3. Results
rDNA clusters contact genes involved in chromatin organization. The 4C-rDNA reads after trimming of sequences from the viewpoint region of the rDNA were mapped in the dm6 genome as described in the Materials and Methods. We used Illumina read lengths of 150 nucleotides to ensure the robustness of the mapping in the Drosophila genome. As expected, the processed reads possessed EcoRI and/or FaeI sites at their ends. To visualize the mapped 4C-rDNA reads, we used the IGB Browser.
Figure 1A,B shows that here is a good correlation between the replicates for experiments without heat shock treatment or after it. Heat shock treatment was used to test whether nucleoli contacts with different chromosome sites are physiological and could change upon stimulation. Nucleoli are key sensors of cellular stress and are involved in the response to different types of physiological challenges including heat shock treatment, hypoxia, pH fluctuation, and redox stress [
12]. Heat shock leads to the disassembly of the nucleolus and affects rRNA transcription [
12] and, thus, could affect the inter-chromosomal contacts of nucleoli. The 3.5-Mb region from the 3R chromosome possesses many contacts, and changes in the rDNA-contact pattern were observed after the treatment. The
Drosophila genome is enriched with mobile and repetitive elements of different classes. Although the true rDNA contacts could correspond to these repetitive sequences, we filtered the 4C-rDNA reads using the Dfam database of repetitive DNA families [
20] in order to remove the reads entirely corresponding to
Drosophila repeats without any adjacent sequences coming from unique DNA stretches.
Previously, microscopic analysis of nucleoli contacts with polytene chromosomes revealed the wide distribution of contact frequencies [
5]. In this study, we characterized the most frequent rDNA contacts in the genome, as previously performed in the human genome [
8]. That is why the mapped reads exceeding 100 contacts were selected and genes located within ±2.5 kb around such contact sites in the normal cells and in cells after heat shock treatment were identified. In this way, we selected 588 genes near rDNA-contacting sites. Without the Dfam filtering, 699 genes were detected in this way from S2 cells that were not subjected to heat shock treatment and 1001 genes after treatment. The overlap between these three groups of genes is shown in
Figure 1C and
Table S1. The groups of 81 and 30 genes in the Venn diagram correspond to rDNA contacts with repetitive sequences because they were subtracted by Dfam filtering and, thus, are not present in the group of 588 genes. The heat shock treatment leads to redistribution of the rDNA contacts. However, the group of 149 genes that do not correspond to genomic repeats retained their contacts with rDNA after heat shock treatment.
Table S2 shows that these genes revealed extremely high associations (up to 3.6 × 10
−48) with a number of Gene Ontology (GO) items relating to the nucleosome level of chromatin organization, including histone genes. High correlations with particular GO groups were observed for the selected 588 genes (
Table S3). The top five groups of these genes are associated with nucleosome organization, chromatin organization, chromosome organization, cellular component assembly, and protein-containing subunit organization (
Figure 1D).
Interestingly, the group of 871 genes shown in
Figure 1C includes 36 genes corresponding to another set of particular histone genes that do not overlap either with histone genes in the group of 588 genes lacking repeats or with the group of 699 genes with repeats (
Table S4). These data suggest that heat shock treatment induces a shift of rDNA-contacting sites within the cluster of histone genes in chromosome 2L or changes the frequencies of these contacts. Among this group of 871 genes, there are also 12
Ste genes (involved in the regulation of protein serine/threonine phosphatase activity), 76
lncRNA (long non-coding RNA) genes,
Df31 (encodes a histone-binding protein, which is involved in nucleosome assembly), the
mod(mdg4) gene (encodes a nuclear protein that specifically interacts with various DNA-binding proteins), and several genes specifying co-regulators of transcription (
Taf11,
kis,
tara, and others). These data show that heat shock stress causes changes in the pattern of nucleoli contacts in the genomic regions where genes controlling chromatin organization reside.
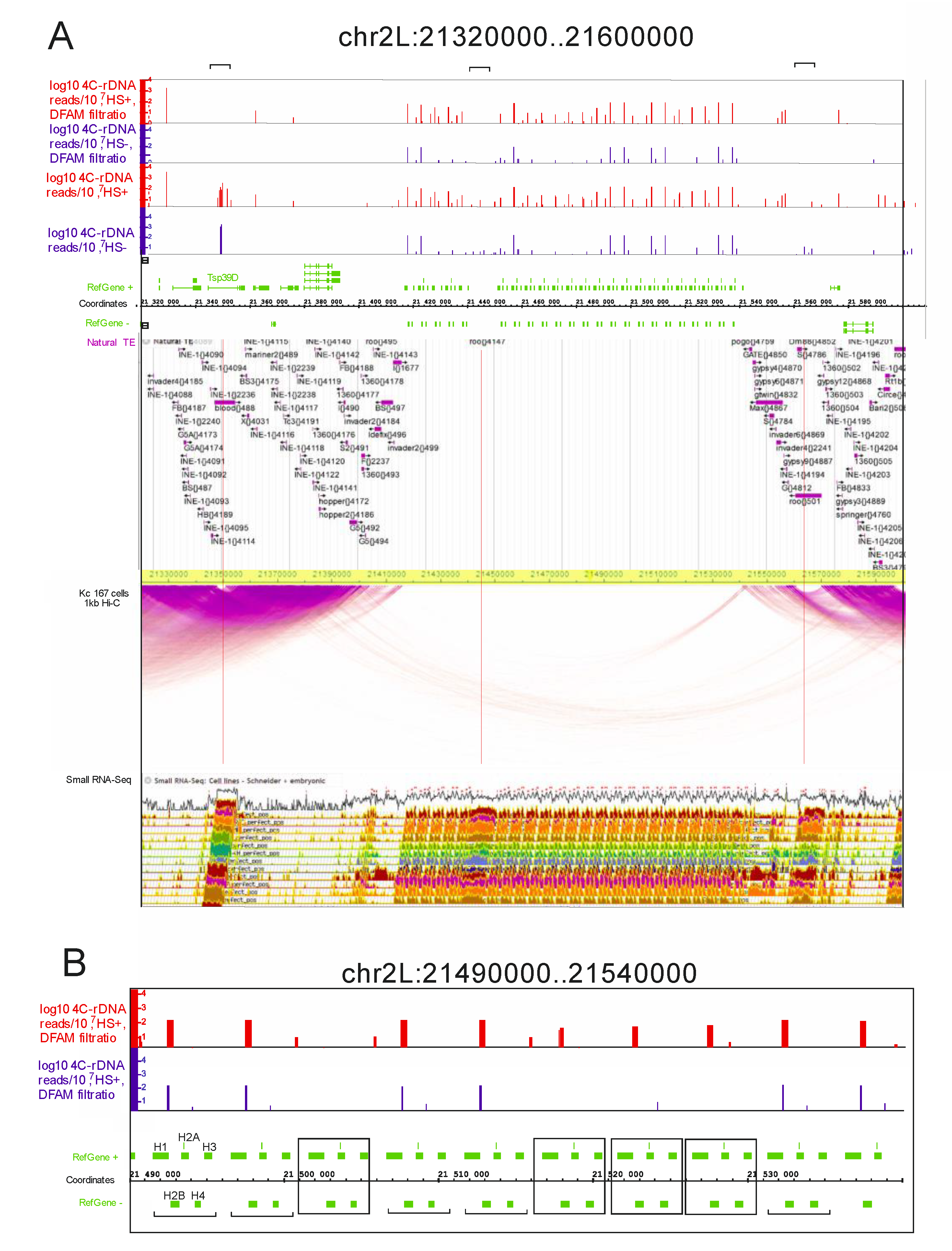
Heat shock treatment intensifies the nucleoli contacts within histone gene clusters. Next, we studied the rDNA contacts inside histone gene clusters in more detail.
Figure 2 shows the 280-kb region from chromosome 2L that possesses a histone gene cluster, which is located in the center of the region.
There are many transposable elements of different classes that are found mainly on the flanks of the region. At the top of
Figure 2A, the Dfam-filtered results corresponding to the rDNA contacts in the heat-shock-treated and untreated cells are shown. There are many rDNA-contacting sites in the region corresponding to a histone gene cluster. The number of individual contacts varies and does not always coincide between the normal and heat-shock-treated cells.
The results are shown in more detail in
Figure 2B. Surprisingly, better resolution revealed that heat shock treatment induces the appearance of new contacts. The distribution of the contact sites varies between different histone repeats. One set of repeats has a similar distribution of rDNA-contacting sites before and after the treatment; these contact sites are located after the
H1 gene. However, other copies of the histone repeat acquire the contacts only after the heat shock treatment. These data confirm our conclusion drawn from the analysis of the Venn diagram shown in
Figure 1 that new rDNA contacts appear after heat shock treatment. The result is partially due to the selection of sites possessing 100 or more mapped reads. Nevertheless, the data indicate that within the histone gene cluster, which spans about 130 kb, there are changes in the rDNA contacts inside rather small regions. Surprisingly, this indicates that the area of contacts of large organelles, such as nucleoli, with a particular chromosome region may be localized.
The Hi-C data, with 1-kb resolution and detected in Kc 167 cells, are shown in
Figure 2A. There is a gap in the intra-chromosomal contacts corresponding to a histone gene cluster. It is not clear whether this is because the mapping of Hi-C reads is hampered by the presence of histone repeats or if the histone gene cluster that is enriched with rDNA contacts is free of intra-chromosomal contacts. An argument in favor of the latter follows from the fact that the dense intra-chromosomal contacts were mapped by the same procedure on the flanks of the histone gene cluster, which are enriched with transposable elements. In these dense regions, two “bubbles” are observed that correspond to
Blood and
Roo LTR-elements on the left and right flanks, respectively.
There is one more
Roo element that resides inside the histone gene cluster. This element, as well the
Blood and
Roo elements located on the flanks of the cluster, exhibits rDNA contacts that disappear after Dfam filtering. This result again demonstrates that mobile elements are the targets in rDNA contacts. Three LTR-elements and the region of the histone gene cluster itself are the sites where small RNAs are detected in S2 and embryonic cells (
Figure 2). The
H1 gene in S2 cells is transcribed throughout the S phase, while the core histone genes are only transcribed in a short pulse during early S phase [
32]. It follows that the histone cluster is silenced and small RNAs and rDNA contacts could be involved in the mechanism of this silencing. Our data on the dynamics of rDNA contacts within the histone gene cluster upon heat shock treatment support this view.
DNA-FISH experiments confirm the contacts between rDNA units and histone genes. 4C-rDNA data demonstrate the existence of contacts between nucleoli and histone genes. In these experiments, we used a bulk of S2 cells, which is why we next determined whether these contacts are stable and could occur either in every S2 cell or in some portion of the unsynchronized cells. To address this question, we used two-color DNA-FISH.
Figure 3D shows that only one-third of the cells possess the inter-chromosomal contacts between rDNA units and histone genes. S2 cells are generally tetraploid [
33], which is why we observed several foci for the histone probe. Interestingly, only one or two histone foci reveal contacts with the nucleoli (
Figure 3). These data independently confirm the presence of contacts between histone genes and rDNA units and suggest that the contacts are dynamic and may be formed in a particular stage of the cell cycle. Currently, we are studying these contacts in more detail by DNA-FISH using synchronized S2 cells and normal diploid
Drosophila cells.
Heat shock treatment induces nucleoli contacts with Ste genes. Histone clusters demonstrated a shift in the rDNA contacts following heat shock treatment. The data shown in
Figure 1C and
Table S4 indicate that 12
Ste genes that did not shape the contacts with rDNA in the normal state are moved to the nucleoli after heat shock treatment. Euchromatin
Ste genes, together with heterochromatin
Su(
Ste) genes, are involved in the maintenance of male fertility, reproduction, and reproductive isolation of
Drosophila melanogaster. The cluster of
Ste genes inside an 85-kb segment of the X chromosome is shown in
Figure 4.
No contacts of rDNA genes with Ste genes were detected in the normal cells even without Dfam filtering and the contacts were detected only after heat shock treatment. Again, similar to the results obtained in the histone gene cluster, there was a lack of Ste transcripts in both the normal and heat-shock-treated cells, and small RNAs exactly corresponding to the region of the Ste genes were detected. A “bubble” in the pattern of intra-chromosomal contacts was observed in the area where the Ste genes reside and coinciding with the region containing rDNA contacts. We suggest that nucleoli contacts with this gene cluster are required to ensure the silent state of Ste genes.
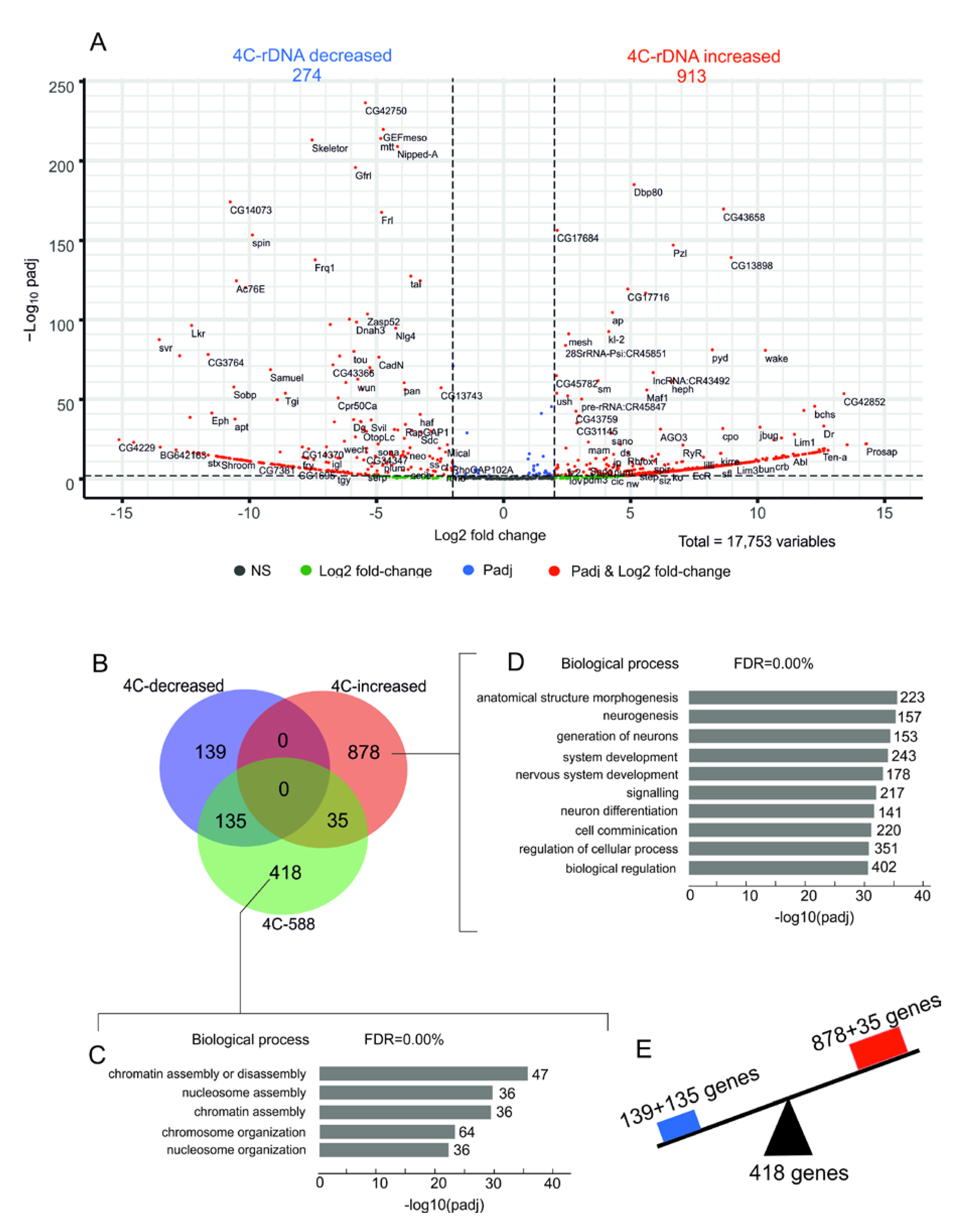
The genes involved in chromosome organization remain in contact with nucleoli while the genes that control morphogenesis show altered contacts after heat stress. The data on rDNA contacts within the histone and
Ste gene clusters prompted us to perform a whole-genome analysis of changes in the nucleoli contacts induced by heat shock treatment using a volcano presentation. This type of visualization allows us to observe changes in the contact frequencies of nucleoli with different genes.
Figure 5B shows the statistically significant results of the differential 4C-rDNA analysis for all detected rDNA contacts in the genome without a threshold of 100 reads that were used before (
Figure 1C). Dfam filtering of the 4C-rDNA reads was used to remove the contacts with transposable elements.
We observed that 274 rDNA-contacting genes decreased the number of contacts with rDNA while 913 genes increased the number of contacts. Among these latter genes are those that did not have contacts with nucleoli before the heat shock treatment, e.g.,
Ste genes. The complete list of the genes is shown in
Table S5. The list of genes from the 4C differentiation analysis was compared with the previously selected list of 588 genes (
Table S1) that revealed 100 or more contacts with nucleoli. The Venn diagram in
Figure 5B shows that the majority of genes (418 out of 588 rDNA-contacting genes) retained their contacts with rDNA upon heat shock treatment and corresponds to genes involved in chromatin assembly (
Figure 5C,
Table S6). Simultaneously, two groups of non-overlapping genes (135 and 139 genes) shown in the Venn diagram in
Figure 5B decreased their number of contacts with nucleoli. Both groups are highly associated with morphogenesis, organ development, and differentiation (
Tables S7 and S8). Among the genes that decreased the number of contacts are the homeotic
ct and
Abd-B genes. It is of interest that the largest group of genes (878 genes in the Venn diagram in
Figure 5B) is also associated with morphogenesis and development (
Figure 5D and
Table S9). These data indicate that heat shock treatment induces alterations (increase or decrease) in the contacts of genes controlling development with nucleoli (
Figure 5E), while the genes responsible for chromosome organization retain the contacts.
A set of rDNA-contacting genes controlling development is upregulated after heat shock treatment. To test the hypothesis that changes in contacts with nucleoli correlate with the changes in expression of rDNA-contacting genes, we analyzed the differential expression of rDNA-contacting genes after heat shock treatment (
Table S10). Our data indicate that contacts with nucleoli do not correlate significantly with changes in expression and that most differentially expressed genes do not change their contact frequency. It is likely that a reversible change in 3D chromatin architecture upon heat shock treatment affects only a subset of rDNA-contacting genes. We found that 42 of 588 rDNA-contacting genes were downregulated while 60 genes were upregulated (
Figure 6A).
Seven downregulated genes revealed a decrease in the number of contacts with rDNA genes (
Figure 6B,
Table S11). Among these, two genes (
Sema1a and
PlexA) are involved in the semaphorin-plexin signaling pathway (
Table S12). Only five of the downregulated genes showed an increase in the number of contacts and the remaining 30 downregulated genes did not alter their contacts with rDNA after the treatment.
In the group of upregulated genes, there are only five that increased the number of contacts with rDNA (
Figure 6C,
Table S13). Among them are the
cnc (cap-n-collar) gene, which encodes a transcription factor that regulates the activation of genes by oxidative stress and is involved in dendrite morphogenesis. Another gene in this group is
chic, which is involved in cell division and cellular morphogenesis.
Nineteen upregulated genes decreased the number of contacts with rDNA. The group includes several genes encoding transcription factors (
tai,
Tgi,
Glut4EF,
slou, and
cwo) and the
Syp gene that specifies an RNA-binding protein that is required for synapse morphology and synaptic transmission (
Figure 6C and
Figure S2,
Table S13). The GO associations of the 60 upregulated genes that regulate development and contact the rDNA are shown in
Figure 6E and
Table S14. These genes regulate development and are involved in cell projection organization and neurogenesis. These data indicate that heat shock treatment induces an epigenetic switch that orchestrates gene expression by decreasing the expression of one set of rDNA-contacting genes while at the same time increasing the expression of another set of genes that regulate development (
Figure 6D). These results demonstrate that the changes in the contacts with nucleoli of about 100
Drosophila genes are coupled with the downregulation or upregulation of these genes. However, only 36 of these genes change their contacts with nucleoli. It should be mentioned that other mechanisms induced by heat shock could regulate rDNA-contacting genes.
After heat shock treatment of S2 cells and recovery for 1 h, rDNA genes are downregulated (
Figure 6F) to about 60% of the 47S pre-rRNA level. Inhibition of rDNA gene expression is likely connected to the observed changes in the contacts of rDNA genes (
Figure 5A). Downregulation of the expression of rDNA units is coupled with a downregulation of the H2B histone gene, which specifies the core histone, by about 30% (
Figure 6F). We suppose that the phase separation mechanism may occur via this coordinated inhibition of rDNA and histone genes and we are currently addressing this question.
Heat shock treatment may induce transition of the nucleoli to epigenetically different regions. Because there are changes in the rDNA contacts in response to heat shock treatment, we studied the profiles of histone marks, small RNAs, Ago1 and Ago2 complexes, and other factors within the genomic regions where the contacts were detected. We used the available data for the normal S2 cells and compared the profiles around rDNA-contacting sites in normal and heat-shock-treated S2 cells. As cells easily recover after a rather short heat shock procedure (1 h incubation at 37 °C followed by recovery for 1 h at 27 °C in our experiments), we assumed that the majority of epigenetic marks would not change. However, this assumption should be experimentally proved, because, for example, brief exposure of human cultured cells to hypoxia leads to an induction of H3K4me3 within hours [
34]. Therefore, we studied the epigenetic states of the regions where rDNA contacts moved to after heat shock treatment in our experiments.
Figure 1C indicates that among 1001 rDNA-contacting sites detected after heat treatment, 230 did not change. However, it is possible to check the putative difference in the contact regions of the remaining 871 sites.
We observed minor differences in the profiles for small RNAs and Ago1 and Ago2 complexes (
Figure 7). Nevertheless, this suggests that the rDNA-contacting sites are near the target sites of Ago1 and Ago2 complexes, which are involved in heterochromatin formation [
35]. No differences were observed for H3K27ac active chromatin marks. These data suggest that there is a prominent set of rDNA contacts within active chromatin regions. In contrast, clear differences were detected for the repressive mark, H3K27me3. The nucleoli after heat shock treatment move from
Polycomb repressive regions to regions that are depleted of this mark and are probably active. This result confirms our previous data suggesting that nucleoli change their pattern of contacts after heat shock treatment (
Figure 1,
Figure 2 and
Figure 4).
Another example that supports our view of dynamic rDNA contacts induced by stress comes from the comparison of profiles of the chromatic modifier GAF and the nucleosome remodeler dMi-2. After heat shock treatment, nucleoli shift from the sites enriched with these factors to other sites that lack them. The same is true for profiles of H3K4me2, which marks
Drosophila PREs and maintains the developmental expression pattern of nearby genes [
36], and the active histone mark H3K4me3, which promotes transcription initiation.
Interestingly, the active H3K9ac mark, which switches from transcription initiation to elongation [
37], and the heterochromatin marker H3K9me3 have contrasting profiles (
Figure 6). After heat shock treatment, rDNA escapes the subset of transcribed regions and moves to inactive or heterochromatin regions. A similar transfer of nucleoli occurs from the active regions marked by H3K18ac to inactive regions.
Taken together, these results independently confirm the above-described data that heat shock treatment leads to changes in nucleoli contacts. Although not all contacts were changed, the data shown in
Figure 7 support this conclusion. Moreover, the search of profiles indicates that the epigenetic features of new contact sites induced by heat stress often correspond to repressed chromatin. The only exception is the transfer of rDNA clusters from the regions marked by HK27me3 to the regions depleted of this mark.
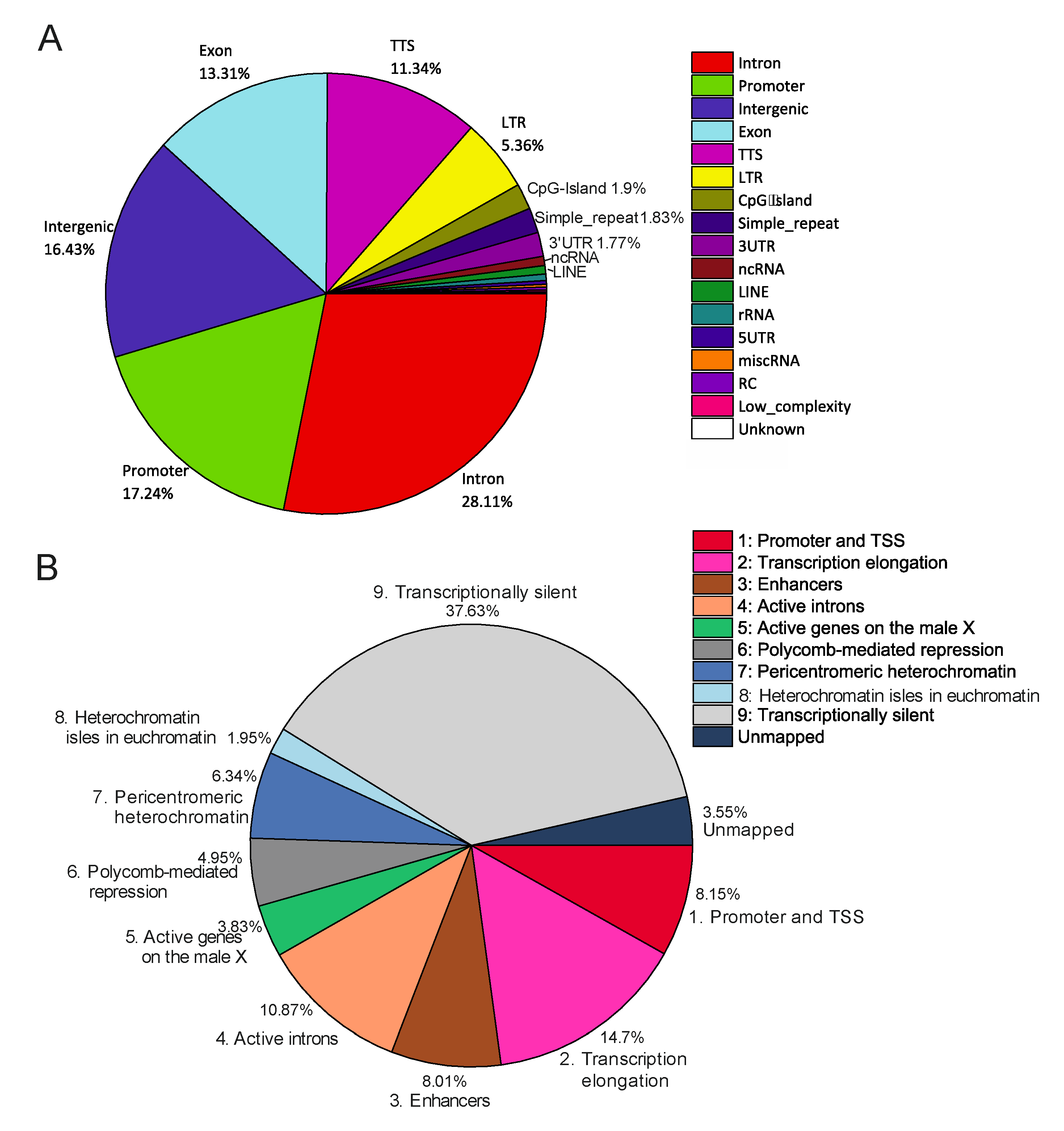
Alterations in rDNA-contacting sites induced by heat shock treatment do not change their proportions of genetic and epigenetic features. To better understand the properties of rDNA-contacting sites, we analyzed the distribution of rDNA-contacting sites in different portions of the
Drosophila genome.
Figure 8A shows that these sites correspond mainly to genes themselves. In reality, 71% of the rDNA-contacting sites correspond to promoters, exons, introns, transcription termination sites (TTS), and untranslated transcribed regions (UTRs).
Intergenic regions and LTR-elements comprise about 16% and 5% of the genome, respectively. It should be mentioned that the values were calculated for Dfam-filtered reads without the threshold of 100 reads per site, which means that at least 5% of 4C-rDNA reads were mapped at the very ends of the LTR-elements because they contained the attached unique sequences from the insertion sites. This class of transposable elements (TEs) comprises about 3.29% of all TEs [
38]. In
Drosophila 20% of the genome consists of TEs [
39] and LTR-elements should make up about 0.7% of the genome. Therefore, these elements are about seven times or more enriched with rDNA contacts than expected from a random distribution of contacts throughout the genome.
A similar distribution of rDNA-contacting sites in different portions of the genome was observed after heat shock treatment (not shown), which was unexpected because the essential changes in the rDNA-contact sites were observed after the treatment (see
Figure 1,
Figure 3 and
Figure 4). Therefore, the data suggest that the shift in the contacts takes place in the same proportions between the promoters, exons, introns, TTS, UTRs, and LTR-elements.
This conclusion is independently supported by the distribution of rDNA contacts among regions with different chromatin states (
Figure 8B). To elucidate the epigenetic states at the nucleoli contacts, we used the search of determined the nine states of chromatin in S2 cells [
40]. The results were similar for the contacts in normal cells (
Figure 8B) and in the cells after heat shock treatment (not shown). It follows that changes in nucleoli contacts after heat shock treatment take place between regions possessing similar epigenetic states. About 44% of the contacts take place with transcriptionally silent or repressed chromatin. This value includes about 5% of the contacts corresponding to Polycomb-mediated repressed regions and heterochromatin isles in euchromatin regions. The regions corresponding to active chromatin comprise about 51% in total (promoters, transcription elongation regions, enhancers, active introns, and active genes on the male X chromosome). A comparison with the genome-wide distribution of the nine chromatin states for S2 cells suggests a non-random distribution of rDNA-contact sites (
p-value < 10
−6). These data suggest a role for nucleoli in both the epigenetic silencing and activation of genes.
rDNA contacts may correspond to gaps in the intra-chromosomal loops.Figure 9 shows the detected rDNA contacts inside the
flamenco locus. Dfam filtering removed almost all the mapped contacts corresponding to the locus. Nevertheless, three contacts occur in the locus only after heat shock treatment and these correspond to the sites targeted by small RNAs.
Again, similar to that observed inside the histone gene cluster, there is a gap in the intra-chromosomal contacts corresponding to the entire
flamenco gene and containing a cluster of LTR-elements (i.e.,
mdg1,
blood,
Stalker,
Doc, and others). The corresponding Hi-C reads were either excluded from the analysis or it was impossible to map them. However, we observed a small gap in the intra-chromosomal loops in the region that does not possess any repeated sequences. There are several protein-coding genes (
CG32500,
CG32819,
CG32857,
CG32820, and
CG33502) in this area, which are shown on the left flank in
Figure 9. In normal S2 cells, this region has three sites of rDNA contacts, which disappear after heat shock treatment. The result suggests that the contacts of rDNA could affect the formation of intra-chromosomal contacts and, as a result, a gap is formed. Gaps in the intra-chromosomal loops were also observed inside the histone gene cluster and in the region corresponding to
Ste genes (
Figure 2 and
Figure 4). In both cases, the gaps coincide with the regions where many rDNA contacts were detected. Taken together with the leftmost gap shown in
Figure 9, these results support our supposition that contacts with nucleoli may prevent the formation of intra-chromosomal loops.
4. Discussion
The nature and mechanisms of inter-chromosome interactions are still unclear. In principle, there should be no differences between inter-chromosomal and long-range intra-chromosomal regulatory interactions. In both cases, interacting chromatin regions could share or shape some epigenetic regulatory elements. However, the main characteristic of intra-chromosomal interactions is that they are predominantly structural and are formed for the compaction of long chromosomal DNA into the limited space of the nucleus. This is why the number of detected inter-chromosomal interactions is two orders of magnitude weaker than the number of intra-chromosomal contacts [
2].
In this study, we examined the most frequent contact sites of rDNA clusters in S2 cells and show that nucleoli form the most stable contacts with genes involved in chromosome organization. In contrast, the association of genes that control morphogenesis and development with nucleoli are altered after heat shock treatment (
Figure 5). This mobility of the contacts argues in favor of their physiological significance. One set of genes controlling differentiation decreases the number of contacts while another non-overlapping set of genes increases them. There is an extremely high association (up to 10
−39) of the latter set with morphogenesis and the generation of neurons (
Figure 5D). S2 cells are derived from late-stage embryos [
33] and after embryonic stages 16–17, there is a global downregulation of genes important for early neuronal development together with a global upregulation of genes necessary for the final differentiation of neurons [
41]. We assume that the upregulation of rDNA-contacting genes involved in neurogenesis may reflect the origin of S2 cells from embryonic neural cells.
Heterochromatin condensation of rDNA clusters initiates the formation of repressed chromatin structures outside of the nucleolus coupled with the transcriptional activation of a set of differentiation genes [
42]. The mechanisms of such global changes in gene expression triggered by rDNA clusters are not known. Nevertheless, the data suggest that nucleoli are involved in the epigenetic regulation of different genomic loci. One possible mechanism of this type of regulation is the formation of direct contacts between rDNA genes and different chromosomal regions. We observed that, upon heat shock treatment, a set of
Drosophila genes involved in differentiation and morphogenesis moved close to nucleoli (
Figure 5) and 60 of these rDNA-contacting genes controlling development were upregulated (
Figure 6). This increase in contacts with nucleoli coupled with upregulation suggests a role for rDNA in the epigenetic regulation of a specific set of genes in S2 cells.
We speculate that the direct contact of active or silenced rDNA units with different genomic regions leads to the activation or silencing of the corresponding genes. Nucleoli, as membraneless organelles, could shape close interactions with particular genomic regions. Our data demonstrate that rDNA contacts are stable with some sets of genes (e.g., histone genes) and dynamic with other sets of genes (e.g.,
Ste genes). Putative trans-activation or trans-silencing produced by nucleoli is reminiscent of the mechanisms of transvection [
6] and Y-linked genetic variation in
Drosophila that modulate the expression of hundreds of genes in the genome [
43].
We observed gaps in the intra-chromosomal contacts within the regions of histone gene repeats and the
Ste locus contacting the rDNA. One possible explanation is that the mapping of Hi-C data is hampered in areas of genomic repeats. An alternative explanation is that the regions that are involved in inter-chromosomal contacts are excluded from the intra-chromosomal folding. The gaps corresponding to histone genes, the
Ste locus, and to mobile elements (
Figure 2 and
Figure 4) are decorated by the mapped small RNAs that probably correspond to siRNAs and piRNAs. We found that LTR-elements comprise about 5% of rDNA-contacting regions (
Figure 8) but at present, we cannot map the corresponding rDNA contacts. It cannot be excluded that mobile elements play some role in the inter-chromosomal contacts with nucleoli.
The nucleoli of human cells shape contacts with genomic regions that often possess strong and wide (up to 50-kb) H3K27ac marks, which are characteristic of super-enhancers [
44,
45]. In S2 cells, we detected an enrichment of this histone mark at the rDNA-contacting regions (
Figure 7 and
Figure S2). At present, the details of the functional anatomy of nucleoli are unknown and we do not understand mechanistically how chromatin loops of rDNA units could expand silenced or active states on the genes at the contact sites. Recently, it was suggested that super-enhancers are involved in phase-separation mechanisms [
46,
47]. Additionally, it was demonstrated recently that RNA promotes the nucleation and sizing of the phase-separated condensates [
12,
48]. We detected that the rDNA-contacting sites are enriched with both small RNAs and the binding sites of Ago1 and Ago2 complexes (
Figure 7). Analysis of Ago2 profiles around rDNA-contacting sites at 42 downregulated or 60 upregulated genes (see
Figure 6 and
Tables S13 and S14) revealed distinct differences in the profiles of these two subgroups of genes. The data suggest that rDNA contacts and heat shock may regulate the activation or repression of the corresponding genes by modulating Ago2 complexes. This conclusion is supported by the recent observation of the involvement of Ago2 in developmental processes by controlling the 3D configuration of the chromatin regions [
49]. Currently, we are analyzing individual rDNA-contacting genes from these lists.
We suppose that RNA-mediated mechanisms could be involved in the targeting of nucleoli to specific genomic regions. Taken together, these data argue in favor of the formation of micro-condensates and phase-separation mechanisms being involved in the targeted epigenetic trans-regulation of different
Drosophila genes that contact the nucleoli. It is unclear how several nucleoli could shape contacts with hundreds of genes. It has been shown previously that eight
Drosophila nucleoli formed contacts with eight different regions in polytene chromosomes [
5]. We suppose that, after heat shock treatment, the disassembled nucleoli rapidly change the pattern of contacts with different chromosomes and that particular sites may differ in individual cells. Our 4C-rDNA procedure, which uses about 34 million cells, could lead to the detection of multiple sites coming from different cells. The current data on cell-to-cell variability support this possibility [
50] and, thus, single-cell analysis of rDNA contacts and the study of phase-separation mechanisms could answer the questions raised by this study.

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








