Dopamine Receptor Activation Modulates the Integrity of the Perisynaptic Extracellular Matrix at Excitatory Synapses

 , , ,
, , ,  , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Antibodies and Drugs
2.2. Primary Neuronal Cultures
2.3. Immunocytochemistry
2.4. Design of shRNA Plasmids
2.5. siRNA Sequences
2.6. Generation of Adeno-Associated Viral Particles
2.7. Knockdown of ECM-Modifying Proteases Using shRNA
2.8. Optogenetic Modulation of cAMP in Dissociated Cortical Neurons
2.9. Cell Lysis
2.10. In Vivo Pharmacology and Subcellular Brain Fractionation
2.11. SDS-PAGE and Western Blot
2.12. Statistical Analysis
2.13. Ethical Statement
3. Results
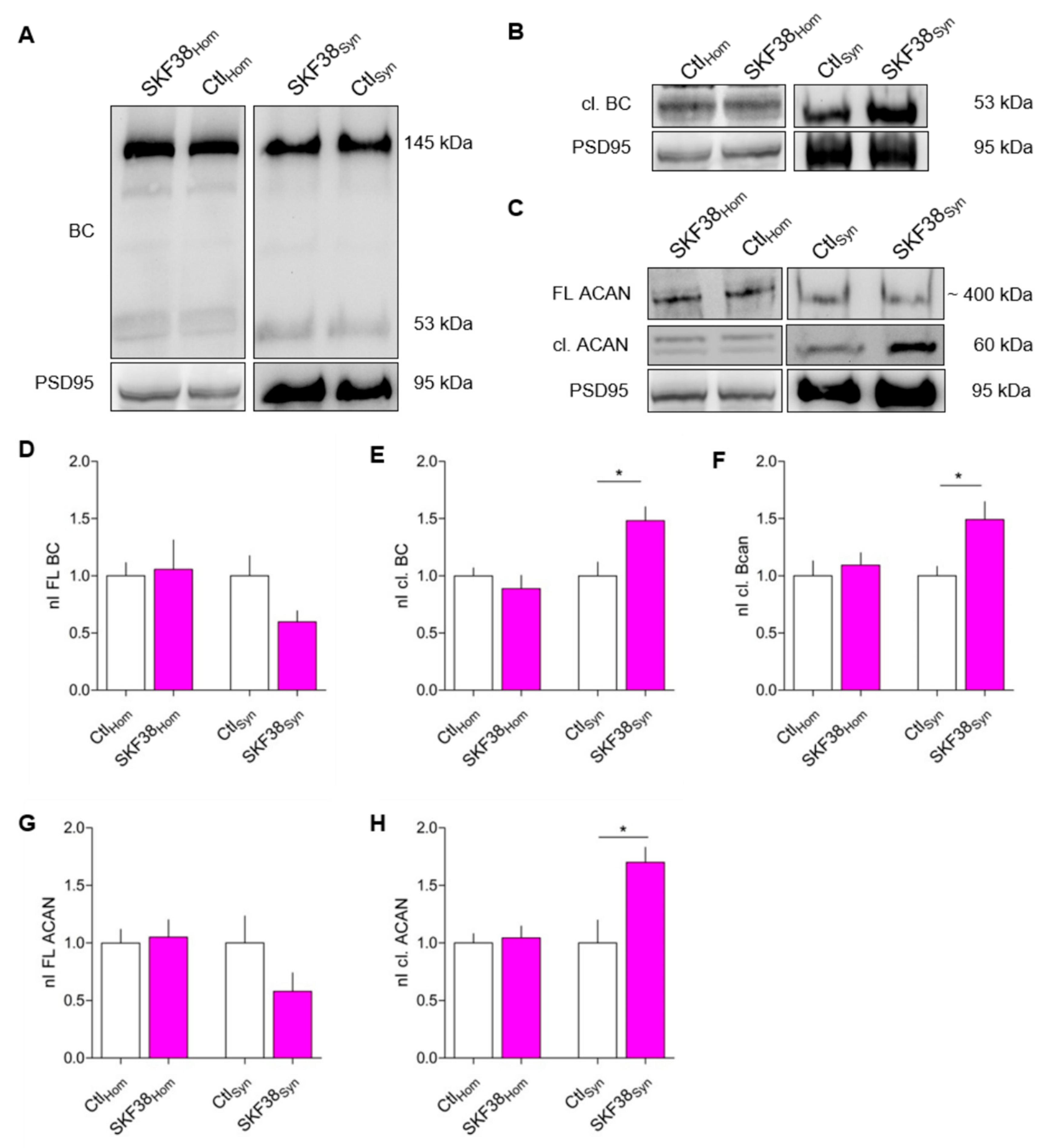
3.1. Increased BC Cleavage in Synaptosomes After D1-like DA Receptor Activation In Vivo
3.2. D1 DA Receptors are Prominently Expressed in Rat Dissociated Cortical Cultures
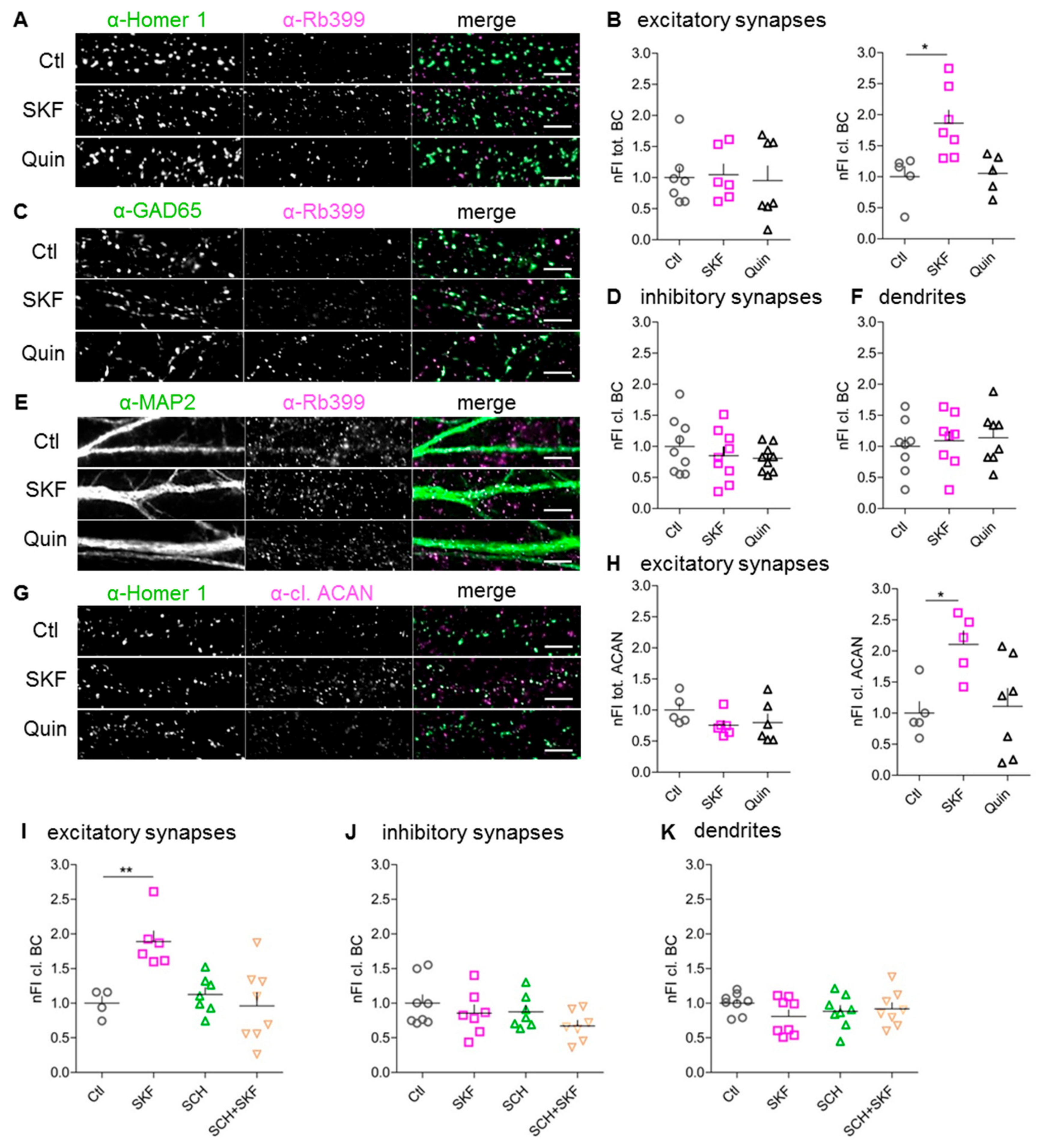
3.3. Stimulation of D1-Like, But Not D2-Like, DA Receptors Augments Perisynaptic BC Cleavage
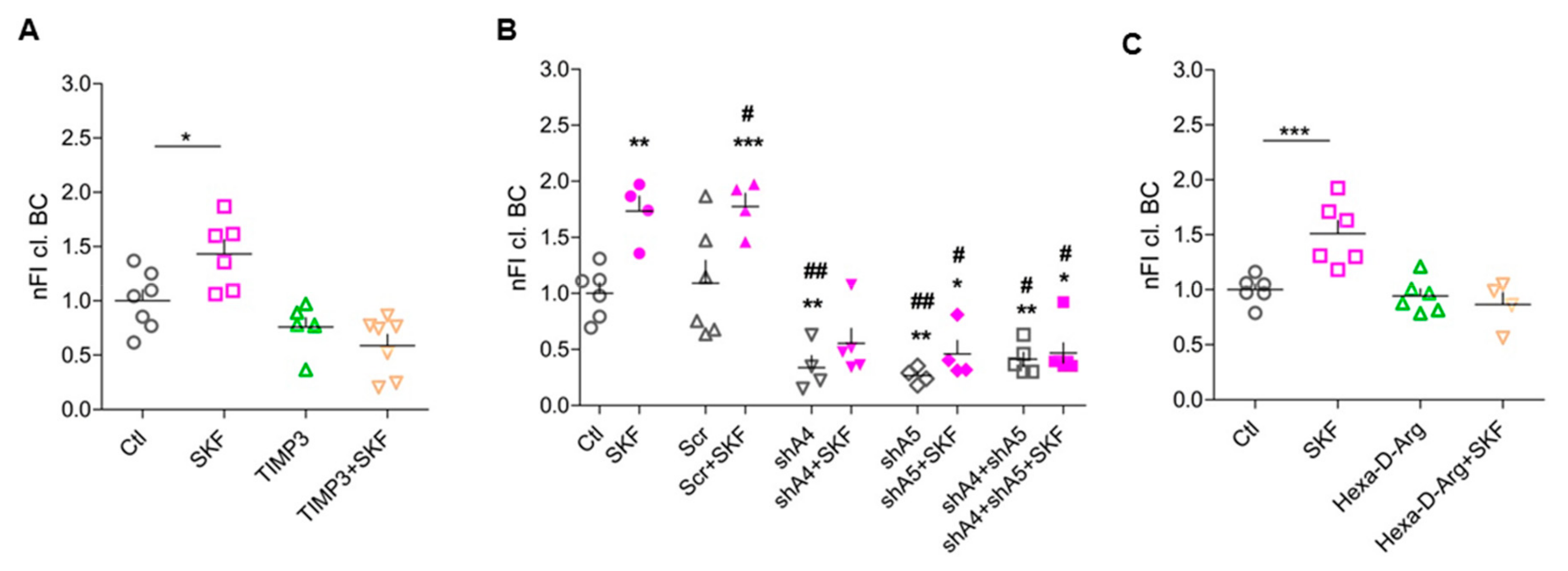
3.4. ADAMTS-4 and ADAMTS-5 are Essential for SKF81297-Induced BC Cleavage
3.5. Pro-Protein Furin-Like Convertases Are Involved in DA-Dependent BC Cleavage
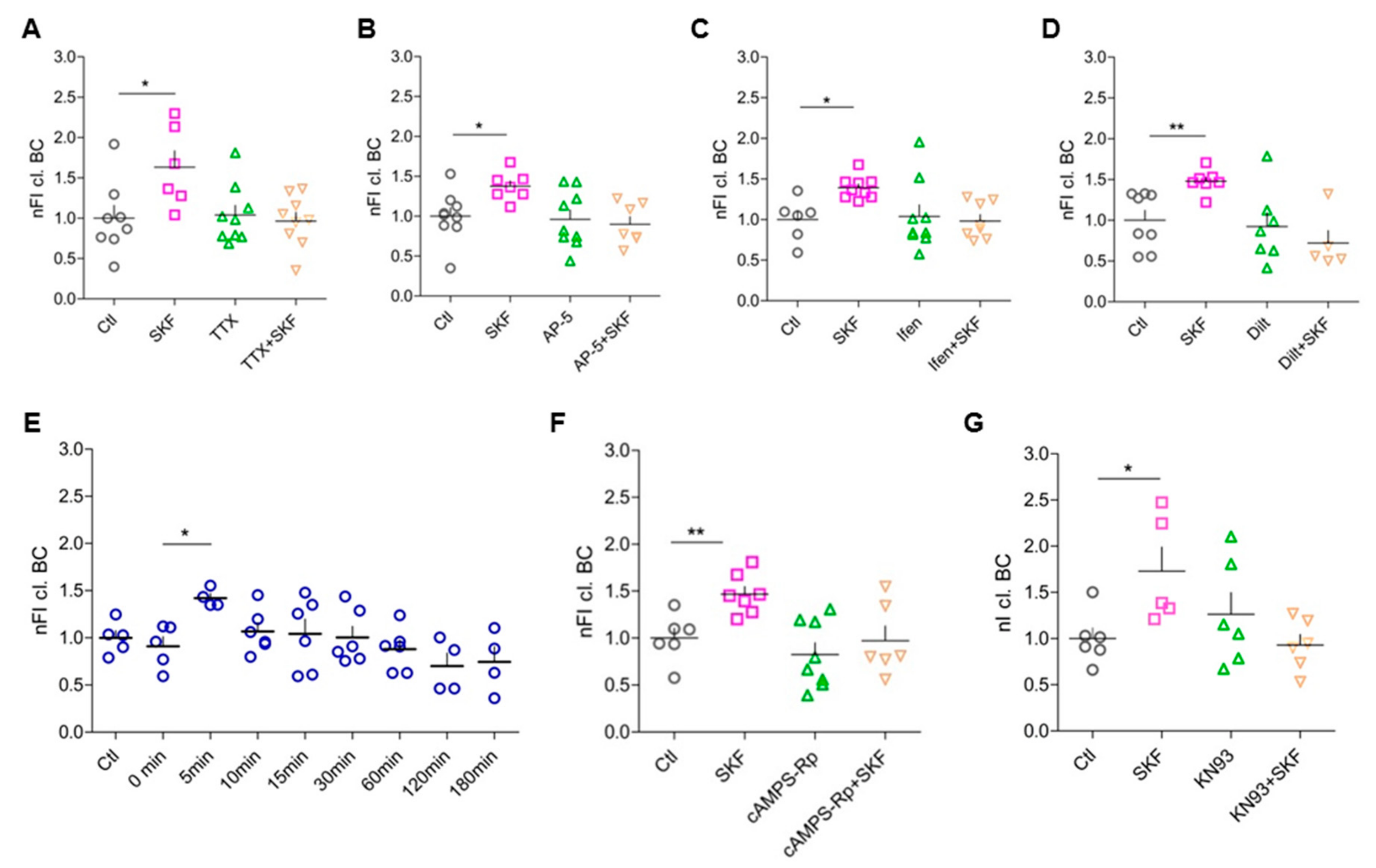
3.6. Network Activity, Activation of Postsynaptic NMDARs, and Opening of L-Type Ca2+ Channels Are Essential for D1-like-DA-Receptor-Induced BC Cleavage
3.7. Elevated Intracellular cAMP Levels Enhance BC Cleavage
3.8. DA-Dependent BC Cleavage Requires Co-Signaling Through PKA and CaMKII
3.9. Increased BC Cleavage at Synapses With More Prominent Homer 1 Expression
4. Discussion
4.1. How Do DA and DA Receptors Contribute to Local Plasticity?
4.2. Induced ECM Cleavage as a Prerequisite for Local Structural and Functional Plasticity?
4.3. Does Protease Activation Occur in a Compartment-Specific Way?
4.4. A Critical View on the Role of Astrocytes
4.5. Loss of Full-Length Lecticans vs. Occurrence of Fragments at Synapses
5. Conclusion: Importance of D1R-Modulated ECM Remodeling for Physiological and Pathological Plasticity
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tritsch, N.X.; Sabatini, B.L. Dopaminergic modulation of synaptic transmission in cortex and striatum. Neuron 2012, 76, 33–50. [Google Scholar] [CrossRef] [PubMed]
- Lisman, J. Glutamatergic synapses are structurally and biochemically complex because of multiple plasticity processes: long-term potentiation, long-term depression, short-term potentiation and scaling. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160260. [Google Scholar] [CrossRef] [PubMed]
- Cannizzaro, C.; Talani, G.; Brancato, A.; Mulas, G.; Spiga, S.; De Luca, M.A.; Sanna, A.; Marino, R.A.M.; Biggio, G.; Sanna, E.; et al. Dopamine Restores Limbic Memory Loss, Dendritic Spine Structure, and NMDAR-Dependent LTD in the Nucleus Accumbens of Alcohol-Withdrawn Rats. J. Neurosci. 2019, 39, 929–943. [Google Scholar] [CrossRef] [PubMed]
- Seeman, P. Dopamine receptors and the dopamine hypothesis of schizophrenia. Synapse 1987, 1, 133–152. [Google Scholar] [CrossRef]
- Tiberi, M.; Jarvie, K.R.; Silvia, C.; Falardeau, P.; Gingrich, J.A.; Godinot, N.; Bertrand, L.; Yang-Feng, T.L.; Fremeau, R.T.; Caron, M.G. Cloning, molecular characterization, and chromosomal assignment of a gene encoding a second D1 dopamine receptor subtype: differential expression pattern in rat brain compared with the D1A receptor. Proc. Natl. Acad. Sci. USA 1991, 88, 7491–7495. [Google Scholar] [CrossRef]
- Dolan, R.J.; Fletcher, P.; Frith, C.D.; Friston, K.J.; Frackowiak, R.S.J.; Grasby, P.M. Dopaminergic modulation of impaired cognitive activation in the anterior cingulate cortex in schizophrenia. Nature 1995, 378, 180–182. [Google Scholar] [CrossRef]
- Beaulieu, J.-M.; Gainetdinov, R.R. The Physiology, Signaling, and Pharmacology of Dopamine Receptors. Pharmacol. Rev. 2011, 63, 182–217. [Google Scholar] [CrossRef]
- Miklosi, A.G.; Del Favero, G.; Bulat, T.; Höger, H.; Shigemoto, R.; Marko, D.; Lubec, G. Super-resolution Microscopical Localization of Dopamine Receptors 1 and 2 in Rat Hippocampal Synaptosomes. Mol. Neurobiol. 2018, 55, 4857–4869. [Google Scholar] [CrossRef]
- Langer, S.Z. 25 Years since the discovery of presynaptic receptors: present knowledge and future perspectives. Trends Pharmacol. Sci. 1997, 18, 95–99. [Google Scholar] [CrossRef]
- Kruse, M.S.; Prémont, J.; Krebs, M.-O.; Jay, T.M. Interaction of dopamine D1 with NMDA NR1 receptors in rat prefrontal cortex. Eur. Neuropsychopharmacol. 2009, 19, 296–304. [Google Scholar] [CrossRef]
- Ladépêche, L.; Dupuis, J.P.; Bouchet, D.; Doudnikoff, E.; Yang, L.; Campagne, Y.; Bézard, E.; Hosy, E.; Groc, L. Single-molecule imaging of the functional crosstalk between surface NMDA and dopamine D1 receptors. Proc. Natl. Acad. Sci. USA 2013, 110, 18005–18010. [Google Scholar] [CrossRef] [PubMed]
- Nai, Q.; Li, S.; Wang, S.-H.; Liu, J.; Lee, F.J.; Frankland, P.W.; Liu, F. Uncoupling the D1-N-Methyl-D-Aspartate (NMDA) Receptor Complex Promotes NMDA-Dependent Long-Term Potentiation and Working Memory. Biol. Psychiatry 2010, 67, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Dityatev, A.; Seidenbecher, C.I.; Schachner, M. Compartmentalization from the outside: the extracellular matrix and functional microdomains in the brain. Trends Neurosci. 2010, 33, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Celio, M.R.; Spreafico, R.; De Biasi, S.; Vitellaro-Zuccarello, L. Perineuronal nets: past and present. Trends Neurosci. 1998, 21, 510–515. [Google Scholar] [CrossRef]
- Sonntag, M.; Blosa, M.; Schmidt, S.; Rübsamen, R.; Morawski, M. Perineuronal nets in the auditory system. Hear. Res. 2015, 329, 21–32. [Google Scholar] [CrossRef]
- Miyata, S.; Nishimura, Y.; Hayashi, N.; Oohira, A. Construction of perineuronal net-like structure by cortical neurons in culture. Cell. Neurosci. 2005, 136, 95–104. [Google Scholar] [CrossRef]
- John, N.; Krügel, H.; Frischknecht, R.; Smalla, K.-H.; Schultz, C.; Kreutz, M.R.; Gundelfinger, E.D.; Seidenbecher, C.I. Brevican-containing perineuronal nets of extracellular matrix in dissociated hippocampal primary cultures. Mol. Cell. Neurosci. 2006, 31, 774–784. [Google Scholar] [CrossRef]
- Frischknecht, R.; Heine, M.; Perrais, D.; Seidenbecher, C.I.; Choquet, D.; Gundelfinger, E.D. Brain extracellular matrix affects AMPA receptor lateral mobility and short-term synaptic plasticity. Nat. Neurosci. 2009, 12, 897–904. [Google Scholar] [CrossRef]
- Faissner, A.; Pyka, M.; Geissler, M.; Sobik, T.; Frischknecht, R.; Gundelfinger, E.D.; Seidenbecher, C. Contributions of astrocytes to synapse formation and maturation — Potential functions of the perisynaptic extracellular matrix. Brain Res. Rev. 2010, 63, 26–38. [Google Scholar] [CrossRef]
- Dityatev, A.; Brückner, G.; Dityateva, G.; Grosche, J.; Kleene, R.; Schachner, M. Activity-dependent formation and functions of chondroitin sulfate-rich extracellular matrix of perineuronal nets. Dev. Neurobiol. 2007, 67, 570–588. [Google Scholar] [CrossRef]
- Valenzuela, J.C.; Heise, C.; Franken, G.; Singh, J.; Schweitzer, B.; Seidenbecher, C.I.; Frischknecht, R. Hyaluronan-based extracellular matrix under conditions of homeostatic plasticity. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130606. [Google Scholar] [CrossRef] [PubMed]
- Rauch, U. Extracellular matrix components associated with remodeling processes in brain. Cell. Mol. Life Sci. 2004, 61, 2031–2045. [Google Scholar] [CrossRef] [PubMed]
- Kelwick, R.; Desanlis, I.; Wheeler, G.N.; Edwards, D.R. The ADAMTS (A Disintegrin and Metalloproteinase with Thrombospondin motifs) family. Genome Biol. 2015, 16, 113. [Google Scholar] [CrossRef] [PubMed]
- Gundelfinger, E.D.; Frischknecht, R.; Choquet, D.; Heine, M. Converting juvenile into adult plasticity: A role for the brain’s extracellular matrix. Eur. J. Neurosci. 2010, 31, 2156–2165. [Google Scholar] [CrossRef]
- Zimmermann, D.R.; Dours-Zimmermann, M.T. Extracellular matrix of the central nervous system: From neglect to challenge. Histochem. Cell Biol. 2008, 130, 635–653. [Google Scholar] [CrossRef]
- Murphy, G. Tissue inhibitors of metalloproteinases. Genome Biol. 2011, 12, 233. [Google Scholar] [CrossRef]
- Gottschall, P.E.; Howell, M.D. ADAMTS expression and function in central nervous system injury and disorders. Matrix Biol. 2015, 44, 70–76. [Google Scholar] [CrossRef]
- Ito, M.; Nagai, T.; Mizoguchi, H.; Sato, K.; Hayase, M.; Otsuka, N.; Fukakusa, A.; Kumagai, N.; Kim, H.-C.; Nabeshima, T.; et al. Activation of post-synaptic dopamine D1 receptors promotes the release of tissue plasminogen activator in the nucleus accumbens via PKA signaling. J. Neurochem. 2007, 103, 2589–2596. [Google Scholar]
- Bruno, M.A.; Cuello, A.C. Activity-dependent release of precursor nerve growth factor, conversion to mature nerve growth factor, and its degradation by a protease cascade. Proc. Natl. Acad. Sci. USA 2006, 103, 6735–6740. [Google Scholar] [CrossRef]
- Yepes, M.; Jeanneret, V. Tissue-type plasminogen activator is a homeostatic regulator of synaptic function in the central nervous system. Neural Regen. Res. 2017, 12, 362–365. [Google Scholar] [CrossRef]
- Yuan, W.; Matthews, R.T.; Sandy, J.D.; Gottschall, P.E. Association between protease-specific proteolytic cleavage of brevican and synaptic loss in the dentate gyrus of kainate-treated rats. Neuroscience 2002, 114, 1091–1101. [Google Scholar] [CrossRef]
- Lazarevic, V.; Schöne, C.; Heine, M.; Gundelfinger, E.D.; Fejtova, A. Extensive Remodeling of the Presynaptic Cytomatrix upon Homeostatic Adaptation to Network Activity Silencing. J. Neurosci. 2011, 31, 10189–10200. [Google Scholar] [CrossRef] [PubMed]
- Friedman, H.V.; Bresler, T.; Garner, C.C.; Ziv, N.E. Assembly of new individual excitatory synapses: time course and temporal order of synaptic molecule recruitment. Neuron 2000, 27, 57–69. [Google Scholar] [CrossRef]
- Shapira, M.; Zhai, R.; Dresbach, T.; Bresler, T.; Torres, V.I.; Gundelfinger, E.D.; Ziv, N.E.; Garner, C.C. Unitary Assembly of Presynaptic Active Zones from Piccolo-Bassoon Transport Vesicles. Neuron 2003, 38, 237–252. [Google Scholar] [CrossRef]
- McClure, C.; Cole, K.L.H.; Wulff, P.; Klugmann, M.; Murray, A.J. Production and titering of recombinant adeno-associated viral vectors. J. Vis. Exp. 2011, e3348. [Google Scholar] [CrossRef] [PubMed]
- Schicknick, H.; Schott, B.H.; Budinger, E.; Smalla, K.-H.; Riedel, A.; Seidenbecher, C.I.; Scheich, H.; Gundelfinger, E.D.; Tischmeyer, W. Dopaminergic modulation of auditory cortex-dependent memory consolidation through mTOR. Cereb. Cortex 2008, 18, 2646–2658. [Google Scholar] [CrossRef] [PubMed]
- Niekisch, H.; Steinhardt, J.; Berghäuser, J.; Bertazzoni, S.; Kaschinski, E.; Kasper, J.; Kisse, M.; Mitlöhner, J.; Singh, J.B.; Weber, J.; et al. Learning Induces Transient Upregulation of Brevican in the Auditory Cortex during Consolidation of Long-Term Memories. J. Neurosci. 2019, 39, 7049–7060. [Google Scholar] [CrossRef]
- Seidenbecher, C.I.; Richter, K.; Rauch, U.; Fässler, R.; Garner, C.C.; Gundelfinger, E.D. Brevican, a Chondroitin Sulfate Proteoglycan of Rat Brain, Occurs as Secreted and Cell Surface Glycosylphosphatidylinositol-anchored Isoforms. J. Biol. Chem. 1995, 270, 27206–27212. [Google Scholar] [CrossRef]
- Lendvai, D.; Morawski, M.; Négyessy, L.; Gáti, G.; Jäger, C.; Baksa, G.; Glasz, T.; Attems, J.; Tanila, H.; Arendt, T.; et al. Neurochemical mapping of the human hippocampus reveals perisynaptic matrix around functional synapses in Alzheimer’s disease. Acta Neuropathol. 2013, 125, 215–229. [Google Scholar] [CrossRef]
- Rowlands, D.; Lensjø, K.K.; Dinh, T.; Yang, S.; Andrews, M.R.; Hafting, T.; Fyhn, M.; Fawcett, J.W.; Dick, G. Aggrecan Directs Extracellular Matrix-Mediated Neuronal Plasticity. J. Neurosci. 2018, 38, 10102–10113. [Google Scholar] [CrossRef]
- Hamel, M.G.; Mayer, J.; Gottschall, P.E. Altered production and proteolytic processing of brevican by transforming growth factor beta in cultured astrocytes. J. Neurochem. 2005, 93, 1533–1541. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H. Brevican Is Degraded by Matrix Metalloproteinases and Aggrecanase-1 (ADAMTS4) at Different Sites. J. Biol. Chem. 2000, 275, 38885–38890. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Zhao, Y.; Wolf, M.E. Dopamine Receptor Stimulation Modulates AMPA Receptor Synaptic Insertion in Prefrontal Cortex Neurons. J. Neurosci. 2005, 25, 7342–7351. [Google Scholar] [CrossRef]
- Chen, L.; Bohanick, J.D.; Nishihara, M.; Seamans, J.K.; Yang, C.R. Dopamine D1/5 receptor-mediated long-term potentiation of intrinsic excitability in rat prefrontal cortical neurons: Ca2+-dependent intracellular signaling. J. Neurophysiol. 2007, 97, 2448–2464. [Google Scholar] [CrossRef] [PubMed]
- Dai, R.; Ali, M.K.; Lezcano, N.; Bergson, C. A Crucial Role for cAMP and Protein Kinase A in D1 Dopamine Receptor Regulated Intracellular Calcium Transients. Neurosignals 2008, 16, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Jürgensen, S.; Antonio, L.L.; Mussi, G.E.A.; Brito-Moreira, J.; Bomfim, T.R.; De Felice, F.G.; Garrido-Sanabria, E.R.; Cavalheiro, É.A.; Ferreira, S.T. Activation of D1/D5 Dopamine Receptors Protects Neurons from Synapse Dysfunction Induced by Amyloid-β Oligomers. J. Biol. Chem. 2011, 286, 3270–3276. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Partridge, J.; Berger, C.; Sepulveda-Rodriguez, A.; Vicini, S.; Conant, K. Dopamine increases NMDA-stimulated calcium flux in striatopallidal neurons through a matrix metalloproteinase-dependent mechanism. Eur. J. Neurosci. 2016, 43, 194–203. [Google Scholar] [CrossRef]
- Matthews, R.T.; Gary, S.C.; Zerillo, C.; Pratta, M.; Solomon, K.; Arner, E.C.; Hockfield, S. Brain-enriched Hyaluronan Binding (BEHAB)/Brevican Cleavage in a Glioma Cell Line Is Mediated by a Disintegrin and Metalloproteinase with Thrombospondin Motifs (ADAMTS) Family Member. J. Biol. Chem. 2000, 275, 22695–22703. [Google Scholar] [CrossRef]
- Mayer, J.; Hamel, M.G.; Gottschall, P.E. Evidence for proteolytic cleavage of brevican by the ADAMTSs in the dentate gyrus after excitotoxic lesion of the mouse entorhinal cortex. BMC Neurosci. 2005, 6, 52. [Google Scholar] [CrossRef]
- Viapiano, M.S.; Hockfield, S.; Matthews, R.T. BEHAB/brevican requires ADAMTS-mediated proteolytic cleavage to promote glioma invasion. J. Neurooncol. 2008, 88, 261–272. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef] [PubMed]
- Tortorella, M.D.; Arner, E.C.; Hills, R.; Gormley, J.; Fok, K.; Pegg, L.; Munie, G.; Malfait, A.-M. ADAMTS-4 (aggrecanase-1): N-Terminal activation mechanisms. Arch. Biochem. Biophys. 2005, 444, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Haddad, J.J. N-methyl-D-aspartate (NMDA) and the regulation of mitogen-activated protein kinase (MAPK) signaling pathways: A revolving neurochemical axis for therapeutic intervention? Prog. Neurobiol. 2005, 77, 252–282. [Google Scholar] [CrossRef] [PubMed]
- Missale, C.; Fiorentini, C.; Busi, C.; Collo, G.; Spano, P. The NMDA/D1 Receptor Complex as a New Target in Drug Development. Curr. Top. Med. Chem. 2006, 6, 801–808. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.-L.; Liu, G.; Li, Y.-C.; Gao, W.-J.; Huang, Y.-Q. Dopamine D1 receptor-mediated NMDA receptor insertion depends on Fyn but not Src kinase pathway in prefrontal cortical neurons. Mol. Brain 2010, 3, 20. [Google Scholar] [CrossRef] [PubMed]
- Surmeier, D.; Bargas, J.; Hemmings, H.C.; Nairn, A.C.; Greengard, P. Modulation of calcium currents by a D1 dopaminergic protein kinase/phosphatase cascade in rat neostriatal neurons. Neuron 1995, 14, 385–397. [Google Scholar] [CrossRef]
- Young, C.E.; Yang, C.R. Dopamine D1/D5 Receptor Modulates State-Dependent Switching of Soma-Dendritic Ca2+ Potentials via Differential Protein Kinase A and C Activation in Rat Prefrontal Cortical Neurons. J. Neurosci. 2004, 24, 8–23. [Google Scholar] [CrossRef]
- Stierl, M.; Stumpf, P.; Udwari, D.; Gueta, R.; Hagedorn, R.; Losi, A.; Gärtner, W.; Petereit, L.; Efetova, M.; Schwarzel, M.; et al. Light Modulation of Cellular cAMP by a Small Bacterial Photoactivated Adenylyl Cyclase, bPAC, of the Soil Bacterium Beggiatoa. J. Biol. Chem. 2011, 286, 1181–1188. [Google Scholar] [CrossRef]
- Lee, S.P.; So, C.H.; Rashid, A.J.; Varghese, G.; Cheng, R.; Lança, A.J.; O’Dowd, B.F.; George, S.R. Dopamine D1 and D2 Receptor Co-activation Generates a Novel Phospholipase C-mediated Calcium Signal. J. Biol. Chem. 2004, 279, 35671–35678. [Google Scholar] [CrossRef]
- Krug, M.; Chepkova, A.N.; Geyer, C.; Ott, T. Aminergic blockade modulates long-term potentiation in the dentate gyrus of freely moving rats. Brain Res. Bull. 1983, 11, 1–6. [Google Scholar] [CrossRef]
- Calabresi, P.; Maj, R.; Pisani, A.; Mercuri, N.; Bernardi, G. Long-term synaptic depression in the striatum: physiological and pharmacological characterization. J. Neurosci. 1992, 12, 4224–4233. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Dani, J.A. Dopamine D1 and D5 receptors modulate spike timing-dependent plasticity at medial perforant path to dentate granule cell synapses. J. Neurosci. 2014, 34, 15888–15897. [Google Scholar] [CrossRef] [PubMed]
- Otani, S.; Bai, J.; Blot, K. Dopaminergic modulation of synaptic plasticity in rat prefrontal neurons. Neurosci. Bull. 2015, 31, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Yagishita, S.; Hayashi-Takagi, A.; Ellis-Davies, G.C.; Urakubo, H.; Ishii, S.; Kasai, H. A critical time window for dopamine actions on the structural plasticity of dendritic spines. Science 2014, 345, 1616–1620. [Google Scholar] [CrossRef]
- O’Neill, B.; Patel, J.C.; Rice, M.E. Characterization of Optically and Electrically Evoked Dopamine Release in Striatal Slices from Digenic Knock-in Mice with DAT-Driven Expression of Channelrhodopsin. ACS Chem. Neurosci. 2017, 8, 310–319. [Google Scholar] [CrossRef]
- Gao, C.; Wolf, M.E. Dopamine Alters AMPA Receptor Synaptic Expression and Subunit Composition in Dopamine Neurons of the Ventral Tegmental Area Cultured with Prefrontal Cortex Neurons. J. Neurosci. 2007, 27, 14275–14285. [Google Scholar] [CrossRef]
- Iwakura, Y.; Wang, R.; Abe, Y.; Piao, Y.-S.; Shishido, Y.; Higashiyama, S.; Takei, N.; Nawa, H. Dopamine-dependent ectodomain shedding and release of epidermal growth factor in developing striatum: target-derived neurotrophic signaling (Part 2). J. Neurochem. 2011, 118, 57–68. [Google Scholar] [CrossRef]
- Kochlamazashvili, G.; Henneberger, C.; Bukalo, O.; Dvoretskova, E.; Senkov, O.; Lievens, P.M.-J.; Westenbroek, R.; Engel, A.K.; Catterall, W.A.; Rusakov, D.A.; et al. The extracellular matrix molecule hyaluronic acid regulates hippocampal synaptic plasticity by modulating postsynaptic L-type Ca(2+) channels. Neuron 2010, 67, 116–128. [Google Scholar] [CrossRef]
- Pyka, M.; Wetzel, C.; Aguado, A.; Geissler, M.; Hatt, H.; Faissner, A. Chondroitin sulfate proteoglycans regulate astrocyte-dependent synaptogenesis and modulate synaptic activity in primary embryonic hippocampal neurons. Eur. J. Neurosci. 2011, 33, 2187–2202. [Google Scholar] [CrossRef]
- Sonderegger, P.; Matsumoto-Miyai, K. Activity-controlled proteolytic cleavage at the synapse. Trends Neurosci. 2014, 37, 413–423. [Google Scholar] [CrossRef]
- Kurshan, P.T.; Phan, A.Q.; Wang, G.J.; Crane, M.M.; Lu, H.; Shen, K. Regulation of synaptic extracellular matrix composition is critical for proper synapse morphology. J. Neurosci. 2014, 34, 12678–12689. [Google Scholar] [CrossRef]
- Brakebusch, C.; Seidenbecher, C.I.; Asztely, F.; Rauch, U.; Matthies, H.; Meyer, H.; Krug, M.; Böckers, T.M.; Zhou, X.; Kreutz, M.R.; et al. Brevican-Deficient Mice Display Impaired Hippocampal CA1 Long-Term Potentiation but Show No Obvious Deficits in Learning and Memory. Mol. Cell. Biol. 2002, 22, 7417–7427. [Google Scholar] [CrossRef]
- Lemarchant, S.; Pruvost, M.; Montaner, J.; Emery, E.; Vivien, D.; Kanninen, K.; Koistinaho, J. ADAMTS proteoglycanases in the physiological and pathological central nervous system. J. Neuroinflamm. 2013, 10, 133. [Google Scholar] [CrossRef]
- Flannery, C.R.; Zeng, W.; Corcoran, C.; Collins-Racie, L.A.; Chockalingam, P.S.; Hebert, T.; Mackie, S.A.; McDonagh, T.; Crawford, T.K.; Tomkinson, K.N.; et al. Autocatalytic Cleavage of ADAMTS-4 (Aggrecanase-1) Reveals Multiple Glycosaminoglycan-binding Sites. J. Biol. Chem. 2002, 277, 42775–42780. [Google Scholar] [CrossRef]
- Longpré, J.-M.; McCulloch, D.R.; Koo, B.-H.; Alexander, J.P.; Apte, S.S.; LeDuc, R. Characterization of proADAMTS5 processing by proprotein convertases. Int. J. Biochem. Cell Biol. 2009, 41, 1116–1126. [Google Scholar] [CrossRef]
- Wang, P.; Tortorella, M.; England, K.; Malfait, A.-M.; Thomas, G.; Arner, E.C.; Pei, D. Proprotein convertase furin interacts with and cleaves pro-ADAMTS4 (Aggrecanase-1) in the trans-Golgi network. J. Biol. Chem. 2004, 279, 15434–15440. [Google Scholar] [CrossRef]
- Lemarchant, S.; Pruvost, M.; Hebert, M.; Gauberti, M.; Hommet, Y.; Briens, A.; Maubert, E.; Gueye, Y.; Féron, F.; Petite, D.; et al. tPA promotes ADAMTS-4-induced CSPG degradation, thereby enhancing neuroplasticity following spinal cord injury. Neurobiol. Dis. 2014, 66, 28–42. [Google Scholar] [CrossRef]
- Zanassi, P.; Paolillo, M.; Montecucco, A.; Avvedimento, E.V.; Schinelli, S. Pharmacological and molecular evidence for dopamine D(1) receptor expression by striatal astrocytes in culture. J. Neurosci. Res. 1999, 58, 544–552. [Google Scholar] [CrossRef]
- Miyazaki, I.; Asanuma, M.; Diaz-Corrales, F.J.; Miyoshi, K.; Ogawa, N. Direct evidence for expression of dopamine receptors in astrocytes from basal ganglia. Brain Res. 2004, 1029, 120–123. [Google Scholar] [CrossRef]
- Brito, V.; Beyer, C.; Küppers, E. BDNF-dependent stimulation of dopamine D5receptor expression in developing striatal astrocytes involves PI3-kinase signaling. Glia 2004, 46, 284–295. [Google Scholar] [CrossRef]
- Hösli, E.; Hösli, L. Binding sites for [3H]dopamine and dopamine-antagonists on cultured astrocytes of rat striatum and spinal cord: An autoradiographic study. Neurosci. Lett. 1986, 65, 177–182. [Google Scholar] [CrossRef]
- Vermeulen, R.; Jongenelen, C.A.; Langeveld, C.H.; Wolters, E.C.; Stoof, J.C.; Drukarch, B. Dopamine D1 receptor agonists display a different intrinsic activity in rat, monkey and human astrocytes. Eur. J. Pharmacol. Mol. Pharmacol. 1994, 269, 121–125. [Google Scholar] [CrossRef]
- Requardt, R.P.; Wilhelm, F.; Rillich, J.; Winkler, U.; Hirrlinger, J. The biphasic NAD(P)H fluorescence response of astrocytes to dopamine reflects the metabolic actions of oxidative phosphorylation and glycolysis. J. Neurochem. 2010, 115, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Favuzzi, E.; Marques-Smith, A.; Deogracias, R.; Winterflood, C.M.; Sánchez-Aguilera, A.; Mantoan, L.; Maeso, P.; Fernandes, C.; Ewers, H.; Rico, B. Activity-Dependent Gating of Parvalbumin Interneuron Function by the Perineuronal Net Protein Brevican. Neuron 2017, 95, 639–655. [Google Scholar] [CrossRef]
- Song, I.; Dembitskaya, Y.; Singh, J.; Doronin, M.; Kaushik, R.; Frischknecht, R.; Seidenbecher, C.I.; Semyanov, S.; Dityatev, A. Two forms of plasticity triggered by enzymatic removal of extracellular matrix in the hippocampus. In Proceedings of the 10th FENS Forum of Neuroscience, Copenhagen, Denmark, 2–6 July 2016. [Google Scholar]
- Davis, G.E.; Bayless, K.J.; Davis, M.J.; Meininger, G.A. Regulation of Tissue Injury Responses by the Exposure of Matricryptic Sites within Extracellular Matrix Molecules. Am. J. Pathol. 2000, 156, 1489–1498. [Google Scholar] [CrossRef]
- Hu, B.; Kong, L.L.; Matthews, R.T.; Viapiano, M.S. The Proteoglycan Brevican Binds to Fibronectin after Proteolytic Cleavage and Promotes Glioma Cell Motility. J. Biol. Chem. 2008, 283, 24848–24859. [Google Scholar] [CrossRef]
- Soleman, S.; Filippov, M.; Dityatev, A.; Fawcett, J. Targeting the neural extracellular matrix in neurological disorders. Neuroscience 2013, 253, 194–213. [Google Scholar] [CrossRef]
- Berezin, V.; Walmod, P.S.; Filippov, M.; Dityatev, A. Targeting of ECM molecules and their metabolizing enzymes and receptors for the treatment of CNS diseases. Prog. Brain Res. 2014, 214, 353–388. [Google Scholar]
- DeWitt, D.; Richey, P.; Praprotnik, D.; Silver, J.; Perry, G. Chondroitin sulfate proteoglycans are a common component of neuronal inclusions and astrocytic reaction in neurodegenerative diseases. Brain Res. 1994, 656, 205–209. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, Q.; Tang, M.; Fu, N.; Shao, W.; Zhang, S.; Yin, Y.; Zeng, R.; Wang, X.; Hu, G.; et al. Upregulation of alphaB-crystallin expression in the substantia nigra of patients with Parkinson’s disease. Neurobiol. Aging 2015, 36, 1686–1691. [Google Scholar] [CrossRef]
- Held-Feindt, J.; Paredes, E.B.; Blömer, U.; Seidenbecher, C.; Stark, A.M.; Mehdorn, H.M.; Mentlein, R. Matrix-degrading proteases ADAMTS4 and ADAMTS5 (disintegrins and metalloproteinases with thrombospondin motifs 4 and 5) are expressed in human glioblastomas. Int. J. Cancer 2006, 118, 55–61. [Google Scholar] [CrossRef]
- Oever, M.C.V.D.; Lubbers, B.R.; Goriounova, N.A.; Li, K.W.; Van Der Schors, R.C.; Loos, M.; Riga, D.; Wiskerke, J.; Binnekade, R.; Stegeman, M.; et al. Extracellular Matrix Plasticity and GABAergic Inhibition of Prefrontal Cortex Pyramidal Cells Facilitates Relapse to Heroin Seeking. Neuropsychopharmacology 2010, 35, 2120–2133. [Google Scholar] [CrossRef] [PubMed]
- Lubbers, B.R.; Matos, M.R.; Horn, A.; Visser, E.; Van Der Loo, R.C.; Gouwenberg, Y.; Meerhoff, G.F.; Frischknecht, R.; Seidenbecher, C.I.; Smit, A.B.; et al. The Extracellular Matrix Protein Brevican Limits Time-Dependent Enhancement of Cocaine Conditioned Place Preference. Neuropsychopharmacology 2016, 41, 1907–1916. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rat ADAMTS 4 | shRNA 1: 5′- ATCGTGACCACATCGCTGT -3′ |
| shRNA 2: 5′- TATAGCGCAAGCTGACTGC -3′ | |
| Rat ADAMTS 5 | shRNA 1: 5′- TAGCGCGCATGCTTGACTG -3′ |
| shRNA 2: 5′- ATCCCCGTAAACTCGTTCG -3′ | |
| Control shRNA (Scramble) | scrRNA: 5′- CGGCTGAAACAAGAGTTGG -3′ |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mitlöhner, J.; Kaushik, R.; Niekisch, H.; Blondiaux, A.; Gee, C.E.; Happel, M.F.K.; Gundelfinger, E.; Dityatev, A.; Frischknecht, R.; Seidenbecher, C. Dopamine Receptor Activation Modulates the Integrity of the Perisynaptic Extracellular Matrix at Excitatory Synapses. Cells 2020, 9, 260. https://doi.org/10.3390/cells9020260
Mitlöhner J, Kaushik R, Niekisch H, Blondiaux A, Gee CE, Happel MFK, Gundelfinger E, Dityatev A, Frischknecht R, Seidenbecher C. Dopamine Receptor Activation Modulates the Integrity of the Perisynaptic Extracellular Matrix at Excitatory Synapses. Cells. 2020; 9(2):260. https://doi.org/10.3390/cells9020260
Chicago/Turabian StyleMitlöhner, Jessica, Rahul Kaushik, Hartmut Niekisch, Armand Blondiaux, Christine E. Gee, Max F. K. Happel, Eckart Gundelfinger, Alexander Dityatev, Renato Frischknecht, and Constanze Seidenbecher. 2020. "Dopamine Receptor Activation Modulates the Integrity of the Perisynaptic Extracellular Matrix at Excitatory Synapses" Cells 9, no. 2: 260. https://doi.org/10.3390/cells9020260
APA StyleMitlöhner, J., Kaushik, R., Niekisch, H., Blondiaux, A., Gee, C. E., Happel, M. F. K., Gundelfinger, E., Dityatev, A., Frischknecht, R., & Seidenbecher, C. (2020). Dopamine Receptor Activation Modulates the Integrity of the Perisynaptic Extracellular Matrix at Excitatory Synapses. Cells, 9(2), 260. https://doi.org/10.3390/cells9020260