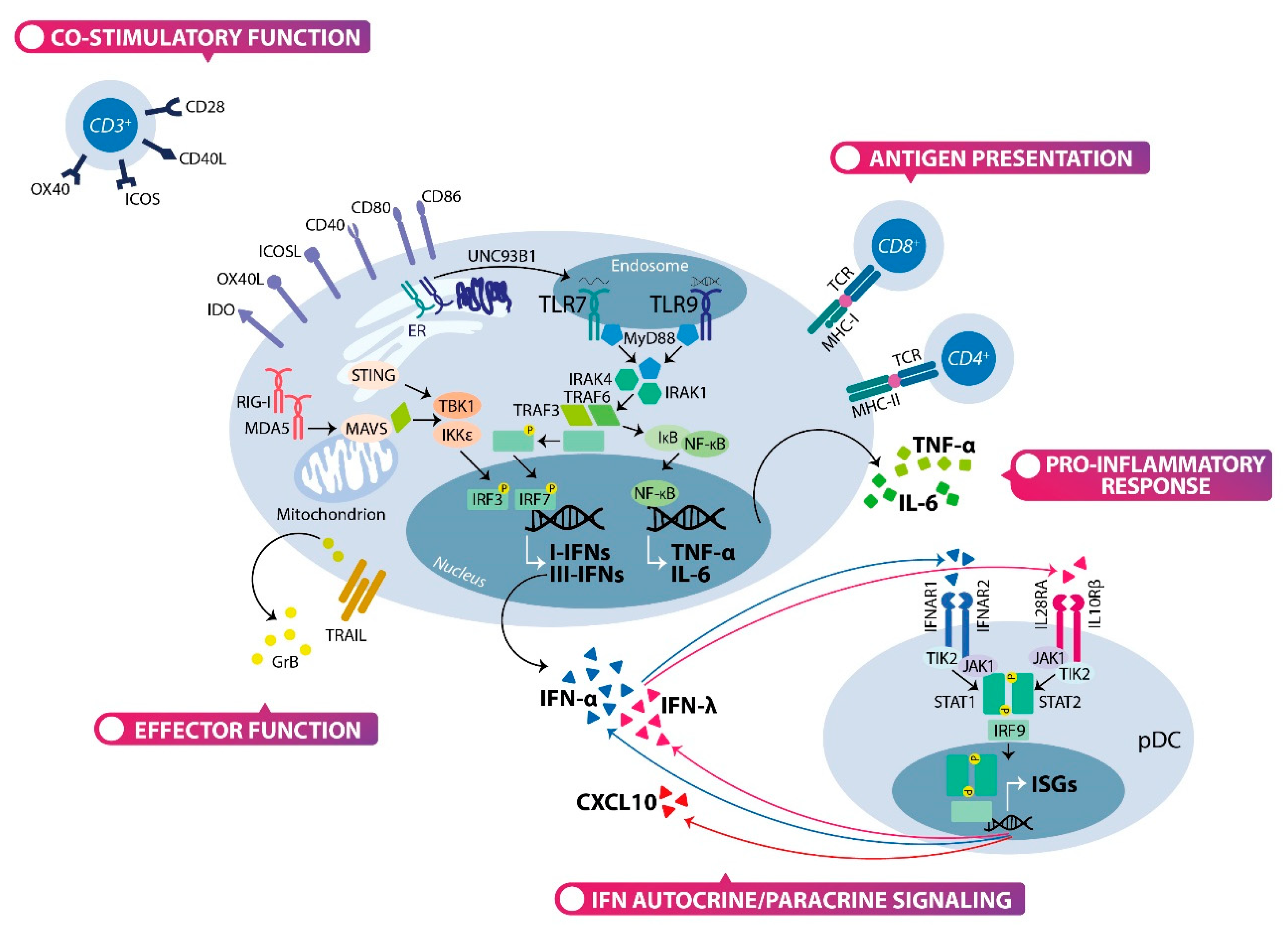{kind=link}
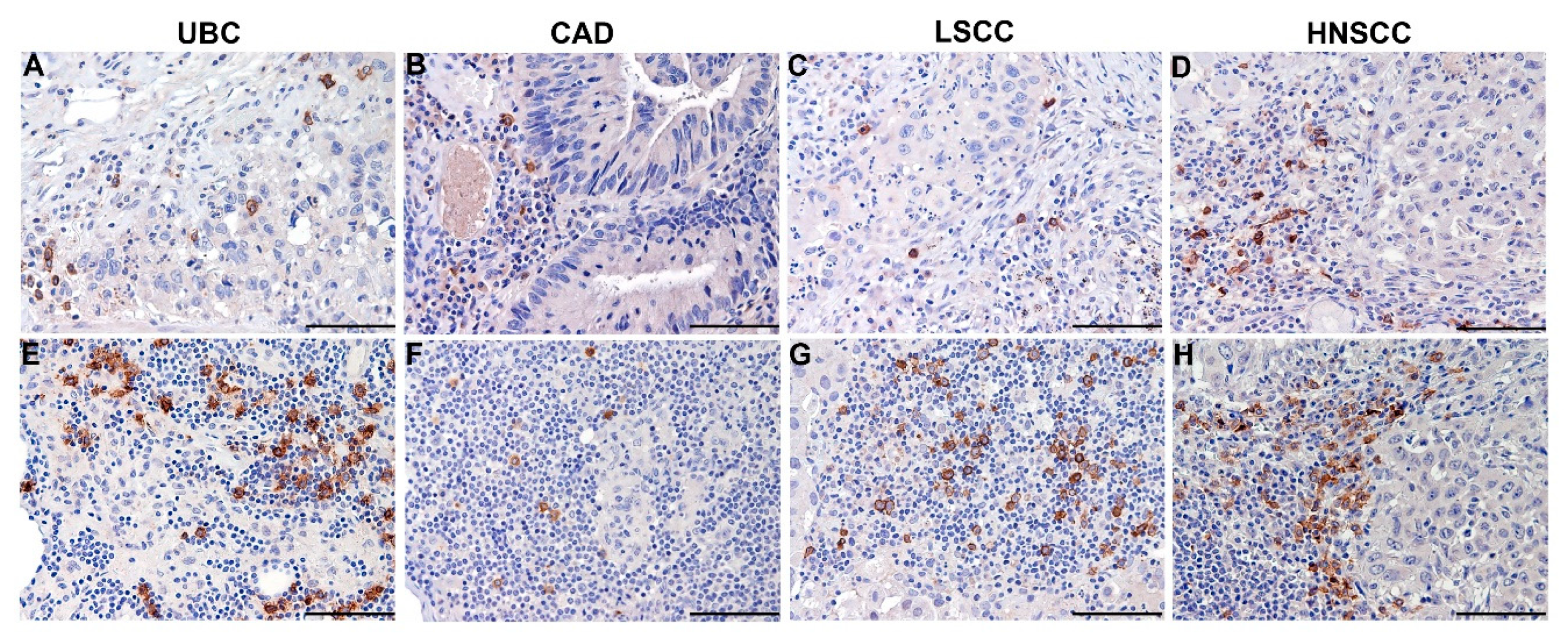{kind=link}
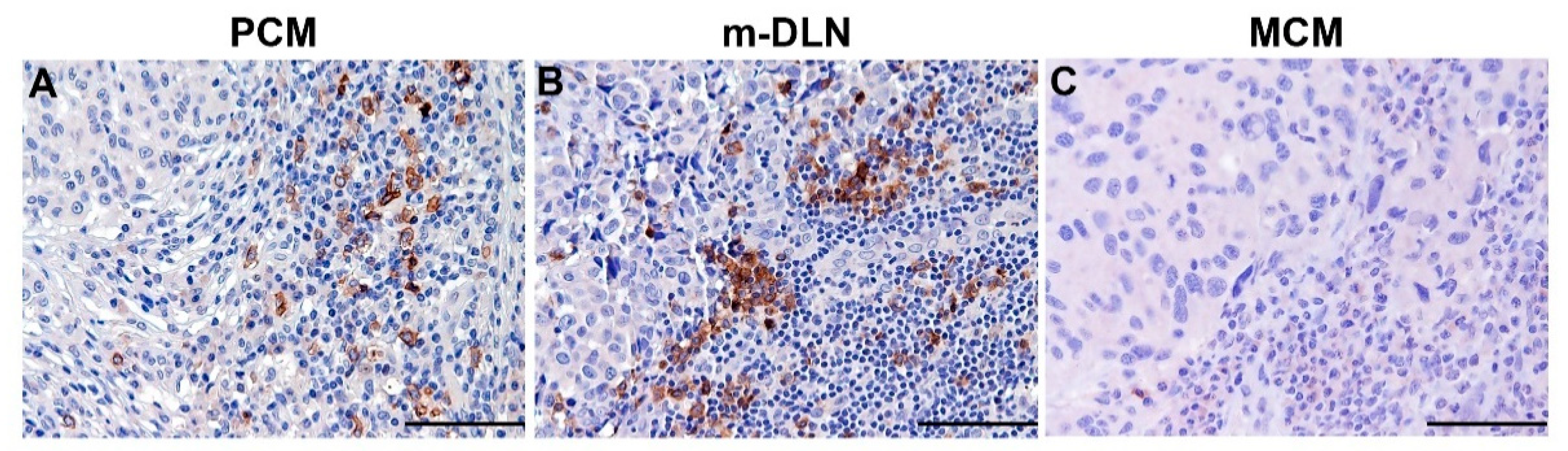{kind=link}
{kind=link}
| Patients’ Sample (N°) | pDC Source | Detection Method | pDC Frequency | pDC Functional State * | Clinical Significance ** | Reference |
|---|---|---|---|---|---|---|
| PCM (5) | FF | IHC; marker: CD123+ BDCA2+ | Increased in PCM vs. sk/nevi | NA | NA | [261] |
| PCM (15); SLN− (2); SLN+ (2) | FF; FFPE | IHC; marker: CD123+ BDCA2+ | Increased in PCM vs. sk/nevi | Defective | NA | [15] |
| r-PCM (14) | CF | IHC; marker: BDCA2+ | Increased in PCM vs. sk | Increased in early-intermediate vs. late r-PCM | Regression of melanoma | [268] |
| SLN− (5); SLN+ (5); mLN (5) | FFPE | IHC; marker: CD123+ | Increased in mLN | - | NA | [263] |
| SLN− (19); SLN+ (5); mLN (5) | FF | IF; marker: BDCA2+ | - | Defective | NA | [263] |
| SLN− (31); SLN+ (8); mLN (9) | Cell suspension | FC; marker: CD123+ BDCA2+ | Increased in SLN+/mLN vs. SLN− | - | NA | [263] |
| SLN− (31); SLN+ (8); mLN (9) | Cell suspension | ELISA | - | Defective | NA | [263] |
| PCM (186) | FFPE | IHC; marker: CD123+ | - | NA | Negative (PFS, OS) | [265] |
| SLN (36) | Cell suspension | FC; marker: CD123+ BDCA2+ | Unchanged in stage I/III | NA | NA | [264] |
| PCM (397); SLN (71); MCM (25) | FFPE | IHC; marker: CD123+ | Decreased in MCM vs. PCM; Increased in BRAFV600E vs. BRAFwt | NA | None | [208] |
| PCM (12); mLN (28) | Cell suspension | FC; marker: CD123+ BDCA2+ | Decreased in mLN vs. PCM | Functional | Negative (OS) | [157] |
| Stage I-IV melanoma | Blood | FC; marker: CD123+ BDCA2+ | Decreased in stage III-IV vs. I-II | Unchanged | NA | [157] |
| PCM (101); SLN (33); MCM (60) | FFPE | IHC; marker: BDCA2+ | Increased in PCM vs. nevi; Increased in SLN vs. PCM; Unchanged in SLN+ vs. SLN−; Decreased in MCM vs. PCM | NA | None | [209] |
| MM (29) | Blood | FC; marker: CD123+ BDCA2+ | Decreased in MM vs. HD | NA | NA | [209] |
| Stage I and IV melanoma | Blood | FC; marker: CD123+ | Increased in stage I vs. HD | NA | NA | [269] |
| Stage I-IV melanoma (69) | Blood | FC; marker: CD123+ | Decreased in stage IV vs. stage I-III | NA | Positive (PFS, OS) | [266] |
| Stage I-IV melanoma (35) | Blood | FC; marker: CD123+ | Decreased in PT vs. HD; Decreased in stage IV vs. I-II | NA | NA | [267] |
| Therapeutic Approach | Compounds | Type of Study | Number of Patients | Reference |
|---|---|---|---|---|
| IFN-α therapy + CPi | IFNα-2b + pembrolizumab | Phase 1 | 30 * | NCT02339324 |
| IFN-α therapy + CPi | PegIFN-2b + pembrolizumab | Phase 1/2 | 293 *† | NCT02089685 [274,290] |
| TLR9-agonist + CPi | IMO-2125 + ipilimumab or pembrolizumab | Phase 1/2 | 53 | NCT02644967 |
| TLR9-agonist + CPi | IMO-2125 + ipilimumab | Phase 3 | 454 * | NCT03445533 |
| TLR9-agonist | IMO-2125 | Phase 1 | 54 † | NCT03052205 |
| TLR9-agonist + CPi | CMP-001 + pembrolizumab | Phase 1 | 106 * | NCT03084640 |
| TLR9-agonist + CPi | CMP-001 + nivolumab | Phase 2 | 32 * | NCT03618641 |
| TLR9-agonist + CPi | CMP-001 + pembrolizumab | Phase 1 | 199 * | NCT02680184 |
| TLR9-agonist + CPi | SD-101 + pembrolizumab | Phase 1/2 | 227 † | NCT02521870 |
| TLR9-agonist | PF-3512676 | Phase 2 | 20 | [281] |
| TLR9-agonist | PF-3512676 | Phase 1 | 10 † | [282] |
| TLR7-agonist | IMQ + monobenzone | Phase 2 | 25 | [284] |
| TLR7-agonist + peptide-base vaccination | IMQ + NY-ESO-1 | Phase 1 | 9 | NCT00142454 [286] |
| pDC-based vaccination | HLA-A2.1+ pDC or mDC | Phase 1 | 30 | NCT01690377 [287] |
| pDC-based vaccination | HLA-A0201+ pDC | Pre-clinical | 12 | [288] |
| pDC-based vaccination | HLA-A0201+ pDC | Pre-clinical | 16 | [289] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Monti, M.; Consoli, F.; Vescovi, R.; Bugatti, M.; Vermi, W. Human Plasmacytoid Dendritic Cells and Cutaneous Melanoma. Cells 2020, 9, 417. https://doi.org/10.3390/cells9020417
Monti M, Consoli F, Vescovi R, Bugatti M, Vermi W. Human Plasmacytoid Dendritic Cells and Cutaneous Melanoma. Cells. 2020; 9(2):417. https://doi.org/10.3390/cells9020417
Chicago/Turabian StyleMonti, Matilde, Francesca Consoli, Raffaella Vescovi, Mattia Bugatti, and William Vermi. 2020. "Human Plasmacytoid Dendritic Cells and Cutaneous Melanoma" Cells 9, no. 2: 417. https://doi.org/10.3390/cells9020417
APA StyleMonti, M., Consoli, F., Vescovi, R., Bugatti, M., & Vermi, W. (2020). Human Plasmacytoid Dendritic Cells and Cutaneous Melanoma. Cells, 9(2), 417. https://doi.org/10.3390/cells9020417