Neuron-Astrocyte Interactions in Parkinson’s Disease



Abstract
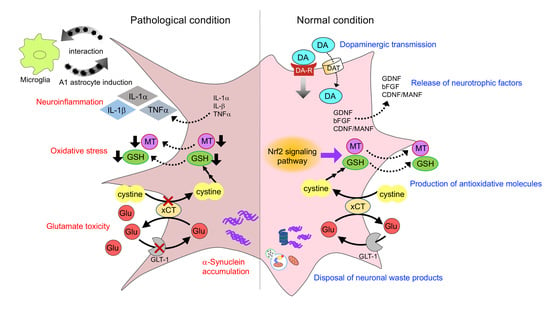
{kind=link}
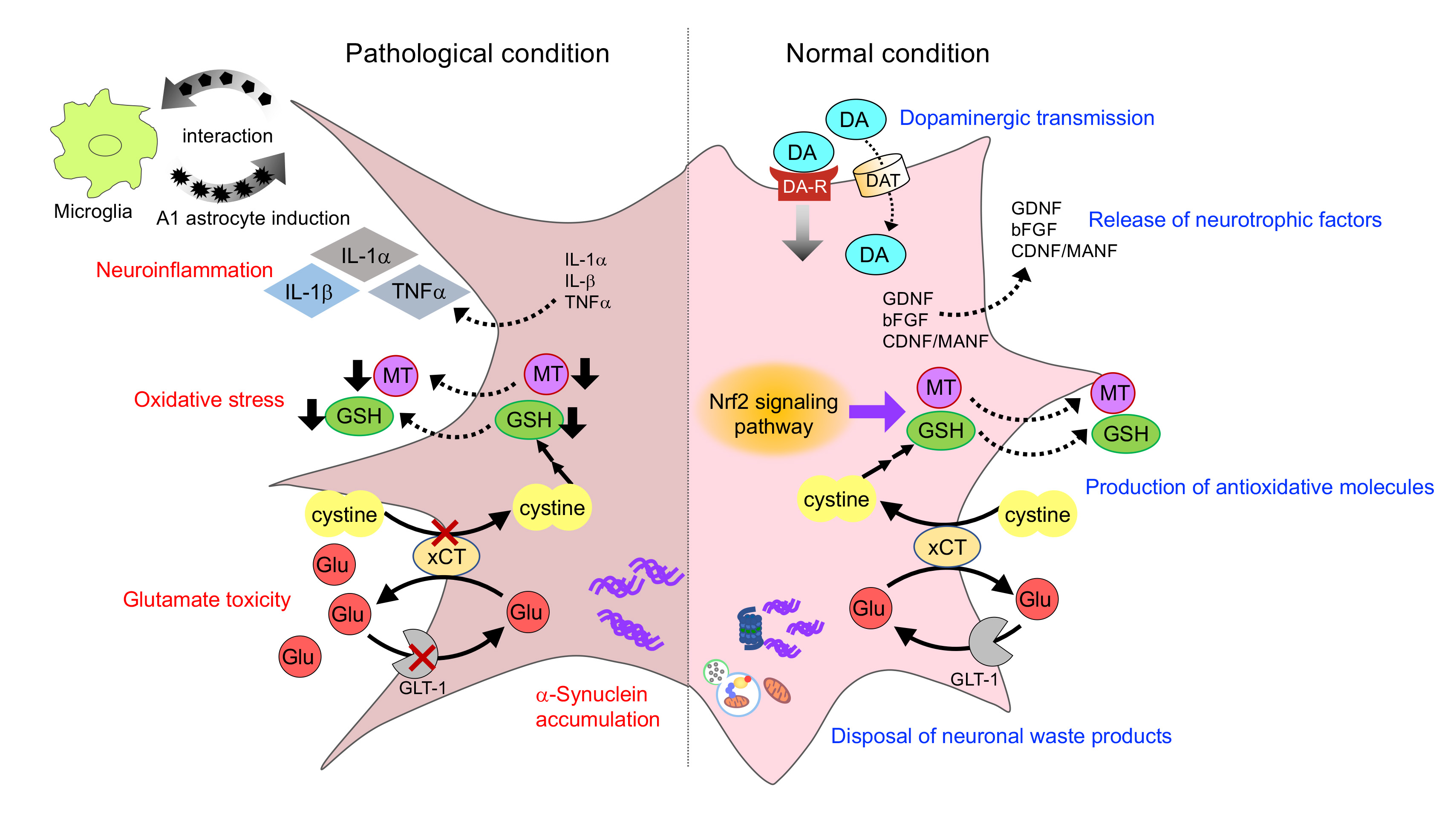
{kind=link}
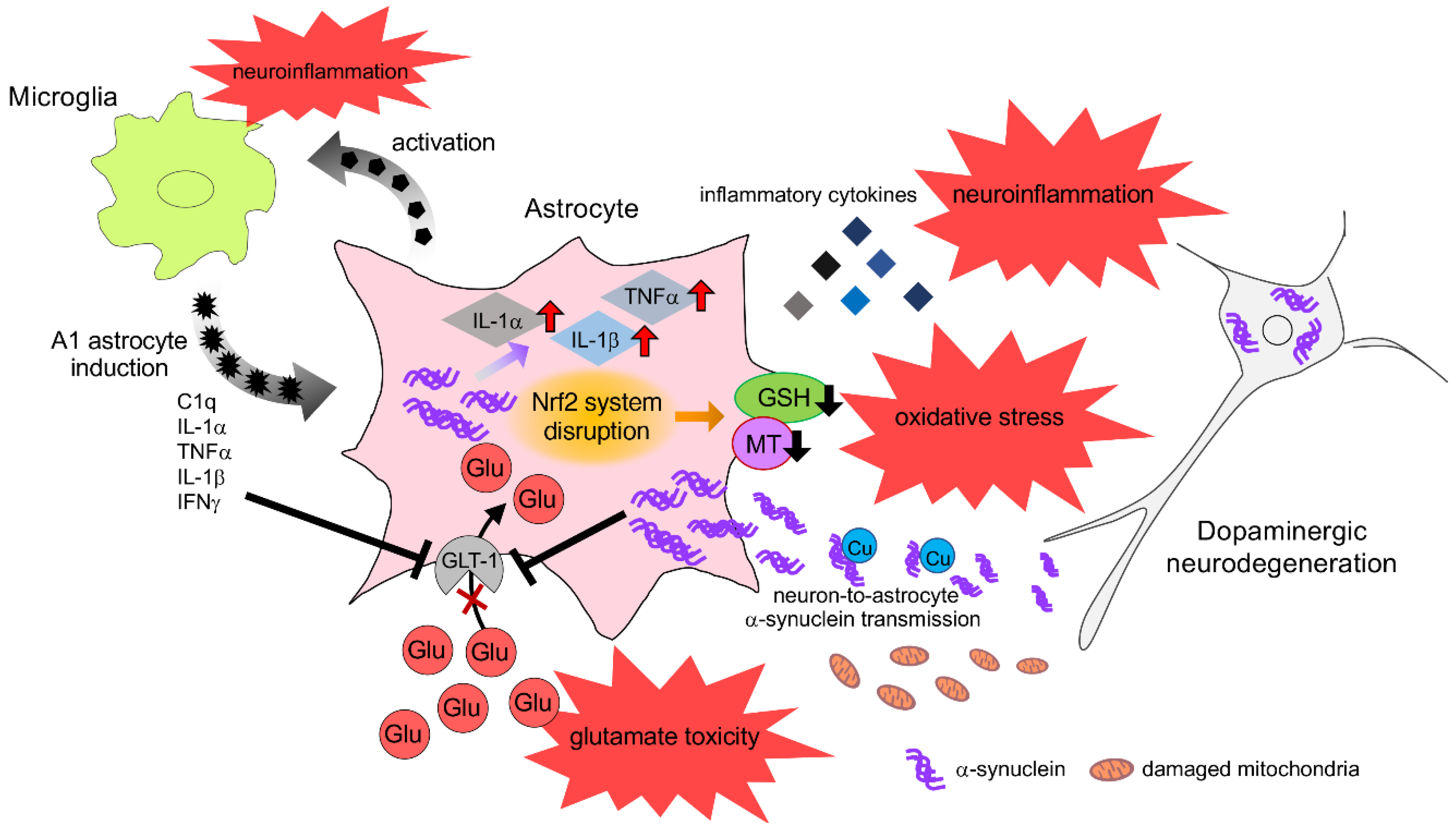
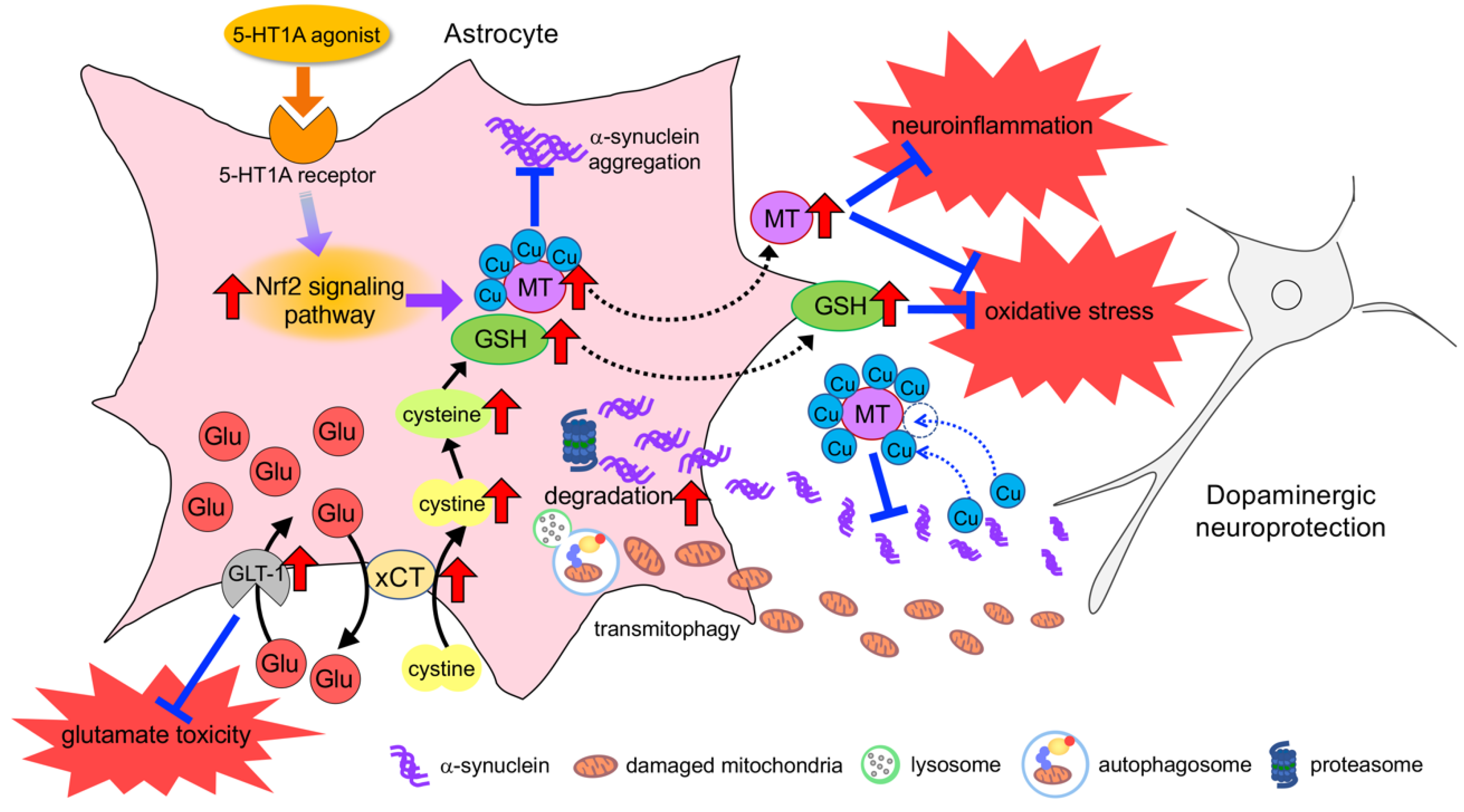{kind=link}
1. Introduction
2. Interactions of Astrocytes and Dopaminergic Neurons
2.1. Dopaminergic Transmission
2.2. Release of Neurotrophic Factors
2.3. Production of Antioxidative Molecules
2.4. Rescue of Mitochondria in Dopaminergic Neurons
2.5. Disposal of Neuronal Waste Products
3. Histology of Astrocyte in Parkinsonism
3.1. Astrocyte Reaction
3.2. Expression and Inclusion of α-Synuclein in Astrocytes
4. Neurotoxic Role of Astrocyte in Parkinsonism
4.1. Neuroinflammation
4.2. α-Synuclein Toxicity
5. Astrocyte Dysfunction in Parkinsonism
6. Neuroprotective Effects of Astrocyte and Therapeutic Strategy for PD
6.1. Neuroprotective Response of Astrocytes against Oxidative Stress
6.2. Upregulation of Neuroprotective Properties in Astrocytes
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Schapira, A.H.V.; Chaudhuri, K.R.; Jenner, P. Non-motor features of Parkinson disease. Nat. Rev. Neurosci. 2017, 18, 435–450. [Google Scholar] [CrossRef]
- Braak, H.; de Vos, R.A.; Bohl, J.; Del Tredici, K. Gastric alpha-synuclein immunoreactive inclusions in Meissner’s and Auerbach’s plexuses in cases staged for Parkinson’s disease-related brain pathology. Neurosci. Lett. 2006, 396, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Orimo, S.; Uchihara, T.; Nakamura, A.; Mori, F.; Kakita, A.; Wakabayashi, K.; Takahashi, H. Axonal alpha-synuclein aggregates herald centripetal degeneration of cardiac sympathetic nerve in Parkinson’s disease. Brain 2008, 131, 642–650. [Google Scholar] [CrossRef] [PubMed]
- Hawkes, C.H.; Del Tredici, K.; Braak, H. A timeline for Parkinson’s disease. Parkinsonism Relat. Disord. 2010, 16, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Klingelhoefer, L.; Reichmann, H. Pathogenesis of Parkinson disease--the gut-brain axis and environmental factors. Nat. Rev. Neurol. 2015, 11, 625–636. [Google Scholar] [CrossRef]
- Gleichman, A.J.; Carmichael, S.T. Glia in neurodegeneration: Drivers of disease or along for the ride? Neurobiol. Dis. 2020, 142, 104957. [Google Scholar] [CrossRef]
- Halliday, G.M.; Holton, J.L.; Revesz, T.; Dickson, D.W. Neuropathology underlying clinical variability in patients with synucleinopathies. Acta Neuropathol. 2011, 122, 187–204. [Google Scholar] [CrossRef]
- Maragakis, N.J.; Rothstein, J.D. Mechanisms of Disease: Astrocytes in neurodegenerative disease. Nat. Clin. Pract. Neurol. 2006, 2, 679–689. [Google Scholar] [CrossRef]
- Miyazaki, I.; Asanuma, M. Therapeutic Strategy of Targeting Astrocytes for Neuroprotection in Parkinson’s Disease. Curr. Pharm. Des. 2017, 23, 4936–4947. [Google Scholar] [CrossRef]
- Tremblay, M.E.; Cookson, M.R.; Civiero, L. Glial phagocytic clearance in Parkinson’s disease. Mol. Neurodegener. 2019, 14, 16. [Google Scholar] [CrossRef]
- Sofroniew, M.V. Astrocyte Reactivity: Subtypes, States, and Functions in CNS Innate Immunity. Trends Immunol. 2020, 41, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, I.; Asanuma, M.; Kikkawa, Y.; Takeshima, M.; Murakami, S.; Miyoshi, K.; Sogawa, N.; Kita, T. Astrocyte-derived metallothionein protects dopaminergic neurons from dopamine quinone toxicity. Glia 2011, 59, 435–451. [Google Scholar] [CrossRef] [PubMed]
- Perea, G.; Navarrete, M.; Araque, A. Tripartite synapses: Astrocytes process and control synaptic information. Trends Neurosci. 2009, 32, 421–431. [Google Scholar] [CrossRef] [PubMed]
- Perez-Alvarez, A.; Araque, A. Astrocyte-neuron interaction at tripartite synapses. Curr. Drug Targets 2013, 14, 1220–1224. [Google Scholar] [CrossRef] [PubMed]
- Asanuma, M.; Miyazaki, I.; Murakami, S.; Diaz-Corrales, F.J.; Ogawa, N. Striatal astrocytes act as a reservoir for L-DOPA. PLoS ONE 2014, 9, e106362. [Google Scholar] [CrossRef]
- Miyazaki, I.; Asanuma, M.; Diaz-Corrales, F.J.; Miyoshi, K.; Ogawa, N. Direct evidence for expression of dopamine receptors in astrocytes from basal ganglia. Brain Res. 2004, 1029, 120–123. [Google Scholar] [CrossRef]
- Cervetto, C.; Venturini, A.; Passalacqua, M.; Guidolin, D.; Genedani, S.; Fuxe, K.; Borroto-Esquela, D.O.; Cortelli, P.; Woods, A.; Maura, G.; et al. A2A-D2 receptor-receptor interaction modulates gliotransmitter release from striatal astrocyte processes. J. Neurochem. 2017, 140, 268–279. [Google Scholar] [CrossRef]
- Pelassa, S.; Guidolin, D.; Venturini, A.; Averna, M.; Frumento, G.; Campanini, L.; Bernardi, R.; Cortelli, P.; Buonaura, G.C.; Maura, G.; et al. A2A-D2 Heteromers on Striatal Astrocytes: Biochemical and Biophysical Evidence. Int. J. Mol. Sci. 2019, 20, 2457. [Google Scholar] [CrossRef]
- d’Anglemont de Tassigny, X.; Pascual, A.; Lopez-Barneo, J. GDNF-based therapies, GDNF-producing interneurons, and trophic support of the dopaminergic nigrostriatal pathway. Implications for Parkinson’s disease. Front. Neuroanat. 2015, 9, 10. [Google Scholar] [CrossRef]
- Lin, L.F.; Doherty, D.H.; Lile, J.D.; Bektesh, S.; Collins, F. GDNF: A glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science 1993, 260, 1130–1132. [Google Scholar] [CrossRef] [PubMed]
- Barker, R.A.; Bjorklund, A.; Gash, D.M.; Whone, A.; Van Laar, A.; Kordower, J.H.; Bankiewicz, K.; Kieburtz, K.; Saarma, M.; Booms, S.; et al. GDNF and Parkinson’s Disease: Where Next? A Summary from a Recent Workshop. J. Parkinsons Dis. 2020, 10, 875–891. [Google Scholar] [CrossRef] [PubMed]
- Duarte Azevedo, M.; Sander, S.; Tenenbaum, L. GDNF, A Neuron-Derived Factor Upregulated in Glial Cells during Disease. J. Clin. Med. 2020, 9, 456. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, H.; Itakura, M.; Sato, K.; Nakamura, S.; Azuma, Y.T.; Takeuchi, T. Extracellular poly(ADP-ribose) is a neurotrophic signal that upregulates glial cell line-derived neurotrophic factor (GDNF) levels in vitro and in vivo. Biochem. Biophys. Res. Commun. 2017, 484, 385–389. [Google Scholar] [CrossRef]
- Kinor, N.; Geffen, R.; Golomb, E.; Zinman, T.; Yadid, G. Dopamine increases glial cell line-derived neurotrophic factor in human fetal astrocytes. Glia 2001, 33, 143–150. [Google Scholar] [CrossRef]
- Kuric, E.; Wieloch, T.; Ruscher, K. Dopamine receptor activation increases glial cell line-derived neurotrophic factor in experimental stroke. Exp. Neurol. 2013, 247, 202–208. [Google Scholar] [CrossRef]
- Bouvier, M.M.; Mytilineou, C. Basic fibroblast growth factor increases division and delays differentiation of dopamine precursors in vitro. J. Neurosci. 1995, 15, 7141–7149. [Google Scholar] [CrossRef]
- Chadi, G.; Moller, A.; Rosen, L.; Janson, A.M.; Agnati, L.A.; Goldstein, M.; Ogren, S.O.; Pettersson, R.F.; Fuxe, K. Protective actions of human recombinant basic fibroblast growth factor on MPTP-lesioned nigrostriatal dopamine neurons after intraventricular infusion. Exp. Brain Res. 1993, 97, 145–158. [Google Scholar] [CrossRef]
- Forget, C.; Stewart, J.; Trudeau, L.E. Impact of basic FGF expression in astrocytes on dopamine neuron synaptic function and development. Eur. J. Neurosci. 2006, 23, 608–616. [Google Scholar] [CrossRef]
- Hou, J.G.; Cohen, G.; Mytilineou, C. Basic fibroblast growth factor stimulation of glial cells protects dopamine neurons from 6-hydroxydopamine toxicity: Involvement of the glutathione system. J. Neurochem. 1997, 69, 76–83. [Google Scholar] [CrossRef]
- Engele, J.; Bohn, M.C. The neurotrophic effects of fibroblast growth factors on dopaminergic neurons in vitro are mediated by mesencephalic glia. J. Neurosci. 1991, 11, 3070–3078. [Google Scholar] [CrossRef] [PubMed]
- Reuss, B.; Unsicker, K. Survival and differentiation of dopaminergic mesencephalic neurons are promoted by dopamine-mediated induction of FGF-2 in striatal astroglial cells. Mol. Cell Neurosci. 2000, 16, 781–792. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Guo, H.; Luo, X.; Sheng, J.; Yang, S.; Yin, Y.; Zhou, J.; Zhou, J. Apomorphine-induced activation of dopamine receptors modulates FGF-2 expression in astrocytic cultures and promotes survival of dopaminergic neurons. FASEB J. 2006, 20, 1263–1265. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhou, Z.; Wang, D.; Li, A.; Yin, Y.; Gu, X.; Ding, F.; Zhen, X.; Zhou, J. Activation of phosphatidylinositol-linked D1-like receptor modulates FGF-2 expression in astrocytes via IP3-dependent Ca2+ signaling. J. Neurosci. 2009, 29, 7766–7775. [Google Scholar] [CrossRef]
- Proia, P.; Schiera, G.; Mineo, M.; Ingrassia, A.M.; Santoro, G.; Savettieri, G.; Di Liegro, I. Astrocytes shed extracellular vesicles that contain fibroblast growth factor-2 and vascular endothelial growth factor. Int. J. Mol. Med. 2008, 21, 63–67. [Google Scholar] [CrossRef]
- Shin, S.Y.; Song, H.; Kim, C.G.; Choi, Y.K.; Lee, K.S.; Lee, S.J.; Lee, H.J.; Lim, Y.; Lee, Y.H. Egr-1 is necessary for fibroblast growth factor-2-induced transcriptional activation of the glial cell line-derived neurotrophic factor in murine astrocytes. J. Biol. Chem. 2009, 284, 30583–30593. [Google Scholar] [CrossRef]
- Petrova, P.; Raibekas, A.; Pevsner, J.; Vigo, N.; Anafi, M.; Moore, M.K.; Peaire, A.E.; Shridhar, V.; Smith, D.I.; Kelly, J.; et al. MANF: A new mesencephalic, astrocyte-derived neurotrophic factor with selectivity for dopaminergic neurons. J. Mol. Neurosci. 2003, 20, 173–188. [Google Scholar] [CrossRef]
- Lindholm, P.; Voutilainen, M.H.; Lauren, J.; Peranen, J.; Leppanen, V.M.; Andressoo, J.O.; Lindahl, M.; Janhunen, S.; Kalkkinen, N.; Timmusk, T.; et al. Novel neurotrophic factor CDNF protects and rescues midbrain dopamine neurons in vivo. Nature 2007, 448, 73–77. [Google Scholar] [CrossRef]
- Voutilainen, M.H.; Back, S.; Porsti, E.; Toppinen, L.; Lindgren, L.; Lindholm, P.; Peranen, J.; Saarma, M.; Tuominen, R.K. Mesencephalic astrocyte-derived neurotrophic factor is neurorestorative in rat model of Parkinson’s disease. J. Neurosci. 2009, 29, 9651–9659. [Google Scholar] [CrossRef]
- Airavaara, M.; Harvey, B.K.; Voutilainen, M.H.; Shen, H.; Chou, J.; Lindholm, P.; Lindahl, M.; Tuominen, R.K.; Saarma, M.; Hoffer, B.; et al. CDNF protects the nigrostriatal dopamine system and promotes recovery after MPTP treatment in mice. Cell Transpl. 2012, 21, 1213–1223. [Google Scholar] [CrossRef]
- Huttunen, H.J.; Saarma, M. CDNF Protein Therapy in Parkinson’s Disease. Cell Transpl. 2019, 28, 349–366. [Google Scholar] [CrossRef] [PubMed]
- Jantti, M.; Harvey, B.K. Trophic activities of endoplasmic reticulum proteins CDNF and MANF. Cell Tissue Res. 2020, 382, 83–100. [Google Scholar] [CrossRef] [PubMed]
- Voutilainen, M.H.; Arumae, U.; Airavaara, M.; Saarma, M. Therapeutic potential of the endoplasmic reticulum located and secreted CDNF/MANF family of neurotrophic factors in Parkinson’s disease. FEBS Lett. 2015, 589, 3739–3748. [Google Scholar] [CrossRef] [PubMed]
- Dringen, R.; Pfeiffer, B.; Hamprecht, B. Synthesis of the antioxidant glutathione in neurons: Supply by astrocytes of CysGly as precursor for neuronal glutathione. J. Neurosci. 1999, 19, 562–569. [Google Scholar] [CrossRef] [PubMed]
- Kranich, O.; Hamprecht, B.; Dringen, R. Different preferences in the utilization of amino acids for glutathione synthesis in cultured neurons and astroglial cells derived from rat brain. Neurosci. Lett. 1996, 219, 211–214. [Google Scholar] [CrossRef]
- Hirrlinger, J.; Konig, J.; Keppler, D.; Lindenau, J.; Schulz, J.B.; Dringen, R. The multidrug resistance protein MRP1 mediates the release of glutathione disulfide from rat astrocytes during oxidative stress. J. Neurochem. 2001, 76, 627–636. [Google Scholar] [CrossRef]
- Wang, X.F.; Cynader, M.S. Astrocytes provide cysteine to neurons by releasing glutathione. J. Neurochem. 2000, 74, 1434–1442. [Google Scholar] [CrossRef]
- Dringen, R.; Gutterer, J.M.; Hirrlinger, J. Glutathione metabolism in brain metabolic interaction between astrocytes and neurons in the defense against reactive oxygen species. Eur. J. Biochem. 2000, 267, 4912–4916. [Google Scholar] [CrossRef]
- Shih, A.Y.; Erb, H.; Sun, X.; Toda, S.; Kalivas, P.W.; Murphy, T.H. Cystine/glutamate exchange modulates glutathione supply for neuroprotection from oxidative stress and cell proliferation. J. Neurosci. 2006, 26, 10514–10523. [Google Scholar] [CrossRef]
- Dringen, R.; Gutterer, J.M.; Gros, C.; Hirrlinger, J. Aminopeptidase N mediates the utilization of the GSH precursor CysGly by cultured neurons. J. Neurosci. Res. 2001, 66, 1003–1008. [Google Scholar] [CrossRef]
- Sulzer, D.; Bogulavsky, J.; Larsen, K.E.; Behr, G.; Karatekin, E.; Kleinman, M.H.; Turro, N.; Krantz, D.; Edwards, R.H.; Greene, L.A.; et al. Neuromelanin biosynthesis is driven by excess cytosolic catecholamines not accumulated by synaptic vesicles. Proc. Natl. Acad. Sci. USA 2000, 97, 11869–11874. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.G. Oxidative pathways for catecholamines in the genesis of neuromelanin and cytotoxic quinones. Mol. Pharm. 1978, 14, 633–643. [Google Scholar]
- Tse, D.C.; McCreery, R.L.; Adams, R.N. Potential oxidative pathways of brain catecholamines. J. Med. Chem. 1976, 19, 37–40. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, D.M.; Arthur, R.E., Jr.; Thomas, D.M.; Elferink, L.A. Tyrosine hydroxylase is inactivated by catechol-quinones and converted to a redox-cycling quinoprotein: Possible relevance to Parkinson’s disease. J. Neurochem. 1999, 73, 1309–1317. [Google Scholar] [CrossRef] [PubMed]
- LaVoie, M.J.; Ostaszewski, B.L.; Weihofen, A.; Schlossmacher, M.G.; Selkoe, D.J. Dopamine covalently modifies and functionally inactivates parkin. Nat. Med. 2005, 11, 1214–1221. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, R.E.; Ferrer, J.V.; Javitch, J.A.; Justice, J.B. Reaction of oxidized dopamine with endogenous cysteine residues in the human dopamine transporter. J. Neurochem. 2001, 76, 1242–1251. [Google Scholar] [CrossRef]
- Xu, Y.; Stokes, A.H.; Roskoski, R., Jr.; Vrana, K.E. Dopamine, in the presence of tyrosinase, covalently modifies and inactivates tyrosine hydroxylase. J. Neurosci. Res. 1998, 54, 691–697. [Google Scholar] [CrossRef]
- Miyazaki, I.; Asanuma, M. Dopaminergic neuron-specific oxidative stress caused by dopamine itself. Acta Med. Okayama 2008, 62, 141–150. [Google Scholar] [CrossRef]
- Zhou, Z.D.; Lim, T.M. Glutathione conjugates with dopamine-derived quinones to form reactive or non-reactive glutathione-conjugates. Neurochem. Res. 2010, 35, 1805–1818. [Google Scholar] [CrossRef]
- Penkowa, M. Metallothioneins are multipurpose neuroprotectants during brain pathology. FEBS J. 2006, 273, 1857–1870. [Google Scholar] [CrossRef]
- Aschner, M. Metallothionein (MT) isoforms in the central nervous system (CNS): Regional and cell-specific distribution and potential functions as an antioxidant. Neurotoxicology 1998, 19, 653–660. [Google Scholar] [PubMed]
- Cuajungco, M.P.; Lees, G.J. Zinc metabolism in the brain: Relevance to human neurodegenerative disorders. Neurobiol. Dis. 1997, 4, 137–169. [Google Scholar] [CrossRef] [PubMed]
- Aschner, M. Astrocyte metallothioneins (MTs) and their neuroprotective role. Ann. N. Y. Acad. Sci. 1997, 825, 334–347. [Google Scholar] [CrossRef] [PubMed]
- Michael, G.J.; Esmailzadeh, S.; Moran, L.B.; Christian, L.; Pearce, R.K.; Graeber, M.B. Up-regulation of metallothionein gene expression in parkinsonian astrocytes. Neurogenetics 2011, 12, 295–305. [Google Scholar] [CrossRef]
- Chung, R.S.; Penkowa, M.; Dittmann, J.; King, C.E.; Bartlett, C.; Asmussen, J.W.; Hidalgo, J.; Carrasco, J.; Leung, Y.K.; Walker, A.K.; et al. Redefining the role of metallothionein within the injured brain: Extracellular metallothioneins play an important role in the astrocyte-neuron response to injury. J. Biol. Chem. 2008, 283, 15349–15358. [Google Scholar] [CrossRef]
- Chung, R.S.; West, A.K. A role for extracellular metallothioneins in CNS injury and repair. Neuroscience 2004, 123, 595–599. [Google Scholar] [CrossRef]
- Miyazaki, I.; Asanuma, M.; Hozumi, H.; Miyoshi, K.; Sogawa, N. Protective effects of metallothionein against dopamine quinone-induced dopaminergic neurotoxicity. FEBS Lett. 2007, 581, 5003–5008. [Google Scholar] [CrossRef]
- West, A.K.; Leung, J.Y.; Chung, R.S. Neuroprotection and regeneration by extracellular metallothionein via lipoprotein-receptor-related proteins. J. Biol. Inorg. Chem. 2011, 16, 1115–1122. [Google Scholar] [CrossRef]
- Shih, A.Y.; Johnson, D.A.; Wong, G.; Kraft, A.D.; Jiang, L.; Erb, H.; Johnson, J.A.; Murphy, T.H. Coordinate regulation of glutathione biosynthesis and release by Nrf2-expressing glia potently protects neurons from oxidative stress. J. Neurosci. 2003, 23, 3394–3406. [Google Scholar] [CrossRef]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef]
- Kang, M.I.; Kobayashi, A.; Wakabayashi, N.; Kim, S.G.; Yamamoto, M. Scaffolding of Keap1 to the actin cytoskeleton controls the function of Nrf2 as key regulator of cytoprotective phase 2 genes. Proc. Natl. Acad. Sci. USA 2004, 101, 2046–2051. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Cole, R.N.; Itoh, K.; Wakabayashi, N.; Katoh, Y.; Yamamoto, M.; Talalay, P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc. Natl. Acad. Sci. USA 2002, 99, 11908–11913. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, N.; Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Kang, M.I.; Kobayashi, A.; Yamamoto, M.; Kensler, T.W.; Talalay, P. Protection against electrophile and oxidant stress by induction of the phase 2 response: Fate of cysteines of the Keap1 sensor modified by inducers. Proc. Natl. Acad. Sci. USA 2004, 101, 2040–2045. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.C.; Vargas, M.R.; Pani, A.K.; Smeyne, R.J.; Johnson, D.A.; Kan, Y.W.; Johnson, J.A. Nrf2-mediated neuroprotection in the MPTP mouse model of Parkinson’s disease: Critical role for the astrocyte. Proc. Natl. Acad. Sci. USA 2009, 106, 2933–2938. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, I.; Asanuma, M.; Murakami, S.; Takeshima, M.; Torigoe, N.; Kitamura, Y.; Miyoshi, K. Targeting 5-HT(1A) receptors in astrocytes to protect dopaminergic neurons in Parkinsonian models. Neurobiol. Dis. 2013, 59, 244–256. [Google Scholar] [CrossRef]
- Vargas, M.R.; Johnson, J.A. The Nrf2-ARE cytoprotective pathway in astrocytes. Expert Rev. Mol. Med. 2009, 11, e17. [Google Scholar] [CrossRef]
- Bonifati, V.; Rizzu, P.; Squitieri, F.; Krieger, E.; Vanacore, N.; van Swieten, J.C.; Brice, A.; van Duijn, C.M.; Oostra, B.; Meco, G.; et al. DJ-1( PARK7), a novel gene for autosomal recessive, early onset parkinsonism. Neurol. Sci. 2003, 24, 159–160. [Google Scholar] [CrossRef]
- Kahle, P.J.; Waak, J.; Gasser, T. DJ-1 and prevention of oxidative stress in Parkinson’s disease and other age-related disorders. Free Radic. Biol. Med. 2009, 47, 1354–1361. [Google Scholar] [CrossRef]
- Bandopadhyay, R.; Kingsbury, A.E.; Cookson, M.R.; Reid, A.R.; Evans, I.M.; Hope, A.D.; Pittman, A.M.; Lashley, T.; Canet-Aviles, R.; Miller, D.W.; et al. The expression of DJ-1 (PARK7) in normal human CNS and idiopathic Parkinson’s disease. Brain 2004, 127, 420–430. [Google Scholar] [CrossRef]
- Mullett, S.J.; Hamilton, R.L.; Hinkle, D.A. DJ-1 immunoreactivity in human brain astrocytes is dependent on infarct presence and infarct age. Neuropathology 2009, 29, 125–131. [Google Scholar] [CrossRef]
- Yanagida, T.; Tsushima, J.; Kitamura, Y.; Yanagisawa, D.; Takata, K.; Shibaike, T.; Yamamoto, A.; Taniguchi, T.; Yasui, H.; Taira, T.; et al. Oxidative stress induction of DJ-1 protein in reactive astrocytes scavenges free radicals and reduces cell injury. Oxid. Med. Cell. Longev. 2009, 2, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Clements, C.M.; McNally, R.S.; Conti, B.J.; Mak, T.W.; Ting, J.P. DJ-1, a cancer- and Parkinson’s disease-associated protein, stabilizes the antioxidant transcriptional master regulator Nrf2. Proc. Natl. Acad. Sci. USA 2006, 103, 15091–15096. [Google Scholar] [CrossRef] [PubMed]
- Hao, L.Y.; Giasson, B.I.; Bonini, N.M. DJ-1 is critical for mitochondrial function and rescues PINK1 loss of function. Proc. Natl. Acad. Sci. USA 2010, 107, 9747–9752. [Google Scholar] [CrossRef] [PubMed]
- Thomas, K.J.; McCoy, M.K.; Blackinton, J.; Beilina, A.; van der Brug, M.; Sandebring, A.; Miller, D.; Maric, D.; Cedazo-Minguez, A.; Cookson, M.R. DJ-1 acts in parallel to the PINK1/parkin pathway to control mitochondrial function and autophagy. Hum. Mol. Genet. 2011, 20, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Du, F.; Yu, Q.; Chen, A.; Chen, D.; Yan, S.S. Astrocytes Attenuate Mitochondrial Dysfunctions in Human Dopaminergic Neurons Derived from iPSC. Stem Cell Rep. 2018, 10, 366–374. [Google Scholar] [CrossRef]
- Cheng, X.Y.; Biswas, S.; Li, J.; Mao, C.J.; Chechneva, O.; Chen, J.; Li, K.; Li, J.; Zhang, J.R.; Liu, C.F.; et al. Human iPSCs derived astrocytes rescue rotenone-induced mitochondrial dysfunction and dopaminergic neurodegeneration in vitro by donating functional mitochondria. Transl. Neurodegener. 2020, 9, 13. [Google Scholar] [CrossRef]
- Arima, K.; Ueda, K.; Sunohara, N.; Arakawa, K.; Hirai, S.; Nakamura, M.; Tonozuka-Uehara, H.; Kawai, M. NACP/alpha-synuclein immunoreactivity in fibrillary components of neuronal and oligodendroglial cytoplasmic inclusions in the pontine nuclei in multiple system atrophy. Acta Neuropathol. 1998, 96, 439–444. [Google Scholar] [CrossRef]
- Baba, M.; Nakajo, S.; Tu, P.H.; Tomita, T.; Nakaya, K.; Lee, V.M.; Trojanowski, J.Q.; Iwatsubo, T. Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson’s disease and dementia with Lewy bodies. Am. J. Pathol. 1998, 152, 879–884. [Google Scholar]
- Lopes da Fonseca, T.; Villar-Pique, A.; Outeiro, T.F. The Interplay between Alpha-Synuclein Clearance and Spreading. Biomolecules 2015, 5, 435–471. [Google Scholar] [CrossRef]
- Dunning, C.J.; George, S.; Brundin, P. What’s to like about the prion-like hypothesis for the spreading of aggregated alpha-synuclein in Parkinson disease? Prion 2013, 7, 92–97. [Google Scholar] [CrossRef]
- Kovacs, G.G.; Breydo, L.; Green, R.; Kis, V.; Puska, G.; Lorincz, P.; Perju-Dumbrava, L.; Giera, R.; Pirker, W.; Lutz, M.; et al. Intracellular processing of disease-associated alpha-synuclein in the human brain suggests prion-like cell-to-cell spread. Neurobiol. Dis. 2014, 69, 76–92. [Google Scholar] [CrossRef] [PubMed]
- Stefanis, L.; Emmanouilidou, E.; Pantazopoulou, M.; Kirik, D.; Vekrellis, K.; Tofaris, G.K. How is alpha-synuclein cleared from the cell? J. Neurochem. 2019, 150, 577–590. [Google Scholar] [CrossRef] [PubMed]
- Choi, I.; Zhang, Y.; Seegobin, S.P.; Pruvost, M.; Wang, Q.; Purtell, K.; Zhang, B.; Yue, Z. Microglia clear neuron-released alpha-synuclein via selective autophagy and prevent neurodegeneration. Nat. Commun. 2020, 11, 1386. [Google Scholar] [CrossRef] [PubMed]
- Morales, I.; Sanchez, A.; Puertas-Avendano, R.; Rodriguez-Sabate, C.; Perez-Barreto, A.; Rodriguez, M. Neuroglial transmitophagy and Parkinson’s disease. Glia 2020, 68, 2277–2299. [Google Scholar] [CrossRef] [PubMed]
- Hua, J.; Yin, N.; Xu, S.; Chen, Q.; Tao, T.; Zhang, J.; Ding, J.; Fan, Y.; Hu, G. Enhancing the Astrocytic Clearance of Extracellular alpha-Synuclein Aggregates by Ginkgolides Attenuates Neural Cell Injury. Cell. Mol. Neurobiol. 2019, 39, 1017–1028. [Google Scholar] [CrossRef]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef]
- Shiba-Fukushima, K.; Inoshita, T.; Hattori, N.; Imai, Y. PINK1-mediated phosphorylation of Parkin boosts Parkin activity in Drosophila. PLoS Genet. 2014, 10, e1004391. [Google Scholar] [CrossRef]
- Springer, W.; Kahle, P.J. Regulation of PINK1-Parkin-mediated mitophagy. Autophagy 2011, 7, 266–278. [Google Scholar] [CrossRef]
- Davis, C.H.; Kim, K.Y.; Bushong, E.A.; Mills, E.A.; Boassa, D.; Shih, T.; Kinebuchi, M.; Phan, S.; Zhou, Y.; Bihlmeyer, N.A.; et al. Transcellular degradation of axonal mitochondria. Proc. Natl. Acad. Sci. USA 2014, 111, 9633–9638. [Google Scholar] [CrossRef]
- Morales, I.; Sanchez, A.; Rodriguez-Sabate, C.; Rodriguez, M. Striatal astrocytes engulf dopaminergic debris in Parkinson’s disease: A study in an animal model. PLoS ONE 2017, 12, e0185989. [Google Scholar] [CrossRef]
- McGeer, P.L.; McGeer, E.G. Glial reactions in Parkinson’s disease. Mov. Disord. 2008, 23, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Mirza, B.; Hadberg, H.; Thomsen, P.; Moos, T. The absence of reactive astrocytosis is indicative of a unique inflammatory process in Parkinson’s disease. Neuroscience 2000, 95, 425–432. [Google Scholar] [CrossRef]
- Knott, C.; Wilkin, G.P.; Stern, G. Astrocytes and microglia in the substantia nigra and caudate-putamen in Parkinson’s disease. Parkinsonism Relat. Disord. 1999, 5, 115–122. [Google Scholar] [CrossRef]
- Tong, J.; Ang, L.C.; Williams, B.; Furukawa, Y.; Fitzmaurice, P.; Guttman, M.; Boileau, I.; Hornykiewicz, O.; Kish, S.J. Low levels of astroglial markers in Parkinson’s disease: Relationship to alpha-synuclein accumulation. Neurobiol. Dis. 2015, 82, 243–253. [Google Scholar] [CrossRef]
- Damier, P.; Hirsch, E.C.; Zhang, P.; Agid, Y.; Javoy-Agid, F. Glutathione peroxidase, glial cells and Parkinson’s disease. Neuroscience 1993, 52, 1–6. [Google Scholar] [CrossRef]
- Morales, I.; Sanchez, A.; Rodriguez-Sabate, C.; Rodriguez, M. The astrocytic response to the dopaminergic denervation of the striatum. J. Neurochem. 2016, 139, 81–95. [Google Scholar] [CrossRef]
- Zhu, Y.F.; Wang, W.P.; Zheng, X.F.; Chen, Z.; Chen, T.; Huang, Z.Y.; Jia, L.J.; Lei, W.L. Characteristic response of striatal astrocytes to dopamine depletion. Neural Regen. Res. 2020, 15, 724–730. [Google Scholar] [CrossRef]
- Braak, H.; Sastre, M.; Del Tredici, K. Development of alpha-synuclein immunoreactive astrocytes in the forebrain parallels stages of intraneuronal pathology in sporadic Parkinson’s disease. Acta Neuropathol. 2007, 114, 231–241. [Google Scholar] [CrossRef]
- Song, Y.J.; Halliday, G.M.; Holton, J.L.; Lashley, T.; O’Sullivan, S.S.; McCann, H.; Lees, A.J.; Ozawa, T.; Williams, D.R.; Lockhart, P.J.; et al. Degeneration in different parkinsonian syndromes relates to astrocyte type and astrocyte protein expression. J. Neuropathol. Exp. Neurol. 2009, 68, 1073–1083. [Google Scholar] [CrossRef]
- Wakabayashi, K.; Hayashi, S.; Yoshimoto, M.; Kudo, H.; Takahashi, H. NACP/alpha-synuclein-positive filamentous inclusions in astrocytes and oligodendrocytes of Parkinson’s disease brains. Acta Neuropathol. 2000, 99, 14–20. [Google Scholar] [CrossRef]
- Freundt, E.C.; Maynard, N.; Clancy, E.K.; Roy, S.; Bousset, L.; Sourigues, Y.; Covert, M.; Melki, R.; Kirkegaard, K.; Brahic, M. Neuron-to-neuron transmission of alpha-synuclein fibrils through axonal transport. Ann. Neurol. 2012, 72, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Suk, J.E.; Patrick, C.; Bae, E.J.; Cho, J.H.; Rho, S.; Hwang, D.; Masliah, E.; Lee, S.J. Direct transfer of alpha-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J. Biol. Chem. 2010, 285, 9262–9272. [Google Scholar] [CrossRef] [PubMed]
- Di Domenico, A.; Carola, G.; Calatayud, C.; Pons-Espinal, M.; Munoz, J.P.; Richaud-Patin, Y.; Fernandez-Carasa, I.; Gut, M.; Faella, A.; Parameswaran, J.; et al. Patient-Specific iPSC-Derived Astrocytes Contribute to Non-Cell-Autonomous Neurodegeneration in Parkinson’s Disease. Stem Cell Rep. 2019, 12, 213–229. [Google Scholar] [CrossRef] [PubMed]
- Guzman-Martinez, L.; Maccioni, R.B.; Andrade, V.; Navarrete, L.P.; Pastor, M.G.; Ramos-Escobar, N. Neuroinflammation as a Common Feature of Neurodegenerative Disorders. Front. Pharm. 2019, 10, 1008. [Google Scholar] [CrossRef] [PubMed]
- Niranjan, R. The role of inflammatory and oxidative stress mechanisms in the pathogenesis of Parkinson’s disease: Focus on astrocytes. Mol. Neurobiol. 2014, 49, 28–38. [Google Scholar] [CrossRef]
- Collins, L.M.; Toulouse, A.; Connor, T.J.; Nolan, Y.M. Contributions of central and systemic inflammation to the pathophysiology of Parkinson’s disease. Neuropharmacology 2012, 62, 2154–2168. [Google Scholar] [CrossRef]
- McGeer, P.L.; Itagaki, S.; Boyes, B.E.; McGeer, E.G. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 1988, 38, 1285–1291. [Google Scholar] [CrossRef]
- Castano, A.; Herrera, A.J.; Cano, J.; Machado, A. Lipopolysaccharide intranigral injection induces inflammatory reaction and damage in nigrostriatal dopaminergic system. J. Neurochem. 1998, 70, 1584–1592. [Google Scholar] [CrossRef]
- Aloe, L.; Fiore, M. TNF-alpha expressed in the brain of transgenic mice lowers central tyroxine hydroxylase immunoreactivity and alters grooming behavior. Neurosci. Lett. 1997, 238, 65–68. [Google Scholar] [CrossRef]
- Joshi, A.U.; Minhas, P.S.; Liddelow, S.A.; Haileselassie, B.; Andreasson, K.I.; Dorn, G.W., 2nd; Mochly-Rosen, D. Fragmented mitochondria released from microglia trigger A1 astrocytic response and propagate inflammatory neurodegeneration. Nat. Neurosci. 2019, 22, 1635–1648. [Google Scholar] [CrossRef]
- Jo, M.; Kim, J.H.; Song, G.J.; Seo, M.; Hwang, E.M.; Suk, K. Astrocytic Orosomucoid-2 Modulates Microglial Activation and Neuroinflammation. J. Neurosci. 2017, 37, 2878–2894. [Google Scholar] [CrossRef] [PubMed]
- Rohl, C.; Sievers, J. Microglia is activated by astrocytes in trimethyltin intoxication. Toxicol. Appl. Pharm. 2005, 204, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Han, F. Key Mechanisms and Potential Targets of the NLRP3 Inflammasome in Neurodegenerative Diseases. Front. Integr. Neurosci. 2020, 14, 37. [Google Scholar] [CrossRef] [PubMed]
- Freeman, L.; Guo, H.; David, C.N.; Brickey, W.J.; Jha, S.; Ting, J.P. NLR members NLRC4 and NLRP3 mediate sterile inflammasome activation in microglia and astrocytes. J. Exp. Med. 2017, 214, 1351–1370. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Pei, L.; Yao, S.; Wu, Y.; Shang, Y. NLRP3 Inflammasome in Neurological Diseases, from Functions to Therapies. Front. Cell. Neurosci. 2017, 11, 63. [Google Scholar] [CrossRef]
- Zhang, P.; Shao, X.Y.; Qi, G.J.; Chen, Q.; Bu, L.L.; Chen, L.J.; Shi, J.; Ming, J.; Tian, B. Cdk5-Dependent Activation of Neuronal Inflammasomes in Parkinson’s Disease. Mov. Disord. 2016, 31, 366–376. [Google Scholar] [CrossRef]
- Wang, J.; Zheng, B.; Yang, S.; Tang, X.; Wang, J.; Wei, D. The protective effects of phoenixin-14 against lipopolysaccharide-induced inflammation and inflammasome activation in astrocytes. Inflamm. Res. 2020, 69, 779–787. [Google Scholar] [CrossRef]
- Zhu, J.; Hu, Z.; Han, X.; Wang, D.; Jiang, Q.; Ding, J.; Xiao, M.; Wang, C.; Lu, M.; Hu, G. Dopamine D2 receptor restricts astrocytic NLRP3 inflammasome activation via enhancing the interaction of beta-arrestin2 and NLRP3. Cell Death Differ. 2018, 25, 2037–2049. [Google Scholar] [CrossRef]
- Yi, J.H.; Hazell, A.S. Excitotoxic mechanisms and the role of astrocytic glutamate transporters in traumatic brain injury. Neurochem. Int. 2006, 48, 394–403. [Google Scholar] [CrossRef]
- Haroon, E.; Miller, A.H.; Sanacora, G. Inflammation, Glutamate, and Glia: A Trio of Trouble in Mood Disorders. Neuropsychopharmacology 2017, 42, 193–215. [Google Scholar] [CrossRef]
- Tilleux, S.; Hermans, E. Neuroinflammation and regulation of glial glutamate uptake in neurological disorders. J. Neurosci. Res. 2007, 85, 2059–2070. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.Y.; Crews, F.T. TNF alpha potentiates glutamate neurotoxicity by inhibiting glutamate uptake in organotypic brain slice cultures: Neuroprotection by NF kappa B inhibition. Brain Res. 2005, 1034, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Klegeris, A.; Walker, D.G.; McGeer, P.L. Regulation of glutamate in cultures of human monocytic THP-1 and astrocytoma U-373 MG cells. J. Neuroimmunol. 1997, 78, 152–161. [Google Scholar] [CrossRef]
- Takaki, J.; Fujimori, K.; Miura, M.; Suzuki, T.; Sekino, Y.; Sato, K. L-glutamate released from activated microglia downregulates astrocytic L-glutamate transporter expression in neuroinflammation: The ‘collusion’ hypothesis for increased extracellular L-glutamate concentration in neuroinflammation. J. Neuroinflammation. 2012, 9, 275. [Google Scholar] [CrossRef]
- Hammond, S.L.; Bantle, C.M.; Popichak, K.A.; Wright, K.A.; Thompson, D.; Forero, C.; Kirkley, K.S.; Damale, P.U.; Chong, E.K.P.; Tjalkens, R.B. NF-kappaB signaling in astrocytes modulates brain inflammation and neuronal injury following sequential exposure to manganese and MPTP during development and aging. Toxicol. Sci. 2020. [Google Scholar] [CrossRef]
- Kirkley, K.S.; Popichak, K.A.; Hammond, S.L.; Davies, C.; Hunt, L.; Tjalkens, R.B. Genetic suppression of IKK2/NF-kappaB in astrocytes inhibits neuroinflammation and reduces neuronal loss in the MPTP-Probenecid model of Parkinson’s disease. Neurobiol. Dis. 2019, 127, 193–209. [Google Scholar] [CrossRef]
- Montoya, A.; Elgueta, D.; Campos, J.; Chovar, O.; Falcon, P.; Matus, S.; Alfaro, I.; Bono, M.R.; Pacheco, R. Dopamine receptor D3 signalling in astrocytes promotes neuroinflammation. J. Neuroinflammation 2019, 16, 258. [Google Scholar] [CrossRef]
- Gu, X.L.; Long, C.X.; Sun, L.; Xie, C.; Lin, X.; Cai, H. Astrocytic expression of Parkinson’s disease-related A53T alpha-synuclein causes neurodegeneration in mice. Mol. Brain 2010, 3, 12. [Google Scholar] [CrossRef]
- Yun, S.P.; Kam, T.I.; Panicker, N.; Kim, S.; Oh, Y.; Park, J.S.; Kwon, S.H.; Park, Y.J.; Karuppagounder, S.S.; Park, H.; et al. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat. Med. 2018, 24, 931–938. [Google Scholar] [CrossRef]
- Chavarria, C.; Rodriguez-Bottero, S.; Quijano, C.; Cassina, P.; Souza, J.M. Impact of monomeric, oligomeric and fibrillar alpha-synuclein on astrocyte reactivity and toxicity to neurons. Biochem. J. 2018, 475, 3153–3169. [Google Scholar] [CrossRef]
- Diaz, E.F.; Labra, V.C.; Alvear, T.F.; Mellado, L.A.; Inostroza, C.A.; Oyarzun, J.E.; Salgado, N.; Quintanilla, R.A.; Orellana, J.A. Connexin 43 hemichannels and pannexin-1 channels contribute to the alpha-synuclein-induced dysfunction and death of astrocytes. Glia 2019, 67, 1598–1619. [Google Scholar] [CrossRef] [PubMed]
- Sheng, L.; Stewart, T.; Yang, D.; Thorland, E.; Soltys, D.; Aro, P.; Khrisat, T.; Xie, Z.; Li, N.; Liu, Z.; et al. Erythrocytic alpha-synuclein contained in microvesicles regulates astrocytic glutamate homeostasis: A new perspective on Parkinson’s disease pathogenesis. Acta Neuropathol. Commun. 2020, 8, 102. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.T.; Woulfe, J.M. Striatal blood-brain barrier permeability in Parkinson’s disease. J. Cereb. Blood Flow Metab. 2015, 35, 747–750. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Ronnback, L.; Hansson, E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef]
- Tamtaji, O.R.; Behnam, M.; Pourattar, M.A.; Jafarpour, H.; Asemi, Z. Aquaporin 4: A key player in Parkinson’s disease. J. Cell. Physiol. 2019, 234, 21471–21478. [Google Scholar] [CrossRef]
- Xue, X.; Zhang, W.; Zhu, J.; Chen, X.; Zhou, S.; Xu, Z.; Hu, G.; Su, C. Aquaporin-4 deficiency reduces TGF-beta1 in mouse midbrains and exacerbates pathology in experimental Parkinson’s disease. J. Cell. Mol. Med. 2019, 23, 2568–2582. [Google Scholar] [CrossRef]
- Sun, H.; Liang, R.; Yang, B.; Zhou, Y.; Liu, M.; Fang, F.; Ding, J.; Fan, Y.; Hu, G. Aquaporin-4 mediates communication between astrocyte and microglia: Implications of neuroinflammation in experimental Parkinson’s disease. Neuroscience 2016, 317, 65–75. [Google Scholar] [CrossRef]
- Zhang, Y.; Meng, X.; Jiao, Z.; Liu, Y.; Zhang, X.; Qu, S. Generation of a Novel Mouse Model of Parkinson’s Disease via Targeted Knockdown of Glutamate Transporter GLT-1 in the Substantia Nigra. ACS Chem. Neurosci. 2020, 11, 406–417. [Google Scholar] [CrossRef]
- Nandipati, S.; Litvan, I. Environmental Exposures and Parkinson’s Disease. Int. J. Environ. Res. Public Health 2016, 13, 881. [Google Scholar] [CrossRef]
- Mutkus, L.; Aschner, J.L.; Fitsanakis, V.; Aschner, M. The in vitro uptake of glutamate in GLAST and GLT-1 transfected mutant CHO-K1 cells is inhibited by manganese. Biol. Trace Elem. Res. 2005, 107, 221–230. [Google Scholar] [CrossRef]
- Sidoryk-Wegrzynowicz, M.; Lee, E.; Albrecht, J.; Aschner, M. Manganese disrupts astrocyte glutamine transporter expression and function. J. Neurochem. 2009, 110, 822–830. [Google Scholar] [CrossRef]
- Innamorato, N.G.; Jazwa, A.; Rojo, A.I.; Garcia, C.; Fernandez-Ruiz, J.; Grochot-Przeczek, A.; Stachurska, A.; Jozkowicz, A.; Dulak, J.; Cuadrado, A. Different susceptibility to the Parkinson’s toxin MPTP in mice lacking the redox master regulator Nrf2 or its target gene heme oxygenase-1. PLoS ONE 2010, 5, e11838. [Google Scholar] [CrossRef]
- L’Episcopo, F.; Tirolo, C.; Testa, N.; Caniglia, S.; Morale, M.C.; Impagnatiello, F.; Pluchino, S.; Marchetti, B. Aging-induced Nrf2-ARE pathway disruption in the subventricular zone drives neurogenic impairment in parkinsonian mice via PI3K-Wnt/beta-catenin dysregulation. J. Neurosci. 2013, 33, 1462–1485. [Google Scholar] [CrossRef]
- Lastres-Becker, I.; Ulusoy, A.; Innamorato, N.G.; Sahin, G.; Rabano, A.; Kirik, D.; Cuadrado, A. alpha-Synuclein expression and Nrf2 deficiency cooperate to aggravate protein aggregation, neuronal death and inflammation in early-stage Parkinson’s disease. Hum. Mol. Genet. 2012, 21, 3173–3192. [Google Scholar] [CrossRef]
- Chinta, S.J.; Kumar, M.J.; Hsu, M.; Rajagopalan, S.; Kaur, D.; Rane, A.; Nicholls, D.G.; Choi, J.; Andersen, J.K. Inducible alterations of glutathione levels in adult dopaminergic midbrain neurons result in nigrostriatal degeneration. J. Neurosci. 2007, 27, 13997–14006. [Google Scholar] [CrossRef]
- Okita, Y.; Rcom-H’cheo-Gauthier, A.N.; Goulding, M.; Chung, R.S.; Faller, P.; Pountney, D.L. Metallothionein, Copper and Alpha-Synuclein in Alpha-Synucleinopathies. Front. Neurosci. 2017, 11, 114. [Google Scholar] [CrossRef]
- Davies, K.M.; Bohic, S.; Carmona, A.; Ortega, R.; Cottam, V.; Hare, D.J.; Finberg, J.P.; Reyes, S.; Halliday, G.M.; Mercer, J.F.; et al. Copper pathology in vulnerable brain regions in Parkinson’s disease. Neurobiol. Aging 2014, 35, 858–866. [Google Scholar] [CrossRef]
- Rose, F.; Hodak, M.; Bernholc, J. Mechanism of copper(II)-induced misfolding of Parkinson’s disease protein. Sci. Rep. 2011, 1, 11. [Google Scholar] [CrossRef]
- Calvo, J.; Jung, H.; Meloni, G. Copper metallothioneins. Iubmb Life 2017, 69, 236–245. [Google Scholar] [CrossRef]
- McLeary, F.A.; Rcom-H’cheo-Gauthier, A.N.; Goulding, M.; Radford, R.A.W.; Okita, Y.; Faller, P.; Chung, R.S.; Pountney, D.L. Switching on Endogenous Metal Binding Proteins in Parkinson’s Disease. Cells 2019, 8, 179. [Google Scholar] [CrossRef]
- Meloni, G.; Sonois, V.; Delaine, T.; Guilloreau, L.; Gillet, A.; Teissie, J.; Faller, P.; Vasak, M. Metal swap between Zn7-metallothionein-3 and amyloid-beta-Cu protects against amyloid-beta toxicity. Nat. Chem. Biol. 2008, 4, 366–372. [Google Scholar] [CrossRef] [PubMed]
- Asanuma, M.; Miyazaki, I.; Higashi, Y.; Tanaka, K.; Haque, M.E.; Fujita, N.; Ogawa, N. Aggravation of 6-hydroxydopamine-induced dopaminergic lesions in metallothionein-I and -II knock-out mouse brain. Neurosci. Lett. 2002, 327, 61–65. [Google Scholar] [CrossRef]
- Murakami, S.; Miyazaki, I.; Sogawa, N.; Miyoshi, K.; Asanuma, M. Neuroprotective effects of metallothionein against rotenone-induced myenteric neurodegeneration in parkinsonian mice. Neurotox. Res. 2014, 26, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Dolgacheva, L.P.; Berezhnov, A.V.; Fedotova, E.I.; Zinchenko, V.P.; Abramov, A.Y. Role of DJ-1 in the mechanism of pathogenesis of Parkinson’s disease. J. Bioenergy Biomembr. 2019, 51, 175–188. [Google Scholar] [CrossRef]
- Mullett, S.J.; Di Maio, R.; Greenamyre, J.T.; Hinkle, D.A. DJ-1 expression modulates astrocyte-mediated protection against neuronal oxidative stress. J. Mol. Neurosci. 2013, 49, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Larsen, N.J.; Ambrosi, G.; Mullett, S.J.; Berman, S.B.; Hinkle, D.A. DJ-1 knock-down impairs astrocyte mitochondrial function. Neuroscience 2011, 196, 251–264. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lev, N.; Barhum, Y.; Ben-Zur, T.; Melamed, E.; Steiner, I.; Offen, D. Knocking out DJ-1 attenuates astrocytes neuroprotection against 6-hydroxydopamine toxicity. J. Mol. Neurosci. 2013, 50, 542–550. [Google Scholar] [CrossRef]
- Mullett, S.J.; Hinkle, D.A. DJ-1 deficiency in astrocytes selectively enhances mitochondrial Complex I inhibitor-induced neurotoxicity. J. Neurochem. 2011, 117, 375–387. [Google Scholar] [CrossRef]
- Kim, J.M.; Cha, S.H.; Choi, Y.R.; Jou, I.; Joe, E.H.; Park, S.M. DJ-1 deficiency impairs glutamate uptake into astrocytes via the regulation of flotillin-1 and caveolin-1 expression. Sci. Rep. 2016, 6, 28823. [Google Scholar] [CrossRef]
- Waak, J.; Weber, S.S.; Waldenmaier, A.; Gorner, K.; Alunni-Fabbroni, M.; Schell, H.; Vogt-Weisenhorn, D.; Pham, T.T.; Reumers, V.; Baekelandt, V.; et al. Regulation of astrocyte inflammatory responses by the Parkinson’s disease-associated gene DJ-1. FASEB J. 2009, 23, 2478–2489. [Google Scholar] [CrossRef]
- Khasnavis, S.; Pahan, K. Cinnamon treatment upregulates neuroprotective proteins Parkin and DJ-1 and protects dopaminergic neurons in a mouse model of Parkinson’s disease. J. Neuroimmune Pharm. 2014, 9, 569–581. [Google Scholar] [CrossRef]
- Xiong, H.; Wang, D.; Chen, L.; Choo, Y.S.; Ma, H.; Tang, C.; Xia, K.; Jiang, W.; Ronai, Z.; Zhuang, X.; et al. Parkin, PINK1, and DJ-1 form a ubiquitin E3 ligase complex promoting unfolded protein degradation. J. Clin. Investig. 2009, 119, 650–660. [Google Scholar] [CrossRef] [PubMed]
- Ledesma, M.D.; Galvan, C.; Hellias, B.; Dotti, C.; Jensen, P.H. Astrocytic but not neuronal increased expression and redistribution of parkin during unfolded protein stress. J. Neurochem. 2002, 83, 1431–1440. [Google Scholar] [CrossRef] [PubMed]
- Solano, R.M.; Casarejos, M.J.; Menendez-Cuervo, J.; Rodriguez-Navarro, J.A.; Garcia de Yebenes, J.; Mena, M.A. Glial dysfunction in parkin null mice: Effects of aging. J. Neurosci. 2008, 28, 598–611. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Han, K.; Tilve, S.; Wu, K.; Geller, H.M.; Sack, M.N. Parkin targets NOD2 to regulate astrocyte endoplasmic reticulum stress and inflammation. Glia 2018, 66, 2427–2437. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.; Linnartz, B.; Mendritzki, S.; Sczepan, T.; Lubbert, M.; Stichel, C.C.; Lubbert, H. Genetic mouse models for Parkinson’s disease display severe pathology in glial cell mitochondria. Hum. Mol. Genet. 2011, 20, 1197–1211. [Google Scholar] [CrossRef] [PubMed]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [PubMed]
- Choi, I.; Choi, D.J.; Yang, H.; Woo, J.H.; Chang, M.Y.; Kim, J.Y.; Sun, W.; Park, S.M.; Jou, I.; Lee, S.H.; et al. PINK1 expression increases during brain development and stem cell differentiation, and affects the development of GFAP-positive astrocytes. Mol. Brain 2016, 9, 5. [Google Scholar] [CrossRef]
- Choi, I.; Kim, J.; Jeong, H.K.; Kim, B.; Jou, I.; Park, S.M.; Chen, L.; Kang, U.J.; Zhuang, X.; Joe, E.H. PINK1 deficiency attenuates astrocyte proliferation through mitochondrial dysfunction, reduced AKT and increased p38 MAPK activation, and downregulation of EGFR. Glia 2013, 61, 800–812. [Google Scholar] [CrossRef]
- Sun, L.; Shen, R.; Agnihotri, S.K.; Chen, Y.; Huang, Z.; Bueler, H. Lack of PINK1 alters glia innate immune responses and enhances inflammation-induced, nitric oxide-mediated neuron death. Sci. Rep. 2018, 8, 383. [Google Scholar] [CrossRef]
- Roosen, D.A.; Cookson, M.R. LRRK2 at the interface of autophagosomes, endosomes and lysosomes. Mol. Neurodegener. 2016, 11, 73. [Google Scholar] [CrossRef] [PubMed]
- Ho, P.W.; Leung, C.T.; Liu, H.; Pang, S.Y.; Lam, C.S.; Xian, J.; Li, L.; Kung, M.H.; Ramsden, D.B.; Ho, S.L. Age-dependent accumulation of oligomeric SNCA/alpha-synuclein from impaired degradation in mutant LRRK2 knockin mouse model of Parkinson disease: Role for therapeutic activation of chaperone-mediated autophagy (CMA). Autophagy 2020, 16, 347–370. [Google Scholar] [CrossRef] [PubMed]
- MacIsaac, S.; Quevedo Melo, T.; Zhang, Y.; Volta, M.; Farrer, M.J.; Milnerwood, A.J. Neuron-autonomous susceptibility to induced synuclein aggregation is exacerbated by endogenous Lrrk2 mutations and ameliorated by Lrrk2 genetic knock-out. Brain Commun. 2020, 2, fcz052. [Google Scholar] [CrossRef] [PubMed]
- Schapansky, J.; Khasnavis, S.; DeAndrade, M.P.; Nardozzi, J.D.; Falkson, S.R.; Boyd, J.D.; Sanderson, J.B.; Bartels, T.; Melrose, H.L.; LaVoie, M.J. Familial knockin mutation of LRRK2 causes lysosomal dysfunction and accumulation of endogenous insoluble alpha-synuclein in neurons. Neurobiol. Dis. 2018, 111, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Miklossy, J.; Arai, T.; Guo, J.P.; Klegeris, A.; Yu, S.; McGeer, E.G.; McGeer, P.L. LRRK2 expression in normal and pathologic human brain and in human cell lines. J. Neuropathol. Exp. Neurol. 2006, 65, 953–963. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Bandopadhyay, R.; Lashley, T.; Renton, A.E.; Kingsbury, A.E.; Kumaran, R.; Kallis, C.; Vilarino-Guell, C.; O’Sullivan, S.S.; Lees, A.J.; et al. LRRK2 expression in idiopathic and G2019S positive Parkinson’s disease subjects: A morphological and quantitative study. Neuropathol. Appl. Neurobiol. 2011, 37, 777–790. [Google Scholar] [CrossRef]
- Henry, A.G.; Aghamohammadzadeh, S.; Samaroo, H.; Chen, Y.; Mou, K.; Needle, E.; Hirst, W.D. Pathogenic LRRK2 mutations, through increased kinase activity, produce enlarged lysosomes with reduced degradative capacity and increase ATP13A2 expression. Hum. Mol. Genet. 2015, 24, 6013–6028. [Google Scholar] [CrossRef]
- Ramirez, A.; Heimbach, A.; Grundemann, J.; Stiller, B.; Hampshire, D.; Cid, L.P.; Goebel, I.; Mubaidin, A.F.; Wriekat, A.L.; Roeper, J.; et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat. Genet. 2006, 38, 1184–1191. [Google Scholar] [CrossRef]
- Qiao, C.; Yin, N.; Gu, H.Y.; Zhu, J.L.; Ding, J.H.; Lu, M.; Hu, G. Atp13a2 Deficiency Aggravates Astrocyte-Mediated Neuroinflammation via NLRP3 Inflammasome Activation. Cns Neurosci. 2016, 22, 451–460. [Google Scholar] [CrossRef]
- Tsunemi, T.; Ishiguro, Y.; Yoroisaka, A.; Valdez, C.; Miyamoto, K.; Ishikawa, K.; Saiki, S.; Akamatsu, W.; Hattori, N.; Krainc, D. Astrocytes Protect Human Dopaminergic Neurons from alpha-Synuclein Accumulation and Propagation. J. Neurosci. 2020, 40, 8618–8628. [Google Scholar] [CrossRef]
- Sidransky, E.; Nalls, M.A.; Aasly, J.O.; Aharon-Peretz, J.; Annesi, G.; Barbosa, E.R.; Bar-Shira, A.; Berg, D.; Bras, J.; Brice, A.; et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N. Engl. J. Med. 2009, 361, 1651–1661. [Google Scholar] [CrossRef] [PubMed]
- Aflaki, E.; Stubblefield, B.K.; McGlinchey, R.P.; McMahon, B.; Ory, D.S.; Sidransky, E. A characterization of Gaucher iPS-derived astrocytes: Potential implications for Parkinson’s disease. Neurobiol. Dis. 2020, 134, 104647. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.; DeAndrade, M.P.; Novis, H.S.; Lin, S.; Chang, J.; Lengacher, N.; Tomlinson, J.J.; Tansey, M.G.; LaVoie, M.J. Lysosome and Inflammatory Defects in GBA1-Mutant Astrocytes Are Normalized by LRRK2 Inhibition. Mov. Disord. 2020, 35, 760–773. [Google Scholar] [CrossRef] [PubMed]
- Kohutnicka, M.; Lewandowska, E.; Kurkowska-Jastrzebska, I.; Czlonkowski, A.; Czlonkowska, A. Microglial and astrocytic involvement in a murine model of Parkinson’s disease induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Immunopharmacology 1998, 39, 167–180. [Google Scholar] [CrossRef]
- O’Callaghan, J.P.; Miller, D.B.; Reinhard, J.F., Jr. Characterization of the origins of astrocyte response to injury using the dopaminergic neurotoxicant, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Brain Res. 1990, 521, 73–80. [Google Scholar] [CrossRef]
- Brooks, W.J.; Jarvis, M.F.; Wagner, G.C. Astrocytes as a primary locus for the conversion MPTP into MPP+. J. Neural. Transm. 1989, 76, 1–12. [Google Scholar] [CrossRef]
- Sundar Boyalla, S.; Barbara Victor, M.; Roemgens, A.; Beyer, C.; Arnold, S. Sex- and brain region-specific role of cytochrome c oxidase in 1-methyl-4-phenylpyridinium-mediated astrocyte vulnerability. J. Neurosci. Res. 2011, 89, 2068–2082. [Google Scholar] [CrossRef]
- Chuang, J.I.; Chen, T.H. Effect of melatonin on temporal changes of reactive oxygen species and glutathione after MPP(+) treatment in human astrocytoma U373MG cells. J. Pineal Res. 2004, 36, 117–125. [Google Scholar] [CrossRef]
- Blum, D.; Torch, S.; Lambeng, N.; Nissou, M.; Benabid, A.L.; Sadoul, R.; Verna, J.M. Molecular pathways involved in the neurotoxicity of 6-OHDA, dopamine and MPTP: Contribution to the apoptotic theory in Parkinson’s disease. Prog. Neurobiol. 2001, 65, 135–172. [Google Scholar] [CrossRef]
- Gupta, S.; Goswami, P.; Biswas, J.; Joshi, N.; Sharma, S.; Nath, C.; Singh, S. 6-Hydroxydopamine and lipopolysaccharides induced DNA damage in astrocytes: Involvement of nitric oxide and mitochondria. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2015, 778, 22–36. [Google Scholar] [CrossRef]
- Lewerenz, J.; Klein, M.; Methner, A. Cooperative action of glutamate transporters and cystine/glutamate antiporter system Xc- protects from oxidative glutamate toxicity. J. Neurochem. 2006, 98, 916–925. [Google Scholar] [CrossRef] [PubMed]
- Asanuma, M.; Miyazaki, I.; Diaz-Corrales, F.J.; Kimoto, N.; Kikkawa, Y.; Takeshima, M.; Miyoshi, K.; Murata, M. Neuroprotective effects of zonisamide target astrocyte. Ann. Neurol. 2010, 67, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Bentea, E.; Sconce, M.D.; Churchill, M.J.; Van Liefferinge, J.; Sato, H.; Meshul, C.K.; Massie, A. MPTP-induced parkinsonism in mice alters striatal and nigral xCT expression but is unaffected by the genetic loss of xCT. Neurosci. Lett. 2015, 593, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Massie, A.; Schallier, A.; Mertens, B.; Vermoesen, K.; Bannai, S.; Sato, H.; Smolders, I.; Michotte, Y. Time-dependent changes in striatal xCT protein expression in hemi-Parkinson rats. Neuroreport 2008, 19, 1589–1592. [Google Scholar] [CrossRef]
- Massie, A.; Goursaud, S.; Schallier, A.; Vermoesen, K.; Meshul, C.K.; Hermans, E.; Michotte, Y. Time-dependent changes in GLT-1 functioning in striatum of hemi-Parkinson rats. Neurochem. Int. 2010, 57, 572–578. [Google Scholar] [CrossRef]
- Miyazaki, I.; Asanuma, M. Serotonin 1A Receptors on Astrocytes as a Potential Target for the Treatment of Parkinson’s Disease. Curr. Med. Chem. 2016, 23, 686–700. [Google Scholar] [CrossRef]
- Gan, L.; Vargas, M.R.; Johnson, D.A.; Johnson, J.A. Astrocyte-specific overexpression of Nrf2 delays motor pathology and synuclein aggregation throughout the CNS in the alpha-synuclein mutant (A53T) mouse model. J. Neurosci. 2012, 32, 17775–17787. [Google Scholar] [CrossRef]
- Bergstrom, P.; Andersson, H.C.; Gao, Y.; Karlsson, J.O.; Nodin, C.; Anderson, M.F.; Nilsson, M.; Hammarsten, O. Repeated transient sulforaphane stimulation in astrocytes leads to prolonged Nrf2-mediated gene expression and protection from superoxide-induced damage. Neuropharmacology 2011, 60, 343–353. [Google Scholar] [CrossRef]
- Eggler, A.L.; Gay, K.A.; Mesecar, A.D. Molecular mechanisms of natural products in chemoprevention: Induction of cytoprotective enzymes by Nrf2. Mol. Nutr. Food Res. 2008, 52 (Suppl. 1), S84–S94. [Google Scholar] [CrossRef]
- Erlank, H.; Elmann, A.; Kohen, R.; Kanner, J. Polyphenols activate Nrf2 in astrocytes via H2O2, semiquinones, and quinones. Free Radic. Biol. Med. 2011, 51, 2319–2327. [Google Scholar] [CrossRef]
- Isooka, N.; Miyazaki, I.; Kikuoka, R.; Wada, K.; Nakayama, E.; Shin, K.; Yamamoto, D.; Kitamura, Y.; Asanuma, M. Dopaminergic neuroprotective effects of rotigotine via 5-HT1A receptors: Possibly involvement of metallothionein expression in astrocytes. Neurochem. Int. 2020, 132, 104608. [Google Scholar] [CrossRef] [PubMed]
- Monti, D.A.; Zabrecky, G.; Kremens, D.; Liang, T.W.; Wintering, N.A.; Bazzan, A.J.; Zhong, L.; Bowens, B.K.; Chervoneva, I.; Intenzo, C.; et al. N-Acetyl Cysteine Is Associated With Dopaminergic Improvement in Parkinson’s Disease. Clin. Pharm. 2019, 106, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, I.; Murakami, S.; Torigoe, N.; Kitamura, Y.; Asanuma, M. Neuroprotective effects of levetiracetam target xCT in astrocytes in parkinsonian mice. J. Neurochem. 2016, 136, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, M.E.; Moritoyo, T.; Kubo, M.; Kyaw, W.T.; Yabe, H.; Nishikawa, N.; Nagai, M.; Matsuda, S.; Nomoto, M. Zonisamide-induced long-lasting recovery of dopaminergic neurons from MPTP-toxicity. Brain Res. 2011, 1384, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, M.E.; Sugimoto, K.; Kubo, M.; Iwaki, H.; Tsujii, T.; Kyaw, W.T.; Nishikawa, N.; Nagai, M.; Tanaka, J.; Nomoto, M. Zonisamide up-regulated the mRNAs encoding astrocytic anti-oxidative and neurotrophic factors. Eur. J. Pharm. 2012, 689, 72–80. [Google Scholar] [CrossRef]
- Iida, M.; Miyazaki, I.; Tanaka, K.; Kabuto, H.; Iwata-Ichikawa, E.; Ogawa, N. Dopamine D2 receptor-mediated antioxidant and neuroprotective effects of ropinirole, a dopamine agonist. Brain Res. 1999, 838, 51–59. [Google Scholar] [CrossRef]
- Yoshioka, M.; Tanaka, K.; Miyazaki, I.; Fujita, N.; Higashi, Y.; Asanuma, M.; Ogawa, N. The dopamine agonist cabergoline provides neuroprotection by activation of the glutathione system and scavenging free radicals. Neurosci. Res. 2002, 43, 259–267. [Google Scholar] [CrossRef]
- Wei, Y.; Lu, M.; Mei, M.; Wang, H.; Han, Z.; Chen, M.; Yao, H.; Song, N.; Ding, X.; Ding, J.; et al. Pyridoxine induces glutathione synthesis via PKM2-mediated Nrf2 transactivation and confers neuroprotection. Nat. Commun. 2020, 11, 941. [Google Scholar] [CrossRef]
- Takeshima, M.; Miyazaki, I.; Murakami, S.; Kita, T.; Asanuma, M. l-Theanine protects against excess dopamine-induced neurotoxicity in the presence of astrocytes. J. Clin. Biochem. Nutr. 2016, 59, 93–99. [Google Scholar] [CrossRef]
- Du, F.; Li, R.; Huang, Y.; Li, X.; Le, W. Dopamine D3 receptor-preferring agonists induce neurotrophic effects on mesencephalic dopamine neurons. Eur. J. Neurosci. 2005, 22, 2422–2430. [Google Scholar] [CrossRef]
- Ohta, K.; Kuno, S.; Mizuta, I.; Fujinami, A.; Matsui, H.; Ohta, M. Effects of dopamine agonists bromocriptine, pergolide, cabergoline, and SKF-38393 on GDNF, NGF, and BDNF synthesis in cultured mouse astrocytes. Life Sci. 2003, 73, 617–626. [Google Scholar] [CrossRef]
- Imamura, K.; Takeshima, T.; Nakaso, K.; Ito, S.; Nakashima, K. Pramipexole has astrocyte-mediated neuroprotective effects against lactacystin toxicity. Neurosci. Lett. 2008, 440, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Zhao, H.; Zhang, W.; Liu, B.; Liu, Y.; Guo, Y.; Nie, L. Overexpression of conserved dopamine neurotrophic factor (CDNF) in astrocytes alleviates endoplasmic reticulum stress-induced cell damage and inflammatory cytokine secretion. Biochem. Biophys. Res. Commun. 2013, 435, 34–39. [Google Scholar] [CrossRef] [PubMed]
- De Miranda, B.R.; Rocha, E.M.; Bai, Q.; El Ayadi, A.; Hinkle, D.; Burton, E.A.; Timothy Greenamyre, J. Astrocyte-specific DJ-1 overexpression protects against rotenone-induced neurotoxicity in a rat model of Parkinson’s disease. Neurobiol. Dis. 2018, 115, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Rothstein, J.D.; Patel, S.; Regan, M.R.; Haenggeli, C.; Huang, Y.H.; Bergles, D.E.; Jin, L.; Dykes Hoberg, M.; Vidensky, S.; Chung, D.S.; et al. Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature 2005, 433, 73–77. [Google Scholar] [CrossRef]
- Leung, T.C.; Lui, C.N.; Chen, L.W.; Yung, W.H.; Chan, Y.S.; Yung, K.K. Ceftriaxone ameliorates motor deficits and protects dopaminergic neurons in 6-hydroxydopamine-lesioned rats. Acs Chem. Neurosci. 2012, 3, 22–30. [Google Scholar] [CrossRef]
- Hsu, C.Y.; Hung, C.S.; Chang, H.M.; Liao, W.C.; Ho, S.C.; Ho, Y.J. Ceftriaxone prevents and reverses behavioral and neuronal deficits in an MPTP-induced animal model of Parkinson’s disease dementia. Neuropharmacology 2015, 91, 43–56. [Google Scholar] [CrossRef]
- Carbone, M.; Duty, S.; Rattray, M. Riluzole elevates GLT-1 activity and levels in striatal astrocytes. Neurochem. Int. 2012, 60, 31–38. [Google Scholar] [CrossRef]
- Mizuta, I.; Ohta, M.; Ohta, K.; Nishimura, M.; Mizuta, E.; Kuno, S. Riluzole stimulates nerve growth factor, brain-derived neurotrophic factor and glial cell line-derived neurotrophic factor synthesis in cultured mouse astrocytes. Neurosci. Lett. 2001, 310, 117–120. [Google Scholar] [CrossRef]
- Araki, T.; Muramatsu, Y.; Tanaka, K.; Matsubara, M.; Imai, Y. Riluzole (2-amino-6-trifluoromethoxy benzothiazole) attenuates MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) neurotoxicity in mice. Neurosci. Lett. 2001, 312, 50–54. [Google Scholar] [CrossRef]
- Baker, Z.N.; Cobine, P.A.; Leary, S.C. The mitochondrion: A central architect of copper homeostasis. Metallomics 2017, 9, 1501–1512. [Google Scholar] [CrossRef] [PubMed]
- D’Ambrosi, N.; Rossi, L. Copper at synapse: Release, binding and modulation of neurotransmission. Neurochem. Int. 2015, 90, 36–45. [Google Scholar] [CrossRef]
- Pall, H.S.; Williams, A.C.; Blake, D.R.; Lunec, J.; Gutteridge, J.M.; Hall, M.; Taylor, A. Raised cerebrospinal-fluid copper concentration in Parkinson’s disease. Lancet 1987, 2, 238–241. [Google Scholar] [CrossRef]
- Li, Q.; Haney, M.S. The role of glia in protein aggregation. Neurobiol. Dis. 2020, 143, 105015. [Google Scholar] [CrossRef] [PubMed]
- Shu, X.; Sun, Y.; Sun, X.; Zhou, Y.; Bian, Y.; Shu, Z.; Ding, J.; Lu, M.; Hu, G. The effect of fluoxetine on astrocyte autophagy flux and injured mitochondria clearance in a mouse model of depression. Cell Death Dis. 2019, 10, 577. [Google Scholar] [CrossRef]
- Ubhi, K.; Inglis, C.; Mante, M.; Patrick, C.; Adame, A.; Spencer, B.; Rockenstein, E.; May, V.; Winkler, J.; Masliah, E. Fluoxetine ameliorates behavioral and neuropathological deficits in a transgenic model mouse of alpha-synucleinopathy. Exp. Neurol. 2012, 234, 405–416. [Google Scholar] [CrossRef]
- Konno, M.; Hasegawa, T.; Baba, T.; Miura, E.; Sugeno, N.; Kikuchi, A.; Fiesel, F.C.; Sasaki, T.; Aoki, M.; Itoyama, Y.; et al. Suppression of dynamin GTPase decreases alpha-synuclein uptake by neuronal and oligodendroglial cells: A potent therapeutic target for synucleinopathy. Mol. Neurodegener. 2012, 7, 38. [Google Scholar] [CrossRef]
- Sharma, N.; Jamwal, S.; Kumar, P. Beneficial effect of antidepressants against rotenone induced Parkinsonism like symptoms in rats. Pathophysiology 2016, 23, 123–134. [Google Scholar] [CrossRef]
- Dixit, S.; Khan, S.A.; Azad, S. A Case of SSRI Induced Irreversible Parkinsonism. J. Clin. Diagn. Res. 2015, 9, VD01–VD02. [Google Scholar] [CrossRef]
- Clarke, B.E.; Taha, D.M.; Tyzack, G.E.; Patani, R. Regionally encoded functional heterogeneity of astrocytes in health and disease: A perspective. Glia 2020. [Google Scholar] [CrossRef]
- Asanuma, M.; Okumura-Torigoe, N.; Miyazaki, I.; Murakami, S.; Kitamura, Y.; Sendo, T. Region-Specific Neuroprotective Features of Astrocytes against Oxidative Stress Induced by 6-Hydroxydopamine. Int. J. Mol. Sci. 2019, 20, 598. [Google Scholar] [CrossRef] [PubMed]
- Fasano, A.; Visanji, N.P.; Liu, L.W.; Lang, A.E.; Pfeiffer, R.F. Gastrointestinal dysfunction in Parkinson’s disease. Lancet Neurol. 2015, 14, 625–639. [Google Scholar] [CrossRef]
- Jellinger, K.A. Neuropathobiology of non-motor symptoms in Parkinson disease. J. Neural Transm. 2015, 122, 1429–1440. [Google Scholar] [CrossRef] [PubMed]
- Seguella, L.; Capuano, R.; Sarnelli, G.; Esposito, G. Play in advance against neurodegeneration: Exploring enteric glial cells in gut-brain axis during neurodegenerative diseases. Expert Rev. Clin. Pharm. 2019, 12, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, I.; Isooka, N.; Wada, K.; Kikuoka, R.; Kitamura, Y.; Asanuma, M. Effects of Enteric Environmental Modification by Coffee Components on Neurodegeneration in Rotenone-Treated Mice. Cells 2019, 8, 221. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, I.; Isooka, N.; Imafuku, F.; Sun, J.; Kikuoka, R.; Furukawa, C.; Asanuma, M. Chronic Systemic Exposure to Low-Dose Rotenone Induced Central and Peripheral Neuropathology and Motor Deficits in Mice: Reproducible Animal Model of Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 3254. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miyazaki, I.; Asanuma, M. Neuron-Astrocyte Interactions in Parkinson’s Disease. Cells 2020, 9, 2623. https://doi.org/10.3390/cells9122623
Miyazaki I, Asanuma M. Neuron-Astrocyte Interactions in Parkinson’s Disease. Cells. 2020; 9(12):2623. https://doi.org/10.3390/cells9122623
Chicago/Turabian StyleMiyazaki, Ikuko, and Masato Asanuma. 2020. "Neuron-Astrocyte Interactions in Parkinson’s Disease" Cells 9, no. 12: 2623. https://doi.org/10.3390/cells9122623
APA StyleMiyazaki, I., & Asanuma, M. (2020). Neuron-Astrocyte Interactions in Parkinson’s Disease. Cells, 9(12), 2623. https://doi.org/10.3390/cells9122623