Taming the Autophagy as a Strategy for Treating COVID-19



 and
and
Abstract
1. Introduction
2. Brief Overview of the New SARS-CoV-2
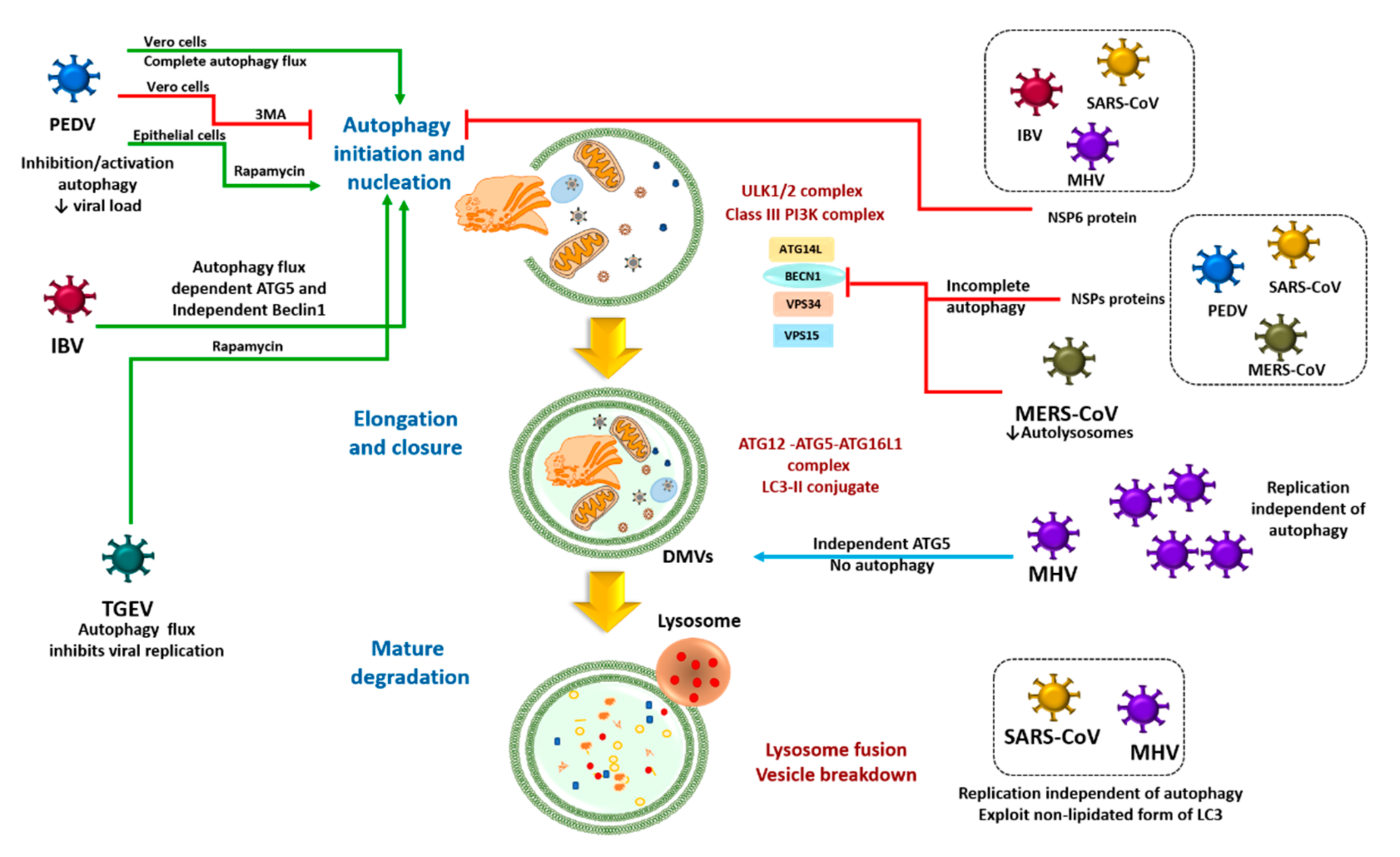
3. The Autophagy-Coronavirus Relationship
3.1. Direct Coronavirus-Autophagy Interplay
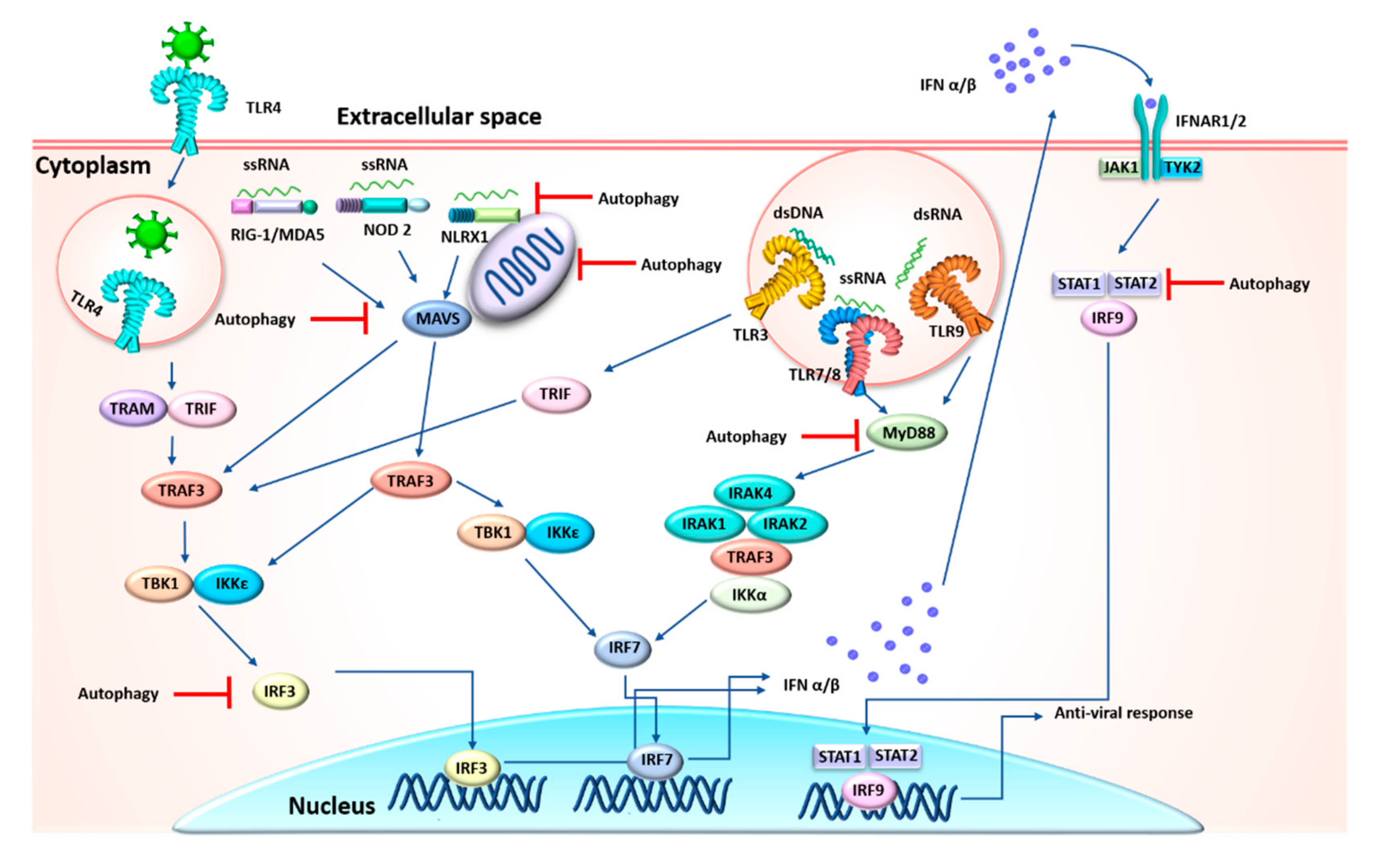
3.2. Indirect Coronavirus–Autophagy Interplay
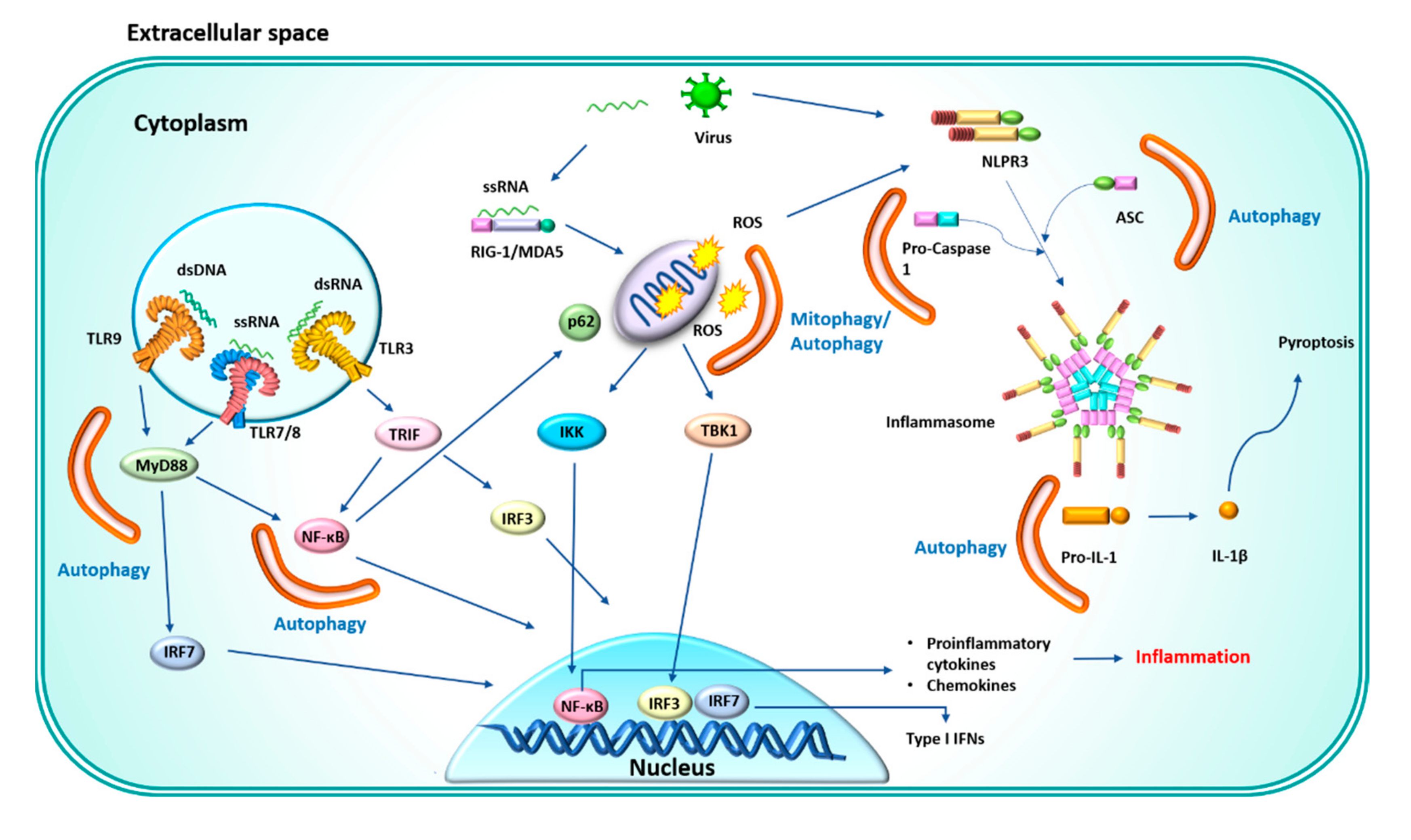
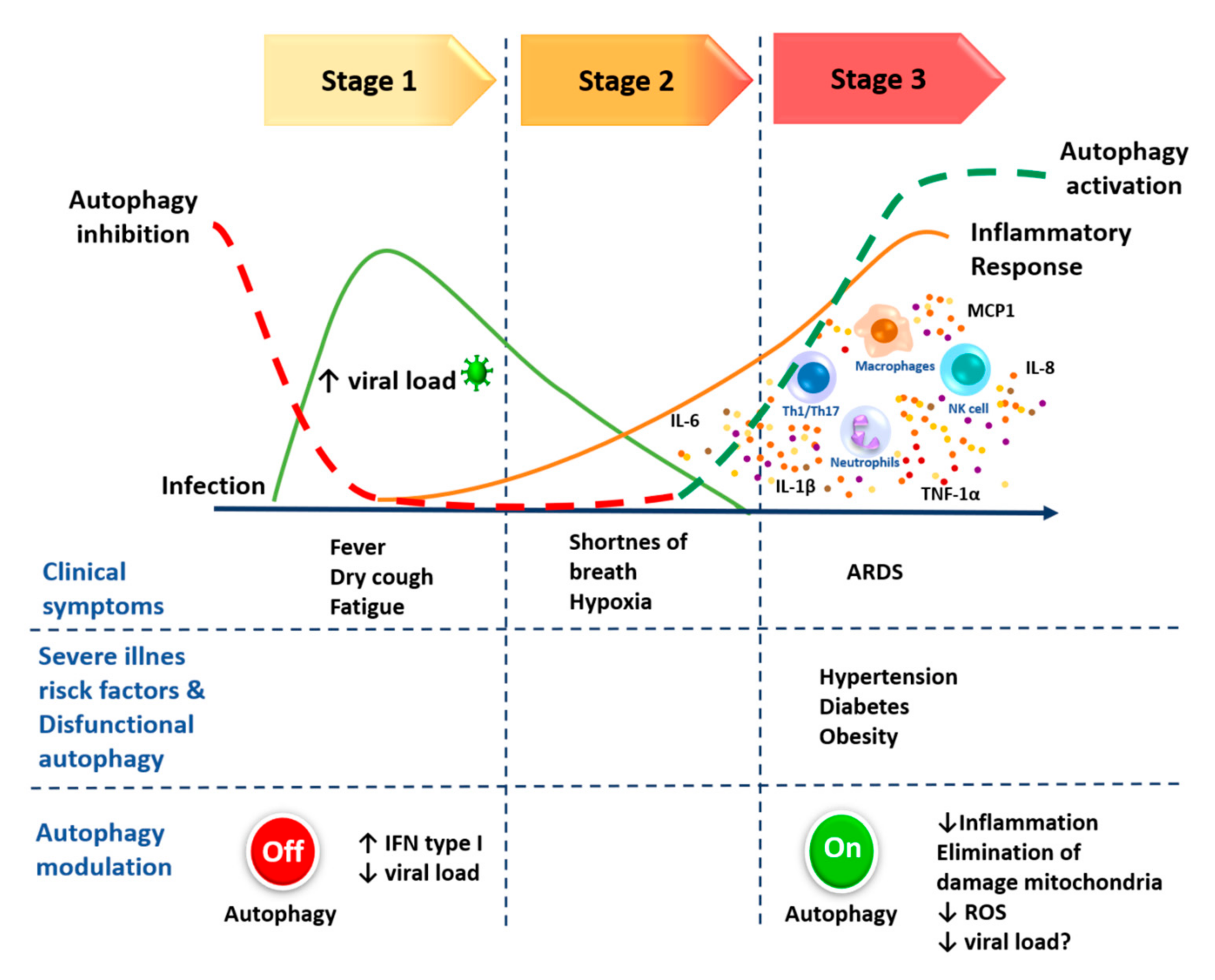
4. Anti-Inflammatory Function of Autophagy
5. Inflammation, Obesity and Autophagy
6. Pharmacological Intervention Targeting Autophagy
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wang, Y.; Zhang, D.; Du, G.; Du, R.; Zhao, J.; Jin, Y.; Fu, S.; Gao, L.; Cheng, Z.; Lu, Q.; et al. Remdesivir in adults with severe COVID-19: A randomised, double-blind, placebo-controlled, multicentre trial. Lancet 2020, 395, 1569–1578. [Google Scholar] [CrossRef]
- Grein, J.; Ohmagari, N.; Shin, D.; Diaz, G.; Asperges, E.; Castagna, A.; Feldt, T.; Green, G.; Green, M.L.; Lescure, F.X.; et al. Compassionate Use of Remdesivir for Patients with Severe Covid-19. N. Engl. J. Med. 2020, 382, 2327–2336. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.; Wang, Y.; Wen, D.; Liu, W.; Wang, J.; Fan, G.; Ruan, L.; Song, B.; Cai, Y.; Wei, M.; et al. A Trial of Lopinavir-Ritonavir in Adults Hospitalized with Severe Covid-19. N. Engl. J. Med. 2020, 382, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Yang, M.; Liu, D.; Chen, J.; Shu, D.; Xia, J.; Liao, X.; Gu, Y.; Cai, Q.; Yang, Y. Experimental treatment with favipiravir for COVID-19: An open-label control study. Engineering 2020. [Google Scholar] [CrossRef]
- Caly, L.; Druce, J.D.; Catton, M.G.; Jans, D.A.; Wagstaff, K.M. The FDA-approved drug ivermectin inhibits the replication of SARS-CoV-2 In Vitro. Antivir. Res. 2020, 178, 104787. [Google Scholar] [CrossRef]
- Chen, J.; Liu, D.; Liu, L.; Liu, P.; Xu, Q.; Xia, L.; Ling, Y.; Huang, D.; Song, S.; Zhang, D. A pilot study of hydroxychloroquine in treatment of patients with common coronavirus disease-19 (COVID-19). Zhejiang Da Xue Xue Bao Yi Xue Ban 2020, 49, 215–219. [Google Scholar] [CrossRef]
- Gautret, P.; Lagier, J.-C.; Parola, P.; Meddeb, L.; Sevestre, J.; Mailhe, M.; Doudier, B.; Aubry, C.; Amrane, S.; Seng, P. Clinical and microbiological effect of a combination of hydroxychloroquine and azithromycin in 80 COVID-19 patients with at least a six-day follow up: A pilot observational study. Travel Med. Infect. Dis. 2020, 34, 101663. [Google Scholar] [CrossRef]
- Meyerowitz, E.A.; Vannier, A.G.L.; Friesen, M.G.N.; Schoenfeld, S.; Gelfand, J.A.; Callahan, M.V.; Kim, A.Y.; Reeves, P.M.; Poznansky, M.C. Rethinking the role of hydroxychloroquine in the treatment of COVID-19. FASEB J. 2020, 34, 6027–6037. [Google Scholar] [CrossRef]
- Jianping, Z. Randomized, Open, Blank Control Study on the Efficacy and Safety of Recombinant Human Interferon Α1β in the Treatment of Patients with New Type of Coronavirus Infection in Wuhan. ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04293887 (accessed on 2 March 2020).
- Deng, L.; Li, C.; Zeng, Q.; Liu, X.; Li, X.; Zhang, H.; Hong, Z.; Xia, J. Arbidol combined with LPV/r versus LPV/r alone against Corona Virus Disease 2019: A retrospective cohort study. J. Infect. 2020, 81, e1–e5. [Google Scholar] [CrossRef]
- Zhu, Z.; Lu, Z.; Xu, T.; Chen, C.; Yang, G.; Zha, T.; Lu, J.; Xue, Y. Arbidol monotherapy is superior to lopinavir/ritonavir in treating COVID-19. J. Infect. 2020, 81, e21–e23. [Google Scholar] [CrossRef]
- Luo, P.; Liu, Y.; Qui, L.; Liu, X.; Liu, D.; Li, J. Tocilizumab treatment in COVID-19: A single center experience. J. Med. Virol. 2020, 92, 814–818. [Google Scholar] [CrossRef] [PubMed]
- Toniati, P.; Piva, S.; Cattalini, M.; Garrafa, E.; Regola, F.; Castelli, F.; Franceschini, F.; Focà, E.; Andreoli, L.; Latronico, N. Tocilizumab for the treatment of severe COVID-19 pneumonia with hyperinflammatory syndrome and acute respiratory failure: A single center study of 100 patients in Brescia, Italy. Autoimmun. Rev. 2020, 19, 102568. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Han, M.; Li, T.; Sun, W.; Wang, D.; Fu, B.; Zhou, Y.; Zheng, X.; Yang, Y.; Li, X. Effective treatment of severe COVID-19 patients with tocilizumab. Proc. Natl. Acad. Sci. USA 2020, 117, 10970–10975. [Google Scholar] [CrossRef] [PubMed]
- Ye, M.; Fu, D.; Ren, Y.; Wang, F.; Wang, D.; Zhang, F.; Xia, X.; Lv, T. Treatment with convalescent plasma for COVID-19 patients in Wuhan, China. J. Med. Virol. 2020, 10. [Google Scholar] [CrossRef]
- Shen, C.; Wang, Z.; Zhao, F.; Yang, Y.; Li, J.; Yuan, J.; Wang, F.; Li, D.; Yang, M.; Xing, L.; et al. Treatment of 5 Critically Ill Patients With COVID-19 with Convalescent Plasma. JAMA 2020, 323, 1582–1589. [Google Scholar] [CrossRef]
- Li, L.; Zhang, W.; Hu, Y.; Tong, X.; Zheng, S.; Yang, J.; Kong, Y.; Ren, L.; Wei, Q.; Mei, H.; et al. Effect of Convalescent Plasma Therapy on Time to Clinical Improvement in Patients with Severe and Life-threatening COVID-19: A Randomized Clinical Trial. JAMA 2020, 324, 460–470. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, Y.; Qi, C.; Shen, L.; Li, J. Clinical trial analysis of 2019-nCoV therapy registered in China. J. Med. Virol. 2020, 92, 540–545. [Google Scholar] [CrossRef]
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef]
- Guan, W.J.; Ni, Z.Y.; Hu, Y.; Liang, W.H.; Ou, C.Q.; He, J.X.; Liu, L.; Shan, H.; Lei, C.L.; Hui, D.S.C.; et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef]
- Xu, Z.; Shi, L.; Wang, Y.; Zhang, J.; Huang, L.; Zhang, C.; Liu, S.; Zhao, P.; Liu, H.; Zhu, L.; et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 2020, 8, 420–422. [Google Scholar] [CrossRef]
- Libby, P.; Lüscher, T. COVID-19 is, in the end, an endothelial disease. Eur. Heart J. 2020, 41, 3038–3044. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Zhou, X.; Qiu, Y.; Song, Y.; Feng, F.; Feng, J.; Song, Q.; Jia, Q.; Wang, J. Clinical characteristics of 82 cases of death from COVID-19. PLoS ONE 2020, 15, e0235458. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Chen, S.; Chen, Z. Advances in COVID-19: The virus, the pathogenesis, and evidence-based control and therapeutic strategies. Front. Med. 2020, 14, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Castrejón-Jiménez, N.S.; Leyva-Paredes, K.; Hernández-González, J.C.; Luna-Herrera, J.; García-Pérez, B.E. The role of autophagy in bacterial infections. Biosci. Trends 2015, 9, 149–159. [Google Scholar] [CrossRef]
- Levine, B.; Deretic, V. Unveiling the roles of autophagy in innate and adaptive immunity. Nat. Rev. Immunol. 2007, 7, 767–777. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef]
- Choi, Y.; Bowman, J.W.; Jung, J.U. Autophagy during viral infection—A double-edged sword. Nat. Rev. Microbiol. 2018, 16, 341–354. [Google Scholar] [CrossRef]
- Choi, A.M.; Ryter, S.W.; Levine, B. Autophagy in human health and disease. N. Engl. J. Med. 2013, 368, 651–662. [Google Scholar] [CrossRef]
- Saha, S.; Panigrahi, D.P.; Patil, S.; Bhutia, S.K. Autophagy in health and disease: A comprehensive review. Biomed. Pharmacother. 2018, 104, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sowers, J.R.; Ren, J. Targeting autophagy in obesity: From pathophysiology to management. Nat. Rev. Endocrinol. 2018, 14, 356–376. [Google Scholar] [CrossRef] [PubMed]
- Heath, R.J.; Xavier, R.J. Autophagy, immunity and human disease. Curr. Opin. Gastroenterol. 2009, 25, 512. [Google Scholar] [CrossRef]
- Barbosa, M.C.; Grosso, R.A.; Fader, C.M. Hallmarks of aging: An autophagic perspective. Front. Endocrinol. 2019, 9, 790. [Google Scholar] [CrossRef] [PubMed]
- Ksiazek, T.G.; Erdman, D.; Goldsmith, C.S.; Zaki, S.R.; Peret, T.; Emery, S.; Tong, S.; Urbani, C.; Comer, J.A.; Lim, W. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 2003, 348, 1953–1966. [Google Scholar] [CrossRef] [PubMed]
- Hijawi, B.; Abdallat, M.; Sayaydeh, A.; Alqasrawi, S.; Haddadin, A.; Jaarour, N.; El Sheikh, S.; Alsanouri, T. Novel coronavirus infections in Jordan, April 2012: Epidemiological findings from a retrospective investigation. East. Mediterr. Health J. 2013, 19, S12–S18. [Google Scholar] [CrossRef]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef]
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef]
- Wu, A.; Peng, Y.; Huang, B.; Ding, X.; Wang, X.; Niu, P.; Meng, J.; Zhu, Z.; Zhang, Z.; Wang, J. Genome composition and divergence of the novel coronavirus (2019-nCoV) originating in China. Cell Host Microbe 2020, 27, 325–328. [Google Scholar] [CrossRef]
- Luan, J.; Lu, Y.; Jin, X.; Zhang, L. Spike protein recognition of mammalian ACE2 predicts the host range and an optimized ACE2 for SARS-CoV-2 infection. Biochem. Biophys. Res. Commun. 2020, 526, 165–169. [Google Scholar] [CrossRef]
- Fang, L.; Karakiulakis, G.; Roth, M. Are patients with hypertension and diabetes mellitus at increased risk for COVID-19 infection? Lancet Respir. Med. 2020, 8, e21. [Google Scholar] [CrossRef]
- Xu, H.; Zhong, L.; Deng, J.; Peng, J.; Dan, H.; Zeng, X.; Li, T.; Chen, Q. High expression of ACE2 receptor of 2019-nCoV on the epithelial cells of oral mucosa. Int. J. Oral Sci. 2020, 12, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef] [PubMed]
- Bestle, D.; Heindl, M.R.; Limburg, H.; Pilgram, O.; Moulton, H.; Stein, D.A.; Hardes, K.; Eickmann, M.; Dolnik, O.; Rohde, C. TMPRSS2 and furin are both essential for proteolytic activation of SARS- CoV-2 in human airway cells. Life Sci. Alliance 2020, 3, e202000786. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.L. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef]
- De Haan, C.A.; Reggiori, F. Are nidoviruses hijacking the autophagy machinery? Autophagy 2008, 4, 276–279. [Google Scholar] [CrossRef]
- Gosert, R.; Kanjanahaluethai, A.; Egger, D.; Bienz, K.; Baker, S.C. RNA replication of mouse hepatitis virus takes place at double-membrane vesicles. J. Virol. 2002, 76, 3697–3708. [Google Scholar] [CrossRef]
- Subauste, C.S. Interplay between Toxoplasma gondii, autophagy, and autophagy proteins. Front. Cell. Infect. Microbiol. 2019, 9, 139. [Google Scholar] [CrossRef]
- Orvedahl, A.; Sumpter, R., Jr.; Xiao, G.; Ng, A.; Zou, Z.; Tang, Y.; Narimatsu, M.; Gilpin, C.; Sun, Q.; Roth, M. Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature 2011, 480, 113–117. [Google Scholar] [CrossRef]
- Mao, J.; Lin, E.; He, L.; Yu, J.; Tan, P.; Zhou, Y. Autophagy and Viral Infection. Adv. Exp. Med. Biol. 2019, 1209, 55–78. [Google Scholar] [CrossRef]
- Prentice, E.; Jerome, W.G.; Yoshimori, T.; Mizushima, N.; Denison, M.R. Coronavirus replication complex formation utilizes components of cellular autophagy. J. Biol. Chem. 2004, 279, 10136–10141. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Thackray, L.B.; Miller, B.C.; Lynn, T.M.; Becker, M.M.; Ward, E.; Mizushima, N.; Denison, M.R.; Virgin, H.W. Coronavirus replication does not require the autophagy gene ATG5. Autophagy 2007, 3, 581–585. [Google Scholar] [CrossRef] [PubMed]
- Fung, T.S.; Liu, D.X. The ER stress sensor IRE1 and MAP kinase ERK modulate autophagy induction in cells infected with coronavirus infectious bronchitis virus. Virology 2019, 533, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Yu, H.; Gu, W.; Luo, X.; Li, R.; Zhang, J.; Xu, Y.; Yang, L.; Shen, N.; Feng, L. Autophagy negatively regulates transmissible gastroenteritis virus replication. Sci. Rep. 2016, 6, 1–14. [Google Scholar] [CrossRef]
- Guo, X.; Zhang, M.; Zhang, X.; Tan, X.; Guo, H.; Zeng, W.; Yan, G.; Memon, A.M.; Li, Z.; Zhu, Y. Porcine epidemic diarrhea virus induces autophagy to benefit its replication. Viruses 2017, 9, 53. [Google Scholar] [CrossRef]
- Ko, S.; Gu, M.J.; Kim, C.G.; Kye, Y.C.; Lim, Y.; Lee, J.E.; Park, B.-C.; Chu, H.; Han, S.H.; Yun, C.H. Rapamycin-induced autophagy restricts porcine epidemic diarrhea virus infectivity in porcine intestinal epithelial cells. Antivir. Res. 2017, 146, 86–95. [Google Scholar] [CrossRef]
- Cottam, E.M.; Maier, H.J.; Manifava, M.; Vaux, L.C.; Chandra-Schoenfelder, P.; Gerner, W.; Britton, P.; Ktistakis, N.T.; Wileman, T. Coronavirus nsp6 proteins generate autophagosomes from the endoplasmic reticulum via an omegasome intermediate. Autophagy 2011, 7, 1335–1347. [Google Scholar] [CrossRef]
- Cottam, E.M.; Whelband, M.C.; Wileman, T. Coronavirus NSP6 restricts autophagosome expansion. Autophagy 2014, 10, 1426–1441. [Google Scholar] [CrossRef]
- Chen, X.; Wang, K.; Xing, Y.; Tu, J.; Yang, X.; Zhao, Q.; Li, K.; Chen, Z. Coronavirus membrane-associated papain-like proteases induce autophagy through interacting with Beclin1 to negatively regulate antiviral innate immunity. Protein Cell 2014, 5, 912–927. [Google Scholar] [CrossRef]
- Gassen, N.C.; Niemeyer, D.; Muth, D.; Corman, V.M.; Martinelli, S.; Gassen, A.; Hafner, K.; Papies, J.; Mösbauer, K.; Zellner, A. SKP2 attenuates autophagy through Beclin1-ubiquitination and its inhibition reduces MERS-Coronavirus infection. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef]
- Monastyrska, I.; Ulasli, M.; Rottier, P.J.; Guan, J.L.; Reggiori, F.; De Haan, C.A. An autophagy-independent role for LC3 in equine arteritis virus replication. Autophagy 2013, 9, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Reggiori, F.; Monastyrska, I.; Verheije, M.H.; Calì, T.; Ulasli, M.; Bianchi, S.; Bernasconi, R.; De Haan, C.A.; Molinari, M. Coronaviruses Hijack the LC3-I-positive EDEMosomes, ER-derived vesicles exporting short-lived ERAD regulators, for replication. Cell Host Microbe 2010, 7, 500–508. [Google Scholar] [CrossRef] [PubMed]
- Bernasconi, R.; Noack, J.; Molinari, M. Unconventional roles of nonlipidated LC3 in ERAD tuning and coronavirus infection. Autophagy 2012, 8, 1534–1536. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.; Ackermann, K.; Stuart, M.; Wex, C.; Protzer, U.; Schätzl, H.M.; Gilch, S. Severe acute respiratory syndrome coronavirus replication is severely impaired by MG132 due to proteasome-independent inhibition of M-calpain. J. Virol. 2012, 86, 10112–10122. [Google Scholar] [CrossRef] [PubMed]
- Huttenlocher, A.; Palecek, S.P.; Lu, Q.; Zhang, W.; Mellgren, R.L.; Lauffenburger, D.A.; Ginsberg, M.H.; Horwitz, A.F. Regulation of cell migration by the calcium-dependent protease calpain. J. Biol. Chem. 1997, 272, 32719–32722. [Google Scholar] [CrossRef]
- Gutierrez, M.G.; Master, S.S.; Singh, S.B.; Taylor, G.A.; Colombo, M.I.; Deretic, V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 2004, 119, 753–766. [Google Scholar] [CrossRef] [PubMed]
- Campbell, G.R.; Spector, S.A. Hormonally active vitamin D3 (1α, 25-dihydroxycholecalciferol) triggers autophagy in human macrophages that inhibits HIV-1 infection. J. Biol. Chem. 2011, 286, 18890–18902. [Google Scholar] [CrossRef]
- Campbell, G.R.; Bruckman, R.S.; Chu, Y.L.; Spector, S.A. Autophagy induction by histone deacetylase inhibitors inhibits HIV type 1. J. Biol. Chem. 2015, 290, 5028–5040. [Google Scholar] [CrossRef]
- Borel, S.; Espert, L.; Biard-Piechaczyk, M. Macroautophagy regulation during HIV-1 infection of CD4+ T cells and macrophages. Front. Immunol. 2012, 3, 97. [Google Scholar] [CrossRef]
- Tang, S.W.; Ducroux, A.; Jeang, K.T.; Neuveut, C. Impact of cellular autophagy on viruses: Insights from hepatitis B virus and human retroviruses. J. Biomed. Sci. 2012, 19, 1–11. [Google Scholar] [CrossRef]
- Min, J.S.; Kim, D.E.; Jin, Y.-H.; Kwon, S. Kurarinone Inhibits HCoV-OC43 Infection by Impairing the Virus-Induced Autophagic Flux in MRC-5 Human Lung Cells. J. Clin. Med. 2020, 9, 2230. [Google Scholar] [CrossRef] [PubMed]
- Monie, T.P. NLR activation takes a direct route. Trends Biochem. Sci. 2013, 38, 131–139. [Google Scholar] [CrossRef]
- Tian, Y.; Wang, M.L.; Zhao, J. Crosstalk between Autophagy and Type I Interferon Responses in Innate Antiviral Immunity. Viruses 2019, 11, 132. [Google Scholar] [CrossRef] [PubMed]
- Channappanavar, R.; Fehr, A.R.; Zheng, J.; Wohlford-Lenane, C.; Abrahante, J.E.; Mack, M.; Sompallae, R.; McCray, P.B.; Meyerholz, D.K.; Perlman, S. IFN-I response timing relative to virus replication determines MERS coronavirus infection outcomes. J. Clin. Investig. 2019, 129, 3625–3639. [Google Scholar] [CrossRef] [PubMed]
- Alexopoulou, L.; Holt, A.C.; Medzhitov, R.; Flavell, R.A. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 2001, 413, 732–738. [Google Scholar] [CrossRef]
- Diebold, S.S.; Kaisho, T.; Hemmi, H.; Akira, S.; Reis e Sousa, C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science 2004, 303, 1529–1531. [Google Scholar] [CrossRef]
- Heil, F.; Hemmi, H.; Hochrein, H.; Ampenberger, F.; Kirschning, C.; Akira, S.; Lipford, G.; Wagner, H.; Bauer, S. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science 2004, 303, 1526–1529. [Google Scholar] [CrossRef]
- Hornung, V.; Ellegast, J.; Kim, S.; Brzózka, K.; Jung, A.; Kato, H.; Poeck, H.; Akira, S.; Conzelmann, K.K.; Schlee, M.; et al. 5′-Triphosphate RNA is the ligand for RIG-I. Science 2006, 314, 994–997. [Google Scholar] [CrossRef]
- Bauernfeind, F.; Ablasser, A.; Bartok, E.; Kim, S.; Schmid-Burgk, J.; Cavlar, T.; Hornung, V. Inflammasomes: Current understanding and open questions. Cell. Mol. Life Sci. 2011, 68, 765–783. [Google Scholar] [CrossRef]
- Sabbah, A.; Chang, T.H.; Harnack, R.; Frohlich, V.; Tominaga, K.; Dube, P.H.; Xiang, Y.; Bose, S. Activation of innate immune antiviral responses by Nod2. Nat. Immunol. 2009, 10, 1073–1080. [Google Scholar] [CrossRef]
- Tattoli, I.; Carneiro, L.A.; Jéhanno, M.; Magalhaes, J.G.; Shu, Y.; Philpott, D.J.; Arnoult, D.; Girardin, S.E. NLRX1 is a mitochondrial NOD-like receptor that amplifies NF-kappaB and JNK pathways by inducing reactive oxygen species production. EMBO Rep. 2008, 9, 293–300. [Google Scholar] [CrossRef]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-stimulated genes: A complex web of host defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef]
- Menachery, V.D.; Eisfeld, A.J.; Schäfer, A.; Josset, L.; Sims, A.C.; Proll, S.; Fan, S.; Li, C.; Neumann, G.; Tilton, S.C.; et al. Pathogenic influenza viruses and coronaviruses utilize similar and contrasting approaches to control interferon-stimulated gene responses. mBio 2014, 5, e01174-14. [Google Scholar] [CrossRef]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.-C.; Uhl, S.; Hoagland, D.; Møller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181, 1036–1045.e9. [Google Scholar] [CrossRef]
- Chu, H.; Chan, J.F.; Wang, Y.; Yuen, T.T.; Chai, Y.; Hou, Y.; Shuai, H.; Yang, D.; Hu, B.; Huang, X.; et al. Comparative Replication and Immune Activation Profiles of SARS-CoV-2 and SARS-CoV in Human Lungs: An Ex Vivo Study with Implications for the Pathogenesis of COVID-19. Clin. Infect. Dis. 2020, 71, 1400–1409. [Google Scholar] [CrossRef]
- Hadjadj, J.; Yatim, N.; Barnabei, L.; Corneau, A.; Boussier, J.; Smith, N.; Péré, H.; Charbit, B.; Bondet, V.; Chenevier-Gobeaux, C.; et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 2020, 369, 718–724. [Google Scholar] [CrossRef]
- Chang, C.Y.; Liu, H.M.; Chang, M.-F.; Chang, S.C. Middle East Respiratory Syndrome Coronavirus Nucleocapsid Protein Suppresses Type I and Type III Interferon Induction by Targeting RIG-I Signaling. J. Virol. 2020, 94, e00099-20. [Google Scholar] [CrossRef]
- Sa Ribero, M.; Jouvenet, N.; Dreux, M.; Nisole, S. Interplay between SARS-CoV-2 and the type I interferon response. PLoS Pathog. 2020, 16, e1008737. [Google Scholar] [CrossRef]
- Yu, C.F.; Peng, W.M.; Schlee, M.; Barchet, W.; Eis-Hübinger, A.M.; Kolanus, W.; Geyer, M.; Schmitt, S.; Steinhagen, F.; Oldenburg, J.; et al. SOCS1 and SOCS3 Target IRF7 Degradation to Suppress TLR7-Mediated Type I IFN Production of Human Plasmacytoid Dendritic Cells. J. Immunol. 2018, 200, 4024–4035. [Google Scholar] [CrossRef]
- Jin, R.; Zhu, W.; Cao, S.; Chen, R.; Jin, H.; Liu, Y.; Wang, S.; Wang, W.; Xiao, G. Japanese encephalitis virus activates autophagy as a viral immune evasion strategy. PLoS ONE 2013, 8, e52909. [Google Scholar] [CrossRef]
- Ke, P.Y.; Chen, S.S.L. Activation of the unfolded protein response and autophagy after hepatitis C virus infection suppresses innate antiviral immunity in vitro. J. Clin. Investig. 2011, 121, 37–56. [Google Scholar] [CrossRef]
- Jounai, N.; Takeshita, F.; Kobiyama, K.; Sawano, A.; Miyawaki, A.; Xin, K.-Q.; Ishii, K.J.; Kawai, T.; Akira, S.; Suzuki, K. The Atg5–Atg12 conjugate associates with innate antiviral immune responses. Proc. Natl. Acad. Sci. USA 2007, 104, 14050–14055. [Google Scholar] [CrossRef]
- Lei, Y.; Wen, H.; Yu, Y.; Taxman, D.J.; Zhang, L.; Widman, D.G.; Swanson, K.V.; Wen, K.-W.; Damania, B.; Moore, C.B. The mitochondrial proteins NLRX1 and TUFM form a complex that regulates type I interferon and autophagy. Immunity 2012, 36, 933–946. [Google Scholar] [CrossRef]
- Tal, M.C.; Sasai, M.; Lee, H.K.; Yordy, B.; Shadel, G.S.; Iwasaki, A. Absence of autophagy results in reactive oxygen species-dependent amplification of RLR signaling. Proc. Natl. Acad. Sci. USA 2009, 106, 2770–2775. [Google Scholar] [CrossRef]
- Shi, C.S.; Qi, H.Y.; Boularan, C.; Huang, N.N.; Abu-Asab, M.; Shelhamer, J.H.; Kehrl, J.H. SARS-coronavirus open reading frame-9b suppresses innate immunity by targeting mitochondria and the MAVS/TRAF3/TRAF6 signalosome. J. Immunol. 2014, 193, 3080–3089. [Google Scholar] [CrossRef]
- Xing, Y.; Liqi, Z.; Jian, L.; Qinghua, Y.; Qian, Y. Doxycycline induces mitophagy and suppresses production of interferon-β in IPEC-J2 cells. Front. Cell. Infect. Microbiol. 2017, 7, 21. [Google Scholar] [CrossRef]
- Avia, M.; Rojas, J.M.; Miorin, L.; Pascual, E.; Van Rijn, P.A.; Martin, V.; García-Sastre, A.; Sevilla, N. Virus-induced autophagic degradation of STAT 2 as a mechanism for interferon signaling blockade. EMBO Rep. 2019, 20, e48766. [Google Scholar] [CrossRef]
- Song, J.; Hu, Y.; Li, J.; Zheng, H.; Wang, J.; Guo, L.; Shi, H.; Liu, L. Suppression of the toll-like receptor 7-dependent type I interferon production pathway by autophagy resulting from enterovirus 71 and coxsackievirus A16 infections facilitates their replication. Arch. Virol. 2018, 163, 135–144. [Google Scholar] [CrossRef]
- Mason, R.J. Pathogenesis of COVID-19 from a cell biology perspective. Eur. Respir. J. 2020, 55. [Google Scholar] [CrossRef]
- Polak, S.B.; Van Gool, I.C.; Cohen, D.; Jan, H.; van Paassen, J. A systematic review of pathological findings in COVID-19: A pathophysiological timeline and possible mechanisms of disease progression. Mod. Pathol. 2020, 33, 2128–2138. [Google Scholar] [CrossRef]
- Trouillet-Assant, S.; Viel, S.; Gaymard, A.; Pons, S.; Richard, J.-C.; Perret, M.; Villard, M.; Brengel-Pesce, K.; Lina, B.; Mezidi, M. Type I IFN immunoprofiling in COVID-19 patients. J. Allergy Clin. Immunol. 2020, 146, 206–208.e2. [Google Scholar] [CrossRef]
- Silvas, J.A.; Jureka, A.S.; Nicolini, A.M.; Chvatal, S.A.; Basler, C.F. Inhibitors of VPS34 and lipid metabolism suppress SARS-CoV-2 replication. bioRxiv 2020. [Google Scholar] [CrossRef]
- Yuen, C.K.; Wong, W.M.; Mak, L.F.; Wang, X.; Chu, H.; Yuen, K.Y.; Kok, K.H. Suppression of SARS-CoV-2 infection in ex-vivo human lung tissues by targeting class III phosphoinositide 3-kinase. J. Med. Virol. 2020. [Google Scholar] [CrossRef]
- Chen, G.; Wu, D.; Guo, W.; Cao, Y.; Huang, D.; Wang, H.; Wang, T.; Zhang, X.; Chen, H.; Yu, H.; et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J. Clin. Investig. 2020, 130, 2620–2629. [Google Scholar] [CrossRef]
- Nazinitsky, A.; Rosenthal, K.S. Cytokine storms: Systemic disasters of infectious diseases. Infect. Dis. Clin. Pract. 2010, 18, 188–192. [Google Scholar] [CrossRef]
- Merad, M.; Martin, J.C. Pathological inflammation in patients with COVID-19: A key role for monocytes and macrophages. Nat. Rev. Immunol. 2020, 20, 355–362. [Google Scholar] [CrossRef]
- Yen, Y.T.; Liao, F.; Hsiao, C.H.; Kao, C.L.; Chen, Y.C.; Wu-Hsieh, B.A. Modeling the early events of severe acute respiratory syndrome coronavirus infection in vitro. J. Virol. 2006, 80, 2684–2693. [Google Scholar] [CrossRef]
- Lau, S.K.; Lau, C.C.; Chan, K.H.; Li, C.P.; Chen, H.; Jin, D.Y.; Chan, J.F.; Woo, P.C.; Yuen, K.Y. Delayed induction of proinflammatory cytokines and suppression of innate antiviral response by the novel Middle East respiratory syndrome coronavirus: Implications for pathogenesis and treatment. J. Gen. Virol. 2013, 94, 2679–2690. [Google Scholar] [CrossRef]
- Liu, P.P.; Blet, A.; Smyth, D.; Li, H. The science underlying COVID-19: Implications for the cardiovascular system. Circulation 2020, 142, 68–78. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef]
- Yoneyama, M.; Kikuchi, M.; Natsukawa, T.; Shinobu, N.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Akira, S.; Fujita, T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 2004, 5, 730–737. [Google Scholar] [CrossRef]
- Ting, J.P.Y.; Lovering, R.C.; Alnemri, E.S.P.D.; Bertin, J.; Boss, J.M.; Davis, B.; Flavell, R.A.; Girardin, S.E.; Godzik, A.; Harton, J.A. The NLR gene family: An official nomenclature. Immunity 2008, 28, 285. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373. [Google Scholar] [CrossRef]
- O’Neill, L.A.; Bowie, A.G. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat. Rev. Immunol. 2007, 7, 353–364. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. TLR signaling. Semin. Immunol. 2007, 19, 24–32. [Google Scholar] [CrossRef]
- Kawai, T.; Takahashi, K.; Sato, S.; Coban, C.; Kumar, H.; Kato, H.; Ishii, K.J.; Takeuchi, O.; Akira, S. IPS-1, an adaptor triggering RIG-I-and Mda5-mediated type I interferon induction. Nat. Immunol. 2005, 6, 981–988. [Google Scholar] [CrossRef]
- Meylan, E.; Curran, J.; Hofmann, K.; Moradpour, D.; Binder, M.; Bartenschlager, R.; Tschopp, J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 2005, 437, 1167–1172. [Google Scholar] [CrossRef]
- Xu, L.G.; Wang, Y.Y.; Han, K.J.; Li, L.Y.; Zhai, Z.; Shu, H.B. VISA is an adapter protein required for virus-triggered IFN-β signaling. Mol. Cell 2005, 19, 727–740. [Google Scholar] [CrossRef]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef]
- Chavarría-Smith, J.; Vance, R.E. The NLRP1 inflammasomes. Immunol. Rev. 2015, 265, 22–34. [Google Scholar] [CrossRef]
- Martinon, F.; Mayor, A.; Tschopp, J. The inflammasomes: Guardians of the body. Annu. Rev. Immunol. 2009, 27, 229–265. [Google Scholar] [CrossRef]
- Nieto-Torres, J.L.; Verdiá-Báguena, C.; Jimenez-Guardeño, J.M.; Regla-Nava, J.A.; Castaño-Rodriguez, C.; Fernandez-Delgado, R.; Torres, J.; Aguilella, V.M.; Enjuanes, L. Severe acute respiratory syndrome coronavirus E protein transports calcium ions and activates the NLRP3 inflammasome. Virology 2015, 485, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.Y.; Moriyama, M.; Chang, M.F.; Ichinohe, T. Severe acute respiratory syndrome coronavirus viroporin 3a activates the NLRP3 inflammasome. Front. Microbiol. 2019, 10, 50. [Google Scholar] [CrossRef] [PubMed]
- Rioux, J.D.; Xavier, R.J.; Taylor, K.D.; Silverberg, M.S.; Goyette, P.; Huett, A.; Green, T.; Kuballa, P.; Barmada, M.M.; Datta, L.W. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat. Genet. 2007, 39, 596–604. [Google Scholar] [CrossRef]
- Barrett, J.C.; Hansoul, S.; Nicolae, D.L.; Cho, J.H.; Duerr, R.H.; Rioux, J.D.; Brant, S.R.; Silverberg, M.S.; Taylor, K.D.; Barmada, M.M. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat. Genet. 2008, 40, 955–962. [Google Scholar] [CrossRef] [PubMed]
- Parkes, M.; Barrett, J.C.; Prescott, N.J.; Tremelling, M.; Anderson, C.A.; Fisher, S.A.; Roberts, R.G.; Nimmo, E.R.; Cummings, F.R.; Soars, D. Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn’s disease susceptibility. Nat. Genet. 2007, 39, 830–832. [Google Scholar] [CrossRef]
- Saitoh, T.; Fujita, N.; Jang, M.H.; Uematsu, S.; Yang, B.-G.; Satoh, T.; Omori, H.; Noda, T.; Yamamoto, N.; Komatsu, M. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1β production. Nature 2008, 456, 264–268. [Google Scholar] [CrossRef]
- Deretic, V.; Levine, B. Autophagy balances inflammation in innate immunity. Autophagy 2018, 14, 243–251. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Coyne, C.B.; Zeh, H.J.; Lotze, M.T. PAMP s and DAMP s: Signal 0s that spur autophagy and immunity. Immunol. Rev. 2012, 249, 158–175. [Google Scholar] [CrossRef]
- Liu, X.; Cao, H.; Li, J.; Wang, B.; Zhang, P.; Zhang, X.D.; Liu, Z.; Yuan, H.; Zhan, Z. Autophagy induced by DAMPs facilitates the inflammation response in lungs undergoing ischemia-reperfusion injury through promoting TRAF6 ubiquitination. Cell Death Differ. 2017, 24, 683–693. [Google Scholar] [CrossRef]
- Shi, C.S.; Shenderov, K.; Huang, N.-N.; Kabat, J.; Abu-Asab, M.; Fitzgerald, K.A.; Sher, A.; Kehrl, J.H. Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat. Immunol. 2012, 13, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.-J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef]
- Kim, M.J.; Yoon, J.H.; Ryu, J.H. Mitophagy: A balance regulator of NLRP3 inflammasome activation. BMB Rep. 2016, 49, 529. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Bae, S.H.; Ryu, J.C.; Kwon, Y.; Oh, J.H.; Kwon, J.; Moon, J.S.; Kim, K.; Miyawaki, A.; Lee, M.G. SESN2/sestrin2 suppresses sepsis by inducing mitophagy and inhibiting NLRP3 activation in macrophages. Autophagy 2016, 12, 1272–1291. [Google Scholar] [CrossRef] [PubMed]
- Criollo, A.; Senovilla, L.; Authier, H.; Maiuri, M.C.; Morselli, E.; Vitale, I.; Kepp, O.; Tasdemir, E.; Galluzzi, L.; Shen, S. The IKK complex contributes to the induction of autophagy. EMBO J. 2010, 29, 619–631. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Umemura, A.; Sanchez-Lopez, E.; Liang, S.; Shalapour, S.; Wong, J.; He, F.; Boassa, D.; Perkins, G.; Ali, S.R. NF-κB restricts inflammasome activation via elimination of damaged mitochondria. Cell 2016, 164, 896–910. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.; Hartman, M.; Roche, C.; Zeng, S.G.; O’Shea, A.; Sharp, F.A.; Lambe, E.M.; Creagh, E.M.; Golenbock, D.T.; Tschopp, J. Autophagy controls IL-1β secretion by targeting pro-IL-1β for degradation. J. Biol. Chem. 2011, 286, 9587–9597. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.; Lang, T.; Thomas, J.P.; Sukkar, M.B.; Nabar, N.R.; Kehrl, J.H. Autophagy and inflammasomes. Mol. Immunol. 2017, 86, 10–15. [Google Scholar] [CrossRef]
- de Castro, C.P.; Jones, S.A.; Cheallaigh, C.N.; Hearnden, C.A.; Williams, L.; Winter, J.; Lavelle, E.C.; Mills, K.H.; Harris, J. Autophagy regulates IL-23 secretion and innate T cell responses through effects on IL-1 secretion. J. Immunol. 2012, 189, 4144–4153. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Franchi, L.; Toma, C.; Ashida, H.; Ogawa, M.; Yoshikawa, Y.; Mimuro, H.; Inohara, N.; Sasakawa, C.; Nuñez, G. Differential regulation of caspase-1 activation, pyroptosis, and autophagy via Ipaf and ASC in Shigella-infected macrophages. PLoS Pathog. 2007, 3, e111. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Duan, T.; Feng, Y.; Liu, Q.; Lin, M.; Cui, J.; Wang, R.-F. LRRC25 inhibits type I IFN signaling by targeting ISG15-associated RIG-I for autophagic degradation. EMBO J. 2018, 37, 351–366. [Google Scholar] [CrossRef] [PubMed]
- Xian, H.; Yang, S.; Jin, S.; Zhang, Y.; Cui, J. LRRC59 modulates type I interferon signaling by restraining the SQSTM1/p62-mediated autophagic degradation of pattern recognition receptor DDX58/RIG-I. Autophagy 2020, 16, 408–418. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Duan, T.; Du, Y.; Jin, S.; Wang, M.; Cui, J.; Wang, R.-F. LRRC25 functions as an inhibitor of NF-κB signaling pathway by promoting p65/RelA for autophagic degradation. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Mariathasan, S.; Newton, K.; Monack, D.M.; Vucic, D.; French, D.M.; Lee, W.P.; Roose-Girma, M.; Erickson, S.; Dixit, V.M. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 2004, 430, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Into, T.; Inomata, M.; Niida, S.; Murakami, Y.; Shibata, K. Regulation of MyD88 aggregation and the MyD88-dependent signaling pathway by sequestosome 1 and histone deacetylase 6. J. Biol. Chem. 2010, 285, 35759–35769. [Google Scholar] [CrossRef]
- Inomata, M.; Niida, S.; Shibata, K.; Into, T. Regulation of Toll-like receptor signaling by NDP52-mediated selective autophagy is normally inactivated by A20. Cell. Mol. Life Sci. 2012, 69, 963–979. [Google Scholar] [CrossRef]
- Iula, L.; Keitelman, I.A.; Sabbione, F.; Fuentes, F.; Guzman, M.; Galletti, J.G.; Gerber, P.P.; Ostrowski, M.; Geffner, J.R.; Jancic, C.C. Autophagy mediates interleukin-1β secretion in human neutrophils. Front. Immunol. 2018, 9, 269. [Google Scholar] [CrossRef]
- Misumi, I.; Starmer, J.; Uchimura, T.; Beck, M.A.; Magnuson, T.; Whitmire, J.K. Obesity expands a distinct population of T cells in adipose tissue and increases vulnerability to infection. Cell Rep. 2019, 27, 514–524. [Google Scholar] [CrossRef]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Denova-Gutiérrez, E.; Lopez-Gatell, H.; Alomia-Zegarra, J.L.; López-Ridaura, R.; Zaragoza-Jimenez, C.A.; Dyer-Leal, D.D.; Cortés-Alcala, R.; Villa-Reyes, T.; Gutiérrez-Vargas, R.; Rodríguez-González, K. The association between obesity, type 2 diabetes, and hypertension with severe COVID-19 on admission among Mexicans. Obesity (Silver Spring) 2020, 28, 1826–1832. [Google Scholar] [CrossRef]
- Blüher, M. Obesity: Global epidemiology and pathogenesis. Nat. Rev. Endocrinol. 2019, 15, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Bray, G.A. Etiology and pathogenesis of obesity. Clin. Cornerstone 1999, 2, 1–15. [Google Scholar] [CrossRef]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef]
- Sarparanta, J.; Garcia-Macia, M.; Singh, R. Autophagy and mitochondria in obesity and type 2 diabetes. Curr. Diabetes Rev. 2017, 13, 352–369. [Google Scholar] [CrossRef]
- Yang, L.; Li, P.; Fu, S.; Calay, E.S.; Hotamisligil, G.S. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010, 11, 467–478. [Google Scholar] [CrossRef]
- Saltiel, A.R.; Olefsky, J.M. Inflammatory mechanisms linking obesity and metabolic disease. J. Clin. Investig. 2017, 127, 1–4. [Google Scholar] [CrossRef]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef]
- Lumeng, C.N.; Bodzin, J.L.; Saltiel, A.R. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Investig. 2007, 117, 175–184. [Google Scholar] [CrossRef]
- Kang, Y.H.; Cho, M.H.; Kim, J.Y.; Kwon, M.-S.; Peak, J.-J.; Kang, S.W.; Yoon, S.Y.; Song, Y. Impaired macrophage autophagy induces systemic insulin resistance in obesity. Oncotarget 2016, 7, 35577–35591. [Google Scholar] [CrossRef]
- Fitzgibbons, T.P.; Czech, M.P. Emerging evidence for beneficial macrophage functions in atherosclerosis and obesity-induced insulin resistance. J. Mol. Med. 2016, 94, 267–275. [Google Scholar] [CrossRef]
- Liu, K.; Zhao, E.; Ilyas, G.; Lalazar, G.; Lin, Y.; Haseeb, M.; Tanaka, K.E.; Czaja, M.J. Impaired macrophage autophagy increases the immune response in obese mice by promoting proinflammatory macrophage polarization. Autophagy 2015, 11, 271–284. [Google Scholar] [CrossRef]
- Kassir, R. Risk of COVID-19 for patients with obesity. Obes. Rev. 2020, 21, e13034. [Google Scholar] [CrossRef]
- Sindhu, S.; Thomas, R.; Shihab, P.; Sriraman, D.; Behbehani, K.; Ahmad, R. Obesity is a positive modulator of IL-6R and IL-6 expression in the subcutaneous adipose tissue: Significance for metabolic inflammation. PLoS ONE 2015, 10, e0133494. [Google Scholar] [CrossRef]
- Vavougios, G.D. A data-driven hypothesis on the epigenetic dysregulation of host metabolism by SARS coronaviral infection: Potential implications for the SARS-CoV-2 modus operandi. Med. Hypotheses 2020, 140, 109759. [Google Scholar] [CrossRef]
- Yang, N.; Shen, H.-M. Targeting the Endocytic Pathway and Autophagy Process as a Novel Therapeutic Strategy in COVID-19. Int. J. Biol. Sci. 2020, 16, 1724–1731. [Google Scholar] [CrossRef]
- Bello-Perez, M.; Sola, I.; Novoa, B.; Klionsky, D.J.; Falco, A. Canonical and noncanonical autophagy as potential targets for COVID-19. Cells 2020, 9, 1619. [Google Scholar] [CrossRef]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) In Vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef]
- Savarino, A.; Boelaert, J.R.; Cassone, A.; Majori, G.; Cauda, R. Effects of chloroquine on viral infections: An old drug against today’s diseases. Lancet Infect. Dis. 2003, 3, 722–727. [Google Scholar] [CrossRef]
- Gratton, R.; Agrelli, A.; Tricarico, P.M.; Brandão, L.; Crovella, S. Autophagy in Zika virus infection: A possible therapeutic target to counteract viral replication. Int. J. Mol. Sci. 2019, 20, 1048. [Google Scholar] [CrossRef]
- Liu, J.; Cao, R.; Xu, M.; Wang, X.; Zhang, H.; Hu, H.; Li, Y.; Hu, Z.; Zhong, W.; Wang, M. Hydroxychloroquine, a less toxic derivative of chloroquine, is effective in inhibiting SARS-CoV-2 infection In Vitro. Cell Discov. 2020, 6, 16. [Google Scholar] [CrossRef]
- Gao, J.; Tian, Z.; Yang, X. Breakthrough: Chloroquine phosphate has shown apparent efficacy in treatment of COVID-19 associated pneumonia in clinical studies. Biosci. Trends 2020, 14, 72–73. [Google Scholar] [CrossRef]
- Huang, M.; Li, M.; Xiao, F.; Pang, P.; Liang, J.; Tang, T.; Liu, S.; Chen, B.; Shu, J.; You, Y.; et al. Preliminary evidence from a multicenter prospective observational study of the safety and efficacy of chloroquine for the treatment of COVID-19. Natl. Sci. Rev. 2020, 7, 1428–1436. [Google Scholar] [CrossRef]
- Singh, A.K.; Singh, A.; Singh, R.; Misra, A. Hydroxychloroquine in patients with COVID-19: A Systematic Review and meta-analysis. Diabetes Metab. Syndr. 2020, 14, 589–596. [Google Scholar] [CrossRef]
- Mahévas, M.; Tran, V.T.; Roumier, M.; Chabrol, A.; Paule, R.; Guillaud, C.; Fois, E.; Lepeule, R.; Szwebel, T.A.; Lescure, F.X.; et al. Clinical efficacy of hydroxychloroquine in patients with covid-19 pneumonia who require oxygen: Observational comparative study using routine care data. BMJ 2020, 369, m1844. [Google Scholar] [CrossRef]
- Gautret, P.; Lagier, J.C.; Parola, P.; Hoang, V.T.; Meddeb, L.; Mailhe, M.; Doudier, B.; Courjon, J.; Giordanengo, V.; Vieira, V.E.; et al. Hydroxychloroquine and azithromycin as a treatment of COVID-19: Results of an open-label non-randomized clinical trial. Int. J. Antimicrob. Agents 2020, 56, 105949. [Google Scholar] [CrossRef]
- Chen, Z.; Hu, J.; Zhang, Z. Efficacy of hydroxychloroquine in patients with COVID-19: Results of a randomized clinical trial. Version 2. 03.22. [Preprint]. MedRxiv 2020, 20040758. [Google Scholar] [CrossRef]
- Vollmer, J.; Tluk, S.; Schmitz, C.; Hamm, S.; Jurk, M.; Forsbach, A.; Akira, S.; Kelly, K.M.; Reeves, W.H.; Bauer, S.; et al. Immune stimulation mediated by autoantigen binding sites within small nuclear RNAs involves Toll- like receptors 7 and 8. J. Exp. Med. 2005, 202, 1575–1585. [Google Scholar] [CrossRef]
- Zhang, X.; Wu, J.; Du, F.; Xu, H.; Sun, L.; Chen, Z.; Brautigam, C.A.; Zhang, X.; Chen, Z.J. The cytosolic DNA sensor cGAS forms an oligomeric complex with DNA and undergoes switch- like conformational changes in the activation loop. Cell Rep. 2014, 6, 421–430. [Google Scholar] [CrossRef]
- Schrezenmeier, E.; Dörner, T. Mechanisms of action of hydroxychloroquine and chloroquine: Implications for rheumatology. Nat. Rev. Rheumatol. 2020, 16, 155–166. [Google Scholar] [CrossRef]
- de Barros, C.M.; de F. Almeida, C.A.; Pereira, B.; Costa, K.C.M.; Pinheiro, F.A.; Maia, L.D.B.; Trindade, C.M.; Garcia, R.C.T.; Torres, L.H.; Diwan, S.; et al. COVID-19 Pandemic—A Narrative Review of the Potential Roles of Chloroquine and Hydroxychloroquine. Pain Physician 2020, 23, S351–S366. [Google Scholar]
- De Mei, C.; Ercolani, L.; Parodi, C.; Veronesi, M.; Vecchio, C.L.; Bottegoni, G.; Torrente, E.; Scarpelli, R.; Marotta, R.; Ruffili, R. Dual inhibition of REV-ERBβ and autophagy as a novel pharmacological approach to induce cytotoxicity in cancer cells. Oncogene 2015, 34, 2597–2608. [Google Scholar] [CrossRef]
- McAfee, Q.; Zhang, Z.; Samanta, A.; Levi, S.M.; Ma, X.-H.; Piao, S.; Lynch, J.P.; Uehara, T.; Sepulveda, A.R.; Davis, L.E. Autophagy inhibitor Lys05 has single-agent antitumor activity and reproduces the phenotype of a genetic autophagy deficiency. Proc. Natl. Acad. Sci. USA 2012, 109, 8253–8258. [Google Scholar] [CrossRef]
- Dai, J.P.; Zhao, X.F.; Zeng, J.; Wan, Q.Y.; Yang, J.C.; Li, W.Z.; Chen, X.X.; Wang, G.F.; Li, K.S. Drug screening for autophagy inhibitors based on the dissociation of Beclin1-Bcl2 complex using BiFC technique and mechanism of eugenol on anti-influenza A virus activity. PLoS ONE 2013, 8, e61026. [Google Scholar] [CrossRef]
- Dai, J.P.; Li, W.Z.; Zhao, X.F.; Wang, G.F.; Yang, J.C.; Zhang, L.; Chen, X.X.; Xu, Y.X.; Li, K.S. A drug screening method based on the autophagy pathway and studies of the mechanism of evodiamine against influenza A virus. PLoS ONE 2012, 7, e42706. [Google Scholar] [CrossRef]
- Martin, K.R.; Celano, S.L.; Solitro, A.R.; Gunaydin, H.; Scott, M.; O’Hagan, R.C.; Shumway, S.D.; Fuller, P.; MacKeigan, J.P. A potent and selective ULK1 inhibitor suppresses autophagy and sensitizes cancer cells to nutrient stress. iScience 2018, 8, 74–84. [Google Scholar] [CrossRef]
- Lazarus, M.B.; Novotny, C.J.; Shokat, K.M. Structure of the human autophagy initiating kinase ULK1 in complex with potent inhibitors. ACS Chem. Biol. 2015, 10, 257–261. [Google Scholar] [CrossRef]
- Petherick, K.J.; Conway, O.J.; Mpamhanga, C.; Osborne, S.A.; Kamal, A.; Saxty, B.; Ganley, I.G. Pharmacological inhibition of ULK1 kinase blocks mammalian target of rapamycin (mTOR)-dependent autophagy. J. Biol. Chem. 2015, 290, 11376–11383. [Google Scholar] [CrossRef]
- Egan, D.F.; Chun, M.G.; Vamos, M.; Zou, H.; Rong, J.; Miller, C.J.; Lou, H.J.; Raveendra-Panickar, D.; Yang, C.C.; Sheffler, D.J. Small molecule inhibition of the autophagy kinase ULK1 and identification of ULK1 substrates. Mol. Cell 2015, 59, 285–297. [Google Scholar] [CrossRef]
- Seglen, P.O.; Gordon, P.B. 3-Methyladenine: Specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc. Natl. Acad. Sci. USA 1982, 79, 1889–1892. [Google Scholar] [CrossRef]
- Blommaart, E.F.; Krause, U.; Schellens, J.P.; Vreeling-Sindelárová, H.; Meijer, A.J. The phosphatidylinositol 3-kinase inhibitors wortmannin and LY294002 inhibit autophagy in isolated rat hepatocytes. Eur. J. Biochem. 1997, 243, 240–246. [Google Scholar] [CrossRef]
- Miller, S.; Tavshanjian, B.; Oleksy, A.; Perisic, O.; Houseman, B.T.; Shokat, K.M.; Williams, R.L. Shaping development of autophagy inhibitors with the structure of the lipid kinase Vps34. Science 2010, 327, 1638–1642. [Google Scholar] [CrossRef]
- Knight, S.D.; Adams, N.D.; Burgess, J.L.; Chaudhari, A.M.; Darcy, M.G.; Donatelli, C.A.; Luengo, J.I.; Newlander, K.A.; Parrish, C.A.; Ridgers, L.H.; et al. Discovery of GSK2126458, a Highly Potent Inhibitor of PI3K and the Mammalian Target of Rapamycin. ACS Med. Chem. Lett. 2010, 1, 39–43. [Google Scholar] [CrossRef]
- Shao, S.; Li, S.U.; Qin, Y.; Wang, X.; Yang, Y.; Bai, H.; Zhou, L.; Zhao, C.; Wang, C. Spautin-1, a novel autophagy inhibitor, enhances imatinib-induced apoptosis in chronic myeloid leukemia. Int. J. Oncol. 2014, 44, 1661–1668. [Google Scholar] [CrossRef]
- Pasquier, B. SAR405, a PIK3C3/Vps34 inhibitor that prevents autophagy and synergizes with MTOR inhibition in tumor cells. Autophagy 2015, 11, 725–726. [Google Scholar] [CrossRef]
- Pasquier, B.; El-Ahmad, Y.; Filoche-Rommé, B.; Dureuil, C.; Fassy, F.; Abecassis, P.Y.; Mathieu, M.; Bertrand, T.; Benard, T.; Barrière, C.; et al. Discovery of (2S)-8-[(3R)-3-methylmorpholin-4-yl]-1-(3-methyl-2-oxobutyl)-2-(trifluoromethyl)-3,4-dihydro-2H-pyrimido[1,2-a]pyrimidin-6-one: A novel potent and selective inhibitor of Vps34 for the treatment of solid tumors. J. Med. Chem. 2015, 58, 376–400. [Google Scholar] [CrossRef]
- Dowdle, W.E.; Nyfeler, B.; Nagel, J.; Elling, R.A.; Liu, S.; Triantafellow, E.; Menon, S.; Wang, Z.; Honda, A.; Pardee, G.; et al. Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis In Vivo. Nat. Cell Biol. 2014, 16, 1069–1079. [Google Scholar] [CrossRef]
- Scheen, A.J. Metformin and COVID-19: From cellular mechanisms to reduced mortality. Diabetes Metab. 2020, 46, 423–426. [Google Scholar] [CrossRef]
- Gao, C.; Fang, L.; Zhang, H.; Zhang, W.-S.; Li, X.O.; Du, S.Y. Metformin Induces Autophagy via the AMPK-mTOR Signaling Pathway in Human Hepatocellular Carcinoma Cells. Cancer Manag. Res. 2020, 12, 5803–5811. [Google Scholar] [CrossRef]
- Saisho, Y. Metformin and inflammation: Its potential beyond glucose-lowering effect. Endocr. Metab. Immune Disord. Drug Targets 2015, 15, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Yuk, J.M.; Shin, D.M.; Lee, H.-M.; Yang, C.S.; Jin, H.S.; Kim, K.K.; Lee, Z.W.; Lee, S.H.; Kim, J.M.; Jo, E.K. Vitamin D3 induces autophagy in human monocytes/macrophages via cathelicidin. Cell Host Microbe 2009, 6, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Ohaegbulam, K.C.; Swalih, M.; Patel, P.; Smith, M.A.; Perrin, R. Vitamin D Supplementation in COVID-19 Patients: A Clinical Case Series. Am. J. Ther. 2020, 27, e485–e490. [Google Scholar] [CrossRef]
- Puleston, D.J.; Zhang, H.; Powell, T.J.; Lipina, E.; Sims, S.; Panse, I.; Watson, A.S.; Cerundolo, V.; Townsend, A.R.; Klenerman, P. Autophagy is a critical regulator of memory CD8+ T cell formation. Elife 2014, 3, e03706. [Google Scholar] [CrossRef]
- Sacitharan, P.K.; Lwin, S.; Gharios, G.B.; Edwards, J.R. Spermidine restores dysregulated autophagy and polyamine synthesis in aged and osteoarthritic chondrocytes via EP300. Exp. Mol. Med. 2018, 50, 123. [Google Scholar] [CrossRef]
- Huang, F.C.; Kuo, H.C.; Huang, Y.H.; Yu, H.R.; Li, S.C.; Kuo, H.C. Anti-inflammatory effect of resveratrol in human coronary arterial endothelial cells via induction of autophagy: Implication for the treatment of Kawasaki disease. BMC Pharmacol. Toxicol. 2017, 18, 3. [Google Scholar] [CrossRef]
- Filardo, S.; Di Pietro, M.; Mastromarino, P.; Sessa, R. Therapeutic potential of resveratrol against emerging respiratory viral infections. Pharmacol. Ther. 2020, 214, 107613. [Google Scholar] [CrossRef]
- Belzile, J.P.; Sabalza, M.; Craig, M.; Clark, E.; Morello, C.S.; Spector, D.H. Trehalose, an mTOR-independent inducer of autophagy, inhibits human cytomegalovirus infection in multiple cell types. J. Virol. 2016, 90, 1259–1277. [Google Scholar] [CrossRef]
- Martinon, D.; Borges, V.F.; Gomez, A.C.; Shimada, K. Potential fast COVID-19 containment with trehalose. Front. Immunol. 2020, 11, 1623. [Google Scholar] [CrossRef]
- Rossignol, J.F. Nitazoxanide, a new drug candidate for the treatment of Middle East respiratory syndrome coronavirus. J. Infect. Public Health 2016, 9, 227–230. [Google Scholar] [CrossRef]
- Lam, K.K.; Zheng, X.; Forestieri, R.; Balgi, A.D.; Nodwell, M.; Vollett, S.; Anderson, H.J.; Andersen, R.J.; Av-Gay, Y.; Roberge, M. Nitazoxanide stimulates autophagy and inhibits mTORC1 signaling and intracellular proliferation of Mycobacterium tuberculosis. PLoS Pathog. 2012, 8, e1002691. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liang, H.; Chen, C.; Wang, X.; Qu, F.; Wang, H.; Yang, K.; Wang, Q.; Zhao, N.; Meng, J.; et al. Ivermectin induces autophagy-mediated cell death through the AKT/mTOR signaling pathway in glioma cells. BioSci. Rep. 2019, 39, BSR20192489. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Yu, K.M.; Kim, Y.I.; Kim, S.M.; Kim, E.H.; Kim, S.G.; Kim, E.J.; Casel, M.A.B.; Rollon, R.; Jang, S.G.; et al. Antiviral Efficacies of FDA-Approved Drugs against SARS-CoV-2 Infection in Ferrets. mBio 2020, 11, e01114-20. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, M.; Lapierre, J.; Ojha, C.R.; Pawitwar, S.; Karuppan, M.K.M.; Kashanchi, F.; El-Hage, N. Morphine counteracts the antiviral effect of antiretroviral drugs and causes upregulation of p62/SQSTM1 and histone-modifying enzymes in HIV-infected astrocytes. J. Neurovirol. 2019, 25, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, A.; Thangaraj, A.; Chivero, E.T.; Periyasamy, P.; Callen, S.; Burkovetskaya, M.E.; Guo, M.-L.; Buch, S. Antiretroviral-Mediated Microglial Activation Involves Dysregulated Autophagy and Lysosomal Dysfunction. Cells 2019, 8, 1168. [Google Scholar] [CrossRef]
- Sigman, S.A.; Mokmeli, S.; Monici, M.; Vetrici, M.A. A 57-Year-Old African American Man with Severe COVID-19 Pneumonia Who Responded to Supportive Photobiomodulation Therapy (PBMT): First Use of PBMT in COVID-19. Am. J. Case Rep. 2020, 21, e926779-1. [Google Scholar] [CrossRef]
- Rubio, N.; Verrax, J.; Dewaele, M.; Verfaillie, T.; Johansen, T.; Piette, J.; Agostinis, P. p38MAPK-regulated induction of p62 and NBR1 after photodynamic therapy promotes autophagic clearance of ubiquitin aggregates and reduces reactive oxygen species levels by supporting Nrf2–antioxidant signaling. Free Radic. Biol. Med. 2014, 67, 292–303. [Google Scholar] [CrossRef]
- Dewaele, M.; Martinet, W.; Rubio, N.; Verfaillie, T.; de Witte, P.A.; Piette, J.; Agostinis, P. Autophagy pathways activated in response to PDT contribute to cell resistance against ROS damage. J. Cell. Mol. Med. 2011, 15, 1402–1414. [Google Scholar] [CrossRef]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef]
- Wang, Y.X.; Yang, L.; Wang, H.Q.; Zhao, X.Q.; Liu, T.; Li, Y.H.; Zeng, Q.X.; Li, Y.H.; Song, D.Q. Synthesis and Evolution of Berberine Derivatives as a New Class of Antiviral Agents against Enterovirus 71 through the MEK/ERK Pathway and Autophagy. Molecules 2018, 23, 2084. [Google Scholar] [CrossRef]
- Yu, C.; Li, W.B.; Liu, J.B.; Lu, J.W.; Feng, J.F. Autophagy: Novel applications of nonsteroidal anti-inflammatory drugs for primary cancer. Cancer Med. 2018, 7, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.C.; Guan, K.L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Teymoori-Rad, M.; Shokri, F.; Salimi, V.; Marashi, S.M. The interplay between vitamin D and viral infections. Rev. Med. Virol. 2019, 29, e2032. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.L.; Yi, L.; Jin, X.; Liang, X.Y.; Zhou, Y.; Zhang, T.; Xie, Q.; Zhou, X.; Chang, H.; Fu, Y.J.; et al. Resveratrol attenuates vascular endothelial inflammation by inducing autophagy through the cAMP signaling pathway. Autophagy 2013, 9, 2033–2045. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatments | Mechanisms of Action | Type of Study | Main Results | Ref. |
|---|---|---|---|---|
| Remdesivir | A monophosphoramidate prodrug of an adenosine analogue that inhibits viral RNA polymerases | (a) Clinical trial (b) Compassionate use | (a) No association with statistically significant clinical benefits. (b) Clinical improvement in 36 of 53 patients (68%). | (a) [1] (b) [2] |
| Lopinavir/Ritonavir | A co-formulation of two structurally related protease-inhibitors as antiretroviral agents (HIV type 1 aspartate protease inhibitors) | Clinical trial | No significant benefit from the treatment compared to standard care. | [3] |
| Favipiravir plus IFN-α | Inhibits the RNA-dependent RNA polymerase (RdRp) of RNA viruses | Open label control study | Attenuated disease progression and improved viral clearance. | [4] |
| Ivermectin | A synthetic derivative of a macrocyclic lactone antiparasitic agent. Inhibits the nuclear import of host and viral proteins | In-vitro antiviral activity against SARS-CoV-2 | Compared to the DMSO-treated control, a 93% reduction in viral RNA and a 99.9% in cell-associated viral RNA. | [5] |
| Hydroxychloroquine and Chloroquine | Both drugs accumulate in lysosomes, leading to elevated intra-vesicular pH that prevents endosome trafficking and viral fusion. They also interfere with the glycosylation of ACE-2 receptors, which prevents their binding by SARS-CoV-2 and thus infection. | (a) Prospective randomized trial (b) A pilot observational study (c) Discontinued by WHO | (a) No significant difference between patients with hydroxychloroquine + conventional treatment and those with the conventional treatment alone. (b) Clinical improvement in all participating patients receiving co-administration of hydroxychloroquine with azithromycin. | (a) [6] (b) [7] (c) [8] |
| Interferon (IFN)-α | A broad-spectrum antiviral agent | Clinical trials in process | IFN-β1a and IFN-α2b are currently being evaluated as potential candidates for the treatment of patients with COVID-19. | [9] |
| Arbidol/lopinavir/ritonavir | By inhibiting the virus-mediated fusion with the target membrane, arbidol blocks virus entry into the target cells | (a) Retrospective cohort study (b) Cohort of 50 patients in two groups: lopinavir/ritonavir regimen (34 cases) and arbidol alone (16 cases) | (a) A significant increase in the conversion rate from positive to negative results for the coronavirus test on days 7 and 14 for patients receiving arbidol plus lopinavir/ritonavir versus monotherapy with lopinavir/ritonavir. (b) After 14 days of treatment, there was no viral load for the arbidol-treated group, but a 44.1% viral load for the lopinavir/ritonavir-treated group. | (a) [10] (b) [11] |
| Tocilizumab | A humanized anti-interleukin-6-receptor (IL-6R) monoclonal antibody that inhibits IL-6 | (a) Retrospective observational study (b) Cohort of 100 patients (c) Retrospective study | (a) No attenuation of the disease in critically ill patients after a single dose of tocilizumab. (b) A rapid and sustained positive response to tocilizumab treatment. (c) Alleviation of the clinical symptoms and avoidance of severe COVID-19 with tocilizumab treatments. | (a) [12] (b) [13] (c) [14] |
| Convalescent plasma therapy | Appears to exhibit a neutralizing antibody response directed against the viral S protein. The antibodies block SARS-CoV-ACE2 entry. | (a) Evaluation of 6 COVID-19 patients (b) Case series analysis of 5 critically ill patients (c) Open-label, multi-center, randomized clinical trial | (a) Effective in alleviating patient symptoms and ameliorating radiological injuries. (b) Improved clinical status of patients. (c) No statistically significant improvement in the clinical condition of patients. | (a) [15] (b) [16] (c) [17] |
| Corticosteroids | Anti-inflammatory effects are due to a negative regulatory mechanism (transrepression). | Cohort of 41 patients | Suppressed lung inflammation in 21% of patients. | [18] |
| Prezcobix | HIV protease inhibitor | Under clinical trials | The primary endpoints included symptom improvement and virus nucleic acid turning negative, but the optimal endpoint has not been determined. | [19] |
| Oseltamivir | Neuraminidase inhibitor. | (a) COVID-19 patients (75) (b) Non-severe and severe COVID-19 patients (393) | (a) Recovery rate: 31%; Mortality rate: 11%. (b) No significant improvement in the clinical condition of patients. | (a) [20] (b) [21] |
| Compounds | Effect on Autophagy | Mechanism of Action * | FDA Approval | Reference |
|---|---|---|---|---|
| Chloroquine (CQ) and Hydroxychloroquine (HCQ) | Inhibitors | Interfere with autophagosome-lysosome fusion. | Yes | [220] |
| ARN5187 Lys05 | Inhibitors | Block autophagosome maturation. | No Yes | [184,185] |
| Eugenol | Inhibitor | A decline in oxidative stress and activation of ERK1/2, p38MAPK and IKK/NF-κB; downstream, lesser dissociation of the Beclin1-Bcl2 heterodimer and reduced autophagy. | No | [186] |
| Evodiamine | Inhibitor | A decreased formation of the Atg5-Atg12/Atg16 heterotrimer and expression of Atg5, Atg7 and Atg12. | No | [187] |
| Berberine derivatives | Inhibitors | Diminished activation of the MEK/ERK signaling pathway. | No | [221] |
| ULK-100 ULK-101 Compound 6 MRT67307 MRT68921 SBI-0206965 | Inhibitors | Inhibition of the ULK complex. | No | [188,189,190,191] |
| 3-methyladenine Wortmannin LY294002 PT210 GSK-2126458 | Inhibitors | Inhibition of PI3K. | No | [192,193,194,195] |
| VPS34-IN1 VVPS34-IN1 Spautin-1 SAR405 Compound 31 PIK-III | Inhibitors | Inhibition of VPS34. | No | [103,104,196,197,198,199] |
| Spermidine | Activator | An increase in the expression of acetyltransferase EP300, known to bind to crucial autophagy proteins (Beclin1 and LC3) and stimulate autophagy. | Substance registration system | [206] |
| Nonsteroidal anti-inflammatory drugs (NSAIDs): Celecoxib Sodium Salicylate Aspirin Sulfasalazine Piroxicam Indomethacin | Activators | Modulation of autophagy through the signaling pathways of PI3K/Akt/mTOR, MAPK/ERK1/2, P53/DRAM, AMPK/mTOR, Bip/GRP78, CHOP/GADD153 and HGF/MET. | Yes | [222] |
| Rapamycin and derivative compounds (RAD001, CCI-779 and AP23573). AZD8055 Torin 1 Metformin | Activators | Inhibition of mTOR. | Yes No Yes | [223] |
| Vitamin D3 | Activator | Activation of autophagy, though the pathway is unclear. Stimulation of calcium signaling is a proposed mechanism. | No | [224] |
| Resveratrol | Activator | Activation of autophagy by triggering the cAMPPRKA- AMPK-SI RT1 signaling pathway. | Clinical trial on animals (cancer therapy). | [225] |
| Trehalose | Activator | Activity independent of mTOR. | A food additive. | [209] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Pérez, B.E.; González-Rojas, J.A.; Salazar, M.I.; Torres-Torres, C.; Castrejón-Jiménez, N.S. Taming the Autophagy as a Strategy for Treating COVID-19. Cells 2020, 9, 2679. https://doi.org/10.3390/cells9122679
García-Pérez BE, González-Rojas JA, Salazar MI, Torres-Torres C, Castrejón-Jiménez NS. Taming the Autophagy as a Strategy for Treating COVID-19. Cells. 2020; 9(12):2679. https://doi.org/10.3390/cells9122679
Chicago/Turabian StyleGarcía-Pérez, Blanca Estela, Juan Antonio González-Rojas, Ma Isabel Salazar, Carlos Torres-Torres, and Nayeli Shantal Castrejón-Jiménez. 2020. "Taming the Autophagy as a Strategy for Treating COVID-19" Cells 9, no. 12: 2679. https://doi.org/10.3390/cells9122679
APA StyleGarcía-Pérez, B. E., González-Rojas, J. A., Salazar, M. I., Torres-Torres, C., & Castrejón-Jiménez, N. S. (2020). Taming the Autophagy as a Strategy for Treating COVID-19. Cells, 9(12), 2679. https://doi.org/10.3390/cells9122679