The Genomics of Myelodysplastic Syndromes: Origins of Disease Evolution, Biological Pathways, and Prognostic Implications


Abstract
1. Introduction
2. Chromosomal Abnormalities
2.1. Chromosome 5q Deletion [del(5q)]
2.2. Chromosome 7q Deletion [del(7q)], Monosomy 7 (-7)
2.3. Trisomy 8
2.4. Chromosome 20 q Deletion [del(20q)]
2.5. Chromosome 17Abnormalities [17p Deletions, Isochromosome 17q]
2.6. Complex Karyotypes
2.7. Rare Chromosomal Abnormalities
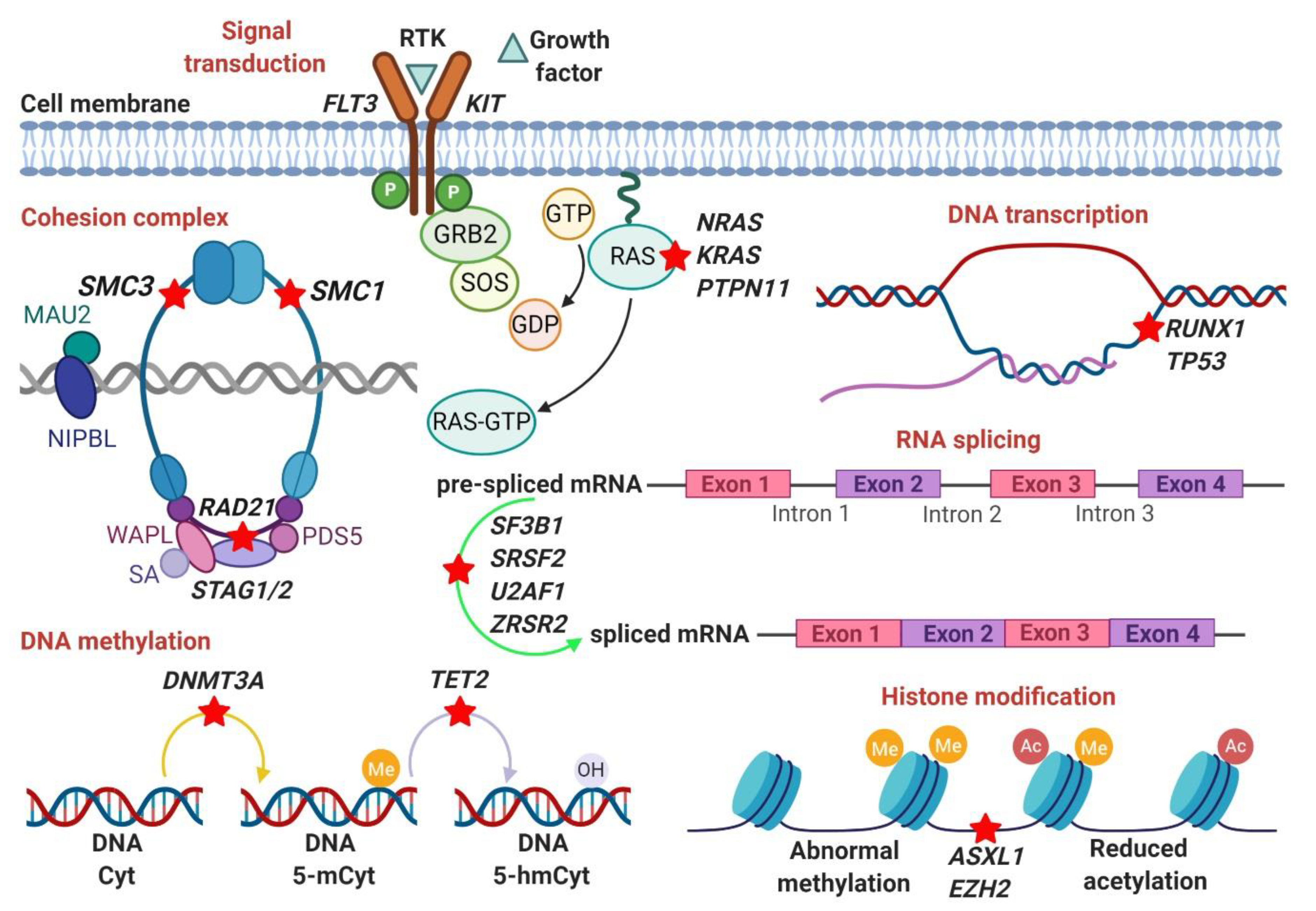
3. Somatic Mutations
3.1. RNA-Splicing Machinery (SF3B1, SRSF2, U2AF1, ZRSR2)
3.2. DNA Methylation (DNMT3A, TET2)
3.3. Histone Modification (ASXL1, EZH2, BCOR/BCORL1)
3.4. DNA Transcription (RUNX1, TP53)
3.5. Signal Transduction (KRAS, NRAS, PTPN11)
3.6. Cohesion Complex (SMC3, SMC1A, RAD21, STAG2)
4. Germline Mutations
4.1. RUNX1
4.2. GATA2
4.3. ETV6
4.4. TP53
4.5. DDX41
4.6. CEBPA
4.7. Genetic Predisposition
5. Clonal Hematopoiesis of Indeterminate Potential (CHIP)
6. Deregulation of Pathways in MDS
7. Prognostic Implications of Chromosomal and Mutational Alterations in MDS
8. Molecular Targeted Therapies
9. External Risk Factors of MDS: “Environmental Exposures”
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Tefferi, A.; Vardiman, J.W. Myelodysplastic Syndromes. N. Engl. J. Med. 2009, 361, 1872–1885. [Google Scholar] [CrossRef] [PubMed]
- Nimer, S.D. Myelodysplastic syndromes. Blood 2008, 111, 4841–4851. [Google Scholar] [CrossRef] [PubMed]
- Montalban-Bravo, G.; Garcia-Manero, G. Myelodysplastic syndromes: 2018 update on diagnosis, risk-stratification and management. Am. J. Hematol. 2018, 93, 129–147. [Google Scholar] [CrossRef] [PubMed]
- Sperling, A.S.; Gibson, C.J.; Ebert, B.L. The genetics of myelodysplastic syndrome: From clonal haematopoiesis to secondary leukaemia. Nat. Rev. Cancer 2017, 17, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Raaijmakers, M.H.G.P.; Mukherjee, S.; Guo, S.; Zhang, S.; Kobayashi, T.; Schoonmaker, J.A.; Ebert, B.L.; Al-Shahrour, F.; Hasserjian, R.P.; Scadden, E.O.; et al. Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature 2010, 464, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Medyouf, H. The microenvironment in human myeloid malignancies: Emerging concepts and therapeutic implications. Blood 2017, 129, 1617–1626. [Google Scholar] [CrossRef]
- Magee, J.A.; Piskounova, E.; Morrison, S.J. Cancer stem cells: Impact, heterogeneity, and uncertainty. Cancer Cell 2012, 21, 283–296. [Google Scholar] [CrossRef]
- Welch, J.S.; Ley, T.J.; Link, D.C.; Miller, C.A.; Larson, D.E.; Koboldt, D.C.; Wartman, L.D.; Lamprecht, T.L.; Liu, F.; Xia, J.; et al. The origin and evolution of mutations in acute myeloid leukemia. Cell 2012, 150, 264–278. [Google Scholar] [CrossRef]
- Nguyen, P.L.; Hasserjian, R.P. 18—Myelodysplastic Syndromes. In Hematopathology, 3rd ed.; Hsi, E.D., Ed.; Foundations in Diagnostic Pathology; Elsevier: Amsterdam, The Netherlands, 2018; pp. 539–563.e2. ISBN 978-0-323-47913-4. [Google Scholar]
- Bejar, R.; Levine, R.; Ebert, B.L. Unraveling the molecular pathophysiology of myelodysplastic syndromes. J. Clin. Oncol. 2011, 29, 504–515. [Google Scholar] [CrossRef]
- Ogawa, S. Genetics of MDS. Blood 2019, 133, 1049–1059. [Google Scholar] [CrossRef]
- Bannon, S.A.; DiNardo, C.D. Hereditary Predispositions to Myelodysplastic Syndrome. Int. J. Mol. Sci. 2016, 17, 838. [Google Scholar] [CrossRef] [PubMed]
- Bejar, R.; Greenberg, P.L. The Impact of Somatic and Germline Mutations in Myelodysplastic Syndromes and Related Disorders. J. Natl. Compr. Cancer Netw. 2017, 15, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Andersen, M.K.; Christiansen, D.H.; Pedersen-Bjergaard, J. Centromeric breakage and highly rearranged chromosome derivatives associated with mutations of TP53 are common in therapy-related MDS and AML after therapy with alkylating agents: An M-FISH study. Genes Chromosomes Cancer 2005, 42, 358–371. [Google Scholar] [CrossRef] [PubMed]
- Pedersen-Bjergaard, J. Radiotherapy- and chemotherapy-induced myelodysplasia and acute myeloid leukemia. A review. Leuk. Res. 1992, 16, 61–65. [Google Scholar] [CrossRef]
- Smith, S.M.; Le Beau, M.M.; Huo, D.; Karrison, T.; Sobecks, R.M.; Anastasi, J.; Vardiman, J.W.; Rowley, J.D.; Larson, R.A. Clinical-cytogenetic associations in 306 patients with therapy-related myelodysplasia and myeloid leukemia: The University of Chicago series. Blood 2003, 102, 43–52. [Google Scholar] [CrossRef]
- Jacobs, R.H.; Cornbleet, M.A.; Vardiman, J.W.; Larson, R.A.; Le Beau, M.M.; Rowley, J.D. Prognostic implications of morphology and karyotype in primary myelodysplastic syndromes. Blood 1986, 67, 1765–1772. [Google Scholar] [CrossRef]
- Zahid, M.F.; Malik, U.A.; Sohail, M.; Hassan, I.N.; Ali, S.; Shaukat, M.H.S. Cytogenetic Abnormalities in Myelodysplastic Syndromes: An Overview. Int. J. Hematol. Oncol. Stem Cell Res. 2017, 11, 231–239. [Google Scholar]
- Kelly, L.; Clark, J.; Gilliland, D.G. Comprehensive genotypic analysis of leukemia: Clinical and therapeutic implications. Curr. Opin. Oncol. 2002, 14, 10–18. [Google Scholar] [CrossRef]
- Herrera, L.A.; Prada, D.; Andonegui, M.A.; Dueñas-González, A. The Epigenetic Origin of Aneuploidy. Curr. Genom. 2008, 9, 43–50. [Google Scholar] [CrossRef]
- Gondek, L.P.; Tiu, R.; O’Keefe, C.L.; Sekeres, M.A.; Theil, K.S.; Maciejewski, J.P. Chromosomal lesions and uniparental disomy detected by SNP arrays in MDS, MDS/MPD, and MDS-derived AML. Blood 2008, 111, 1534–1542. [Google Scholar] [CrossRef]
- Haase, D.; Germing, U.; Schanz, J.; Pfeilstöcker, M.; Nösslinger, T.; Hildebrandt, B.; Kundgen, A.; Lübbert, M.; Kunzmann, R.; Giagounidis, A.A.N.; et al. New insights into the prognostic impact of the karyotype in MDS and correlation with subtypes: Evidence from a core dataset of 2124 patients. Blood 2007, 110, 4385–4395. [Google Scholar] [CrossRef] [PubMed]
- Anastasi, J.; Feng, J.; Le Beau, M.; Larson, R.; Rowley, J.; Vardiman, J. Cytogenetic clonality in myelodysplastic syndromes studied with fluorescence in situ hybridization: Lineage, response to growth factor therapy, and clone expansion. Blood 1993, 81, 1580–1585. [Google Scholar] [CrossRef] [PubMed]
- Komrokji, R.S.; Padron, E.; Ebert, B.L.; List, A.F. Deletion 5q MDS: Molecular and therapeutic implications. Best Pract. Res. Clin. Haematol. 2013, 26, 365–375. [Google Scholar] [CrossRef] [PubMed]
- List, A.; Dewald, G.; Bennett, J.; Giagounidis, A.; Raza, A.; Feldman, E.; Powell, B.; Greenberg, P.; Thomas, D.; Stone, R.; et al. Lenalidomide in the myelodysplastic syndrome with chromosome 5q deletion. N. Engl. J. Med. 2006, 355, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Ebert, B.L. Genetic deletions in AML and MDS. Best Pract. Res. Clin. Haematol. 2010, 23, 457–461. [Google Scholar] [CrossRef][Green Version]
- Jädersten, M.; Saft, L.; Smith, A.; Kulasekararaj, A.; Pomplun, S.; Göhring, G.; Hedlund, A.; Hast, R.; Schlegelberger, B.; Porwit, A.; et al. TP53 mutations in low-risk myelodysplastic syndromes with del(5q) predict disease progression. J. Clin. Oncol. 2011, 29, 1971–1979. [Google Scholar] [CrossRef]
- Ebert, B.L. Molecular dissection of the 5q deletion in myelodysplastic syndrome. Semin. Oncol. 2011, 38, 621–626. [Google Scholar] [CrossRef]
- Pellagatti, A.; Hellström-Lindberg, E.; Giagounidis, A.; Perry, J.; Malcovati, L.; Della Porta, M.G.; Jädersten, M.; Killick, S.; Fidler, C.; Cazzola, M.; et al. Haploinsufficiency of RPS14 in 5q- syndrome is associated with deregulation of ribosomal- and translation-related genes. Br. J. Haematol. 2008, 142, 57–64. [Google Scholar] [CrossRef]
- Ebert, B.L.; Pretz, J.; Bosco, J.; Chang, C.Y.; Tamayo, P.; Galili, N.; Raza, A.; Root, D.E.; Attar, E.; Ellis, S.R.; et al. Identification of RPS14 as a 5q- syndrome gene by RNA interference screen. Nature 2008, 451, 335–339. [Google Scholar] [CrossRef]
- Kumar, M.S.; Narla, A.; Nonami, A.; Mullally, A.; Dimitrova, N.; Ball, B.; McAuley, J.R.; Poveromo, L.; Kutok, J.L.; Galili, N.; et al. Coordinate loss of a microRNA and protein-coding gene cooperate in the pathogenesis of 5q− syndrome. Blood 2011, 118, 4666–4673. [Google Scholar] [CrossRef]
- Schneider, R.K.; Ademà, V.; Heckl, D.; Järås, M.; Mallo, M.; Lord, A.M.; Chu, L.P.; McConkey, M.E.; Kramann, R.; Mullally, A.; et al. Role of casein kinase 1A1 in the biology and targeted therapy of del(5q) MDS. Cancer Cell 2014, 26, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Stoddart, A.; Fernald, A.A.; Wang, J.; Davis, E.M.; Karrison, T.; Anastasi, J.; Le Beau, M.M. Haploinsufficiency of del(5q) genes, Egr1 and Apc, cooperate with Tp53 loss to induce acute myeloid leukemia in mice. Blood 2014, 123, 1069–1078. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.H.-P.; Kambal, A.; Krysiak, K.; Walshauser, M.A.; Raju, G.; Tibbitts, J.F.; Walter, M.J. Knockdown of Hspa9, a del(5q31.2) gene, results in a decrease in hematopoietic progenitors in mice. Blood 2011, 117, 1530–1539. [Google Scholar] [CrossRef] [PubMed]
- Sportoletti, P.; Grisendi, S.; Majid, S.M.; Cheng, K.; Clohessy, J.G.; Viale, A.; Teruya-Feldstein, J.; Pandolfi, P.P. Npm1 is a haploinsufficient suppressor of myeloid and lymphoid malignancies in the mouse. Blood 2008, 111, 3859–3862. [Google Scholar] [CrossRef] [PubMed]
- Hirai, H.; Kobayashi, Y.; Mano, H.; Hagiwara, K.; Maru, Y.; Omine, M.; Mizoguchi, H.; Nishida, J.; Takaku, F. A point mutation at codon 13 of the N-ras oncogene in myelodysplastic syndrome. Nature 1987, 327, 430–432. [Google Scholar] [CrossRef]
- Sashida, G.; Harada, H.; Matsui, H.; Oshima, M.; Yui, M.; Harada, Y.; Tanaka, S.; Mochizuki-Kashio, M.; Wang, C.; Saraya, A.; et al. Ezh2 loss promotes development of myelodysplastic syndrome but attenuates its predisposition to leukaemic transformation. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef]
- Wong, C.C.; Martincorena, I.; Rust, A.G.; Rashid, M.; Alifrangis, C.; Alexandrov, L.B.; Tiffen, J.C.; Kober, C.; Green, A.R.; Massie, C.E.; et al. Inactivating CUX1 mutations promote tumorigenesis. Nat. Genet. 2014, 46, 33–38. [Google Scholar] [CrossRef]
- Chen, C.; Liu, Y.; Rappaport, A.R.; Kitzing, T.; Schultz, N.; Zhao, Z.; Shroff, A.S.; Dickins, R.A.; Vakoc, C.R.; Bradner, J.E.; et al. MLL3 is a haploinsufficient 7q tumor suppressor in acute myeloid leukemia. Cancer Cell 2014, 25, 652–665. [Google Scholar] [CrossRef]
- Nagata, Y.; Narumi, S.; Guan, Y.; Przychodzen, B.P.; Hirsch, C.M.; Makishima, H.; Shima, H.; Aly, M.; Pastor, V.; Kuzmanovic, T.; et al. Germline loss-of-function SAMD9 and SAMD9L alterations in adult myelodysplastic syndromes. Blood 2018, 132, 2309–2313. [Google Scholar] [CrossRef]
- Inaba, T.; Honda, H.; Matsui, H. The enigma of monosomy 7. Blood 2018, 131, 2891–2898. [Google Scholar] [CrossRef]
- Christiansen, D.H.; Andersen, M.K.; Pedersen-Bjergaard, J. Mutations of AML1 are common in therapy-related myelodysplasia following therapy with alkylating agents and are significantly associated with deletion or loss of chromosome arm 7q and with subsequent leukemic transformation. Blood 2004, 104, 1474–1481. [Google Scholar] [CrossRef] [PubMed]
- Kuzmanovic, T.; Patel, B.J.; Sanikommu, S.R.; Nagata, Y.; Awada, H.; Kerr, C.M.; Przychodzen, B.P.; Jha, B.K.; Hiwase, D.; Singhal, D.; et al. Genomics of therapy-related myeloid neoplasms. Haematologica 2020, 105, e98–e101. [Google Scholar] [CrossRef] [PubMed]
- Kere, J.; Ruutu, T.; de la Chapelle, A. Monosomy 7 in granulocytes and monocytes in myelodysplastic syndrome. N. Engl. J. Med. 1987, 316, 499–503. [Google Scholar] [CrossRef] [PubMed]
- Schanz, J.; Steidl, C.; Fonatsch, C.; Pfeilstöcker, M.; Nösslinger, T.; Tuechler, H.; Valent, P.; Hildebrandt, B.; Giagounidis, A.; Aul, C.; et al. Coalesced multicentric analysis of 2351 patients with myelodysplastic syndromes indicates an underestimation of poor-risk cytogenetics of myelodysplastic syndromes in the international prognostic scoring system. J. Clin. Oncol. 2011, 29, 1963–1970. [Google Scholar] [CrossRef] [PubMed]
- Saumell, S.; Florensa, L.; Luño, E.; Sanzo, C.; Cañizo, C.; Hernández, J.M.; Cervera, J.; Gallart, M.A.; Carbonell, F.; Collado, R.; et al. Prognostic value of trisomy 8 as a single anomaly and the influence of additional cytogenetic aberrations in primary myelodysplastic syndromes. Br. J. Haematol. 2012, 159, 311–321. [Google Scholar] [CrossRef]
- Kakosaiou, K.; Panitsas, F.; Daraki, A.; Pagoni, M.; Apostolou, P.; Ioannidou, A.; Vlachadami, I.; Marinakis, T.; Giatra, C.; Vasilatou, D.; et al. ASXL1 mutations in AML are associated with specific clinical and cytogenetic characteristics. Leuk. Lymphoma 2018, 59, 2439–2446. [Google Scholar] [CrossRef]
- Jones, L.; Wei, G.; Sevcikova, S.; Phan, V.; Jain, S.; Shieh, A.; Wong, J.C.Y.; Li, M.; Dubansky, J.; Maunakea, M.L.; et al. Gain of MYC underlies recurrent trisomy of the MYC chromosome in acute promyelocytic leukemia. J. Exp. Med. 2010, 207, 2581–2594. [Google Scholar] [CrossRef]
- Toyonaga, T.; Nakase, H.; Matsuura, M.; Minami, N.; Yamada, S.; Honzawa, Y.; Hukata, N.; Yoshino, T.; Chiba, T.; Okazaki, K. Refractoriness of intestinal Behçet’s disease with myelodysplastic syndrome involving trisomy 8 to medical therapies—Our case experience and review of the literature. Digestion 2013, 88, 217–221. [Google Scholar] [CrossRef]
- Sloand, E.M.; Mainwaring, L.; Fuhrer, M.; Ramkissoon, S.; Risitano, A.M.; Keyvanafar, K.; Lu, J.; Basu, A.; Barrett, A.J.; Young, N.S. Preferential suppression of trisomy 8 compared with normal hematopoietic cell growth by autologous lymphocytes in patients with trisomy 8 myelodysplastic syndrome. Blood 2005, 106, 841–851. [Google Scholar] [CrossRef]
- Bacher, U.; Haferlach, T.; Schnittger, S.; Zenger, M.; Meggendorfer, M.; Jeromin, S.; Roller, A.; Grossmann, V.; Krauth, M.-T.; Alpermann, T.; et al. Investigation of 305 patients with myelodysplastic syndromes and 20q deletion for associated cytogenetic and molecular genetic lesions and their prognostic impact. Br. J. Haematol. 2014, 164, 822–833. [Google Scholar] [CrossRef]
- Braun, T.; de Botton, S.; Taksin, A.-L.; Park, S.; Beyne-Rauzy, O.; Coiteux, V.; Sapena, R.; Lazareth, A.; Leroux, G.; Guenda, K.; et al. Characteristics and outcome of myelodysplastic syndromes (MDS) with isolated 20q deletion: A report on 62 cases. Leuk. Res. 2011, 35, 863–867. [Google Scholar] [CrossRef] [PubMed]
- Shiseki, M.; Ishii, M.; Okada, M.; Ohwashi, M.; Wang, Y.-H.; Osanai, S.; Yoshinaga, K.; Mori, N.; Motoji, T.; Tanaka, J. Expression analysis of genes located within the common deleted region of del(20q) in patients with myelodysplastic syndromes. Leuk. Res. 2019, 84, 106175. [Google Scholar] [CrossRef] [PubMed]
- Stoner, S.A.; Yan, M.; Liu, K.T.H.; Arimoto, K.-I.; Shima, T.; Wang, H.-Y.; Johnson, D.T.; Bejar, R.; Jamieson, C.; Guan, K.-L.; et al. Hippo kinase loss contributes to del(20q) hematologic malignancies through chronic innate immune activation. Blood 2019, 134, 1730–1744. [Google Scholar] [CrossRef] [PubMed]
- Soenen, V.; Preudhomme, C.; Roumier, C.; Daudignon, A.; Laï, J.L.; Fenaux, P. 17p Deletion in acute myeloid leukemia and myelodysplastic syndrome. Analysis of breakpoints and deleted segments by fluorescence in situ. Blood 1998, 91, 1008–1015. [Google Scholar] [CrossRef]
- Sebaa, A.; Ades, L.; Baran-Marzack, F.; Mozziconacci, M.-J.; Penther, D.; Dobbelstein, S.; Stamatoullas, A.; Récher, C.; Prebet, T.; Moulessehoul, S.; et al. Incidence of 17p deletions and TP53 mutation in myelodysplastic syndrome and acute myeloid leukemia with 5q deletion. Genes Chromosomes Cancer 2012, 51, 1086–1092. [Google Scholar] [CrossRef]
- Wong, A.K.; Fang, B.; Zhang, L.; Guo, X.; Lee, S.; Schreck, R. Loss of the Y chromosome: An age-related or clonal phenomenon in acute myelogenous leukemia/myelodysplastic syndrome? Arch. Pathol. Lab. Med. 2008, 132, 1329–1332. [Google Scholar] [CrossRef]
- Abruzzese, E.; Rao, P.N.; Slatkoff, M.; Cruz, J.; Powell, B.L.; Jackle, B.; Pettenati, M.J. Monosomy X as a recurring sole cytogenetic abnormality associated with myelodysplastic diseases. Cancer Genet. Cytogenet. 1997, 93, 140–146. [Google Scholar] [CrossRef]
- Schanz, J.; Tüchler, H.; Solé, F.; Mallo, M.; Luño, E.; Cervera, J.; Granada, I.; Hildebrandt, B.; Slovak, M.L.; Ohyashiki, K.; et al. New comprehensive cytogenetic scoring system for primary myelodysplastic syndromes (MDS) and oligoblastic acute myeloid leukemia after MDS derived from an international database merge. J. Clin. Oncol. 2012, 30, 820–829. [Google Scholar] [CrossRef]
- Kawankar, N.; Vundinti, B.R. Cytogenetic abnormalities in myelodysplastic syndrome: An overview. Hematology 2011, 16, 131–138. [Google Scholar] [CrossRef]
- Kaneko, H.; Misawa, S.; Horiike, S.; Nakai, H.; Kashima, K. TP53 mutations emerge at early phase of myelodysplastic syndrome and are associated with complex chromosomal abnormalities. Blood 1995, 85, 2189–2193. [Google Scholar] [CrossRef]
- Haase, D.; Stevenson, K.E.; Neuberg, D.; Maciejewski, J.P.; Nazha, A.; Sekeres, M.A.; Ebert, B.L.; Garcia-Manero, G.; Haferlach, C.; Haferlach, T.; et al. TP53 mutation status divides myelodysplastic syndromes with complex karyotypes into distinct prognostic subgroups. Leukemia 2019, 33, 1747–1758. [Google Scholar] [CrossRef] [PubMed]
- Makishima, H.; Yoshizato, T.; Yoshida, K.; Sekeres, M.A.; Radivoyevitch, T.; Suzuki, H.; Przychodzen, B.; Nagata, Y.; Meggendorfer, M.; Sanada, M.; et al. Dynamics of clonal evolution in myelodysplastic syndromes. Nat. Genet. 2017, 49, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Haferlach, T.; Nagata, Y.; Grossmann, V.; Okuno, Y.; Bacher, U.; Nagae, G.; Schnittger, S.; Sanada, M.; Kon, A.; Alpermann, T.; et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 2014, 28, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Nagata, Y.; Makishima, H.; Kerr, C.M.; Przychodzen, B.P.; Aly, M.; Goyal, A.; Awada, H.; Asad, M.F.; Kuzmanovic, T.; Suzuki, H.; et al. Invariant patterns of clonal succession determine specific clinical features of myelodysplastic syndromes. Nat. Commun. 2019, 10, 5386. [Google Scholar] [CrossRef] [PubMed]
- Malcovati, L.; Karimi, M.; Papaemmanuil, E.; Ambaglio, I.; Jädersten, M.; Jansson, M.; Elena, C.; Gallì, A.; Walldin, G.; Della Porta, M.G.; et al. SF3B1 mutation identifies a distinct subset of myelodysplastic syndrome with ring sideroblasts. Blood 2015, 126, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Makishima, H.; Visconte, V.; Sakaguchi, H.; Jankowska, A.M.; Abu Kar, S.; Jerez, A.; Przychodzen, B.; Bupathi, M.; Guinta, K.; Afable, M.G.; et al. Mutations in the spliceosome machinery, a novel and ubiquitous pathway in leukemogenesis. Blood 2012, 119, 3203–3210. [Google Scholar] [CrossRef] [PubMed]
- Thol, F.; Kade, S.; Schlarmann, C.; Löffeld, P.; Morgan, M.; Krauter, J.; Wlodarski, M.W.; Kölking, B.; Wichmann, M.; Görlich, K.; et al. Frequency and prognostic impact of mutations in SRSF2, U2AF1, and ZRSR2 in patients with myelodysplastic syndromes. Blood 2012, 119, 3578–3584. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Sanada, M.; Shiraishi, Y.; Nowak, D.; Nagata, Y.; Yamamoto, R.; Sato, Y.; Sato-Otsubo, A.; Kon, A.; Nagasaki, M.; et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 2011, 478, 64–69. [Google Scholar] [CrossRef]
- Graubert, T.A.; Shen, D.; Ding, L.; Okeyo-Owuor, T.; Lunn, C.L.; Shao, J.; Krysiak, K.; Harris, C.C.; Koboldt, D.C.; Larson, D.E.; et al. Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Nat. Genet. 2011, 44, 53–57. [Google Scholar] [CrossRef]
- Delhommeau, F.; Dupont, S.; Della Valle, V.; James, C.; Trannoy, S.; Massé, A.; Kosmider, O.; Le Couedic, J.-P.; Robert, F.; Alberdi, A.; et al. Mutation in TET2 in myeloid cancers. N. Engl. J. Med. 2009, 360, 2289–2301. [Google Scholar] [CrossRef]
- Ko, M.; Huang, Y.; Jankowska, A.M.; Pape, U.J.; Tahiliani, M.; Bandukwala, H.S.; An, J.; Lamperti, E.D.; Koh, K.P.; Ganetzky, R.; et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature 2010, 468, 839–843. [Google Scholar] [CrossRef] [PubMed]
- Awada, H.; Nagata, Y.; Goyal, A.; Asad, M.F.; Patel, B.; Hirsch, C.M.; Kuzmanovic, T.; Guan, Y.; Przychodzen, B.P.; Aly, M.; et al. Invariant phenotype and molecular association of biallelic TET2 mutant myeloid neoplasia. Blood Adv. 2019, 3, 339–349. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, C.M.; Nazha, A.; Kneen, K.; Abazeed, M.E.; Meggendorfer, M.; Przychodzen, B.P.; Nadarajah, N.; Adema, V.; Nagata, Y.; Goyal, A.; et al. Consequences of mutant TET2 on clonality and subclonal hierarchy. Leukemia 2018, 32, 1751–1761. [Google Scholar] [CrossRef] [PubMed]
- Walter, M.J.; Ding, L.; Shen, D.; Shao, J.; Grillot, M.; McLellan, M.; Fulton, R.; Schmidt, H.; Kalicki-Veizer, J.; O’Laughlin, M.; et al. Recurrent DNMT3A mutations in patients with myelodysplastic syndromes. Leukemia 2011, 25, 1153–1158. [Google Scholar] [CrossRef]
- Hou, H.-A.; Kuo, Y.-Y.; Liu, C.-Y.; Chou, W.-C.; Lee, M.C.; Chen, C.-Y.; Lin, L.-I.; Tseng, M.-H.; Huang, C.-F.; Chiang, Y.-C.; et al. DNMT3A mutations in acute myeloid leukemia: Stability during disease evolution and clinical implications. Blood 2012, 119, 559–568. [Google Scholar] [CrossRef]
- Thol, F.; Friesen, I.; Damm, F.; Yun, H.; Weissinger, E.M.; Krauter, J.; Wagner, K.; Chaturvedi, A.; Sharma, A.; Wichmann, M.; et al. Prognostic significance of ASXL1 mutations in patients with myelodysplastic syndromes. J. Clin. Oncol. 2011, 29, 2499–2506. [Google Scholar] [CrossRef]
- Boultwood, J.; Perry, J.; Pellagatti, A.; Fernandez-Mercado, M.; Fernandez-Santamaria, C.; Calasanz, M.J.; Larrayoz, M.J.; Garcia-Delgado, M.; Giagounidis, A.; Malcovati, L.; et al. Frequent mutation of the polycomb-associated gene ASXL1 in the myelodysplastic syndromes and in acute myeloid leukemia. Leukemia 2010, 24, 1062–1065. [Google Scholar] [CrossRef]
- Ernst, T.; Chase, A.J.; Score, J.; Hidalgo-Curtis, C.E.; Bryant, C.; Jones, A.V.; Waghorn, K.; Zoi, K.; Ross, F.M.; Reiter, A.; et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat. Genet. 2010, 42, 722–726. [Google Scholar] [CrossRef]
- Nikoloski, G.; Langemeijer, S.M.C.; Kuiper, R.P.; Knops, R.; Massop, M.; Tönnissen, E.R.L.T.M.; van der Heijden, A.; Scheele, T.N.; Vandenberghe, P.; de Witte, T.; et al. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nat. Genet. 2010, 42, 665–667. [Google Scholar] [CrossRef]
- Abuhadra, N.; Mukherjee, S.; Al-Issa, K.; Adema, V.; Hirsch, C.M.; Advani, A.; Przychodzen, B.; Makhoul, A.; Awada, H.; Maciejewski, J.P.; et al. BCOR and BCORL1 mutations in myelodysplastic syndromes (MDS): Clonal architecture and impact on outcomes. Leuk. Lymphoma 2019, 60, 1587–1590. [Google Scholar] [CrossRef]
- Bejar, R.; Stevenson, K.E.; Caughey, B.; Lindsley, R.C.; Mar, B.G.; Stojanov, P.; Getz, G.; Steensma, D.P.; Ritz, J.; Soiffer, R.; et al. Somatic mutations predict poor outcome in patients with myelodysplastic syndrome after hematopoietic stem-cell transplantation. J. Clin. Oncol. 2014, 32, 2691–2698. [Google Scholar] [CrossRef] [PubMed]
- Bejar, R.; Stevenson, K.E.; Caughey, B.A.; Abdel-Wahab, O.; Steensma, D.P.; Galili, N.; Raza, A.; Kantarjian, H.; Levine, R.L.; Neuberg, D.; et al. Validation of a prognostic model and the impact of mutations in patients with lower-risk myelodysplastic syndromes. J. Clin. Oncol. 2012, 30, 3376–3382. [Google Scholar] [CrossRef]
- Sallman, D.A.; Komrokji, R.; Vaupel, C.; Cluzeau, T.; Geyer, S.M.; McGraw, K.L.; Al Ali, N.H.; Lancet, J.; McGinniss, M.J.; Nahas, S.; et al. Impact of TP53 mutation variant allele frequency on phenotype and outcomes in myelodysplastic syndromes. Leukemia 2016, 30, 666–673. [Google Scholar] [CrossRef] [PubMed]
- Bejar, R.; Stevenson, K.; Abdel-Wahab, O.; Galili, N.; Nilsson, B.; Garcia-Manero, G.; Kantarjian, H.; Raza, A.; Levine, R.L.; Neuberg, D.; et al. Clinical effect of point mutations in myelodysplastic syndromes. N. Engl. J. Med. 2011, 364, 2496–2506. [Google Scholar] [CrossRef] [PubMed]
- Tartaglia, M.; Niemeyer, C.M.; Fragale, A.; Song, X.; Buechner, J.; Jung, A.; Hählen, K.; Hasle, H.; Licht, J.D.; Gelb, B.D. Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat. Genet. 2003, 34, 148–150. [Google Scholar] [CrossRef] [PubMed]
- Hugues, L.; Cavé, H.; Philippe, N.; Pereira, S.; Fenaux, P.; Preudhomme, C. Mutations of PTPN11 are rare in adult myeloid malignancies. Haematologica 2005, 90, 853–854. [Google Scholar] [PubMed]
- Kon, A.; Shih, L.-Y.; Minamino, M.; Sanada, M.; Shiraishi, Y.; Nagata, Y.; Yoshida, K.; Okuno, Y.; Bando, M.; Nakato, R.; et al. Recurrent mutations in multiple components of the cohesin complex in myeloid neoplasms. Nat. Genet. 2013, 45, 1232–1237. [Google Scholar] [CrossRef]
- Thota, S.; Viny, A.D.; Makishima, H.; Spitzer, B.; Radivoyevitch, T.; Przychodzen, B.; Sekeres, M.A.; Levine, R.L.; Maciejewski, J.P. Genetic alterations of the cohesin complex genes in myeloid malignancies. Blood 2014, 124, 1790–1798. [Google Scholar] [CrossRef]
- Yip, B.H.; Steeples, V.; Repapi, E.; Armstrong, R.N.; Llorian, M.; Roy, S.; Shaw, J.; Dolatshad, H.; Taylor, S.; Verma, A.; et al. The U2AF1S34F mutation induces lineage-specific splicing alterations in myelodysplastic syndromes. J. Clin. Investig. 2017, 127, 2206–2221. [Google Scholar] [CrossRef]
- Liang, Y.; Tebaldi, T.; Rejeski, K.; Joshi, P.; Stefani, G.; Taylor, A.; Song, Y.; Vasic, R.; Maziarz, J.; Balasubramanian, K.; et al. SRSF2 mutations drive oncogenesis by activating a global program of aberrant alternative splicing in hematopoietic cells. Leukemia 2018, 32, 2659–2671. [Google Scholar] [CrossRef]
- Ley, T.J.; Ding, L.; Walter, M.J.; McLellan, M.D.; Lamprecht, T.; Larson, D.E.; Kandoth, C.; Payton, J.E.; Baty, J.; Welch, J.; et al. DNMT3A mutations in acute myeloid leukemia. N. Engl. J. Med. 2010, 363, 2424–2433. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.E.; Mohamedali, A.M.; Kulasekararaj, A.; Lim, Z.; Gäken, J.; Lea, N.C.; Przychodzen, B.; Mian, S.A.; Nasser, E.E.; Shooter, C.; et al. Next-generation sequencing of the TET2 gene in 355 MDS and CMML patients reveals low-abundance mutant clones with early origins, but indicates no definite prognostic value. Blood 2010, 116, 3923–3932. [Google Scholar] [CrossRef] [PubMed]
- Kosmider, O.; Gelsi-Boyer, V.; Cheok, M.; Grabar, S.; Della-Valle, V.; Picard, F.; Viguié, F.; Quesnel, B.; Beyne-Rauzy, O.; Solary, E.; et al. TET2 mutation is an independent favorable prognostic factor in myelodysplastic syndromes (MDSs). Blood 2009, 114, 3285–3291. [Google Scholar] [CrossRef] [PubMed]
- Bejar, R. Clinical and genetic predictors of prognosis in myelodysplastic syndromes. Haematologica 2014, 99, 956–964. [Google Scholar] [CrossRef] [PubMed]
- Bejar, R.; Lord, A.; Stevenson, K.; Bar-Natan, M.; Pérez-Ladaga, A.; Zaneveld, J.; Wang, H.; Caughey, B.; Stojanov, P.; Getz, G.; et al. TET2 mutations predict response to hypomethylating agents in myelodysplastic syndrome patients. Blood 2014, 124, 2705–2712. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Liu, L.; Chang, C.-K.; He, Q.; Wu, L.-Y.; Zhang, Z.; Shi, W.-H.; Guo, J.; Zhu, Y.; Zhao, Y.-S.; et al. Genomic loss of EZH2 leads to epigenetic modifications and overexpression of the HOX gene clusters in myelodysplastic syndrome. Oncotarget 2016, 7, 8119–8130. [Google Scholar] [CrossRef][Green Version]
- Babushok, D.V.; Olson, T.S.; Bessler, M. Somatic Mutations and Clonal Hematopoiesis in Aplastic Anemia. N. Engl. J. Med. 2015, 373, 1673. [Google Scholar] [CrossRef]
- Durrani, J.; Awada, H.; Kishtagari, A.; Visconte, V.; Kerr, C.; Adema, V.; Nagata, Y.; Kuzmanovic, T.; Hong, S.; Patel, B.; et al. Large granular lymphocytic leukemia coexists with myeloid clones and myelodysplastic syndrome. Leukemia 2020, 34, 957–962. [Google Scholar] [CrossRef]
- Awada, H.; Rahman, S.; Durrani, J.; Asad, M.F.; Kerr, C.M.; Adema, V.; Kishtagari, A.; Graham, A.; Snider, C.A.; Kongkiatkamon, S.; et al. Leukemia evolving from paroxysmal nocturnal hemoglobinuria. Leukemia 2020, 34, 327–330. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Gerstung, M.; Malcovati, L.; Tauro, S.; Gundem, G.; Van Loo, P.; Yoon, C.J.; Ellis, P.; Wedge, D.C.; Pellagatti, A.; et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 2013, 122, 3616–3627. [Google Scholar] [CrossRef]
- Damm, F.; Chesnais, V.; Nagata, Y.; Yoshida, K.; Scourzic, L.; Okuno, Y.; Itzykson, R.; Sanada, M.; Shiraishi, Y.; Gelsi-Boyer, V.; et al. BCOR and BCORL1 mutations in myelodysplastic syndromes and related disorders. Blood 2013, 122, 3169–3177. [Google Scholar] [CrossRef] [PubMed]
- Watanabe-Okochi, N.; Kitaura, J.; Ono, R.; Harada, H.; Harada, Y.; Komeno, Y.; Nakajima, H.; Nosaka, T.; Inaba, T.; Kitamura, T. AML1 mutations induced MDS and MDS/AML in a mouse BMT model. Blood 2008, 111, 4297–4308. [Google Scholar] [CrossRef] [PubMed]
- Harada, Y.; Inoue, D.; Ding, Y.; Imagawa, J.; Doki, N.; Matsui, H.; Yahata, T.; Matsushita, H.; Ando, K.; Sashida, G.; et al. RUNX1/AML1 mutant collaborates with BMI1 overexpression in the development of human and murine myelodysplastic syndromes. Blood 2013, 121, 3434–3446. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bernard, E.; Nannya, Y.; Hasserjian, R.P.; Devlin, S.M.; Tuechler, H.; Medina-Martinez, J.S.; Yoshizato, T.; Shiozawa, Y.; Saiki, R.; Malcovati, L.; et al. Implications of TP53 allelic state for genome stability, clinical presentation and outcomes in myelodysplastic syndromes. Nat. Med. 2020, 26, 1549–1556. [Google Scholar] [CrossRef]
- Loh, M.L.; Martinelli, S.; Cordeddu, V.; Reynolds, M.G.; Vattikuti, S.; Lee, C.M.; Wulfert, M.; Germing, U.; Haas, P.; Niemeyer, C.; et al. Acquired PTPN11 mutations occur rarely in adult patients with myelodysplastic syndromes and chronic myelomonocytic leukemia. Leuk. Res. 2005, 29, 459–462. [Google Scholar] [CrossRef]
- Chen, C.-Y.; Lin, L.-I.; Tang, J.-L.; Tsay, W.; Chang, H.-H.; Yeh, Y.-C.; Huang, C.-F.; Chiou, R.-J.; Yao, M.; Ko, B.-S.; et al. Acquisition of JAK2, PTPN11, and RAS mutations during disease progression in primary myelodysplastic syndrome. Leukemia 2006, 20, 1155–1158. [Google Scholar] [CrossRef]
- Viny, A.D.; Levine, R.L. Cohesin mutations in myeloid malignancies made simple. Curr. Opin. Hematol. 2018, 25, 61–66. [Google Scholar] [CrossRef]
- Rio-Machin, A.; Vulliamy, T.; Hug, N.; Walne, A.; Tawana, K.; Cardoso, S.; Ellison, A.; Pontikos, N.; Wang, J.; Tummala, H.; et al. The complex genetic landscape of familial MDS and AML reveals pathogenic germline variants. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef]
- Harada, H.; Harada, Y.; Niimi, H.; Kyo, T.; Kimura, A.; Inaba, T. High incidence of somatic mutations in the AML1/RUNX1 gene in myelodysplastic syndrome and low blast percentage myeloid leukemia with myelodysplasia. Blood 2004, 103, 2316–2324. [Google Scholar] [CrossRef]
- Ito, Y.; Bae, S.-C.; Chuang, L.S.H. The RUNX family: Developmental regulators in cancer. Nat. Rev. Cancer 2015, 15, 81–95. [Google Scholar] [CrossRef]
- Hyde, R.K.; Liu, P.P. GATA2 mutations lead to MDS and AML. Nat. Genet. 2011, 43, 926–927. [Google Scholar] [CrossRef] [PubMed]
- Micol, J.-B.; Abdel-Wahab, O. Collaborating constitutive and somatic genetic events in myeloid malignancies: ASXL1 mutations in patients with germline GATA2 mutations. Haematologica 2014, 99, 201–203. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.Y.; Churpek, J.E.; Keel, S.B.; Walsh, T.; Lee, M.K.; Loeb, K.R.; Gulsuner, S.; Pritchard, C.C.; Sanchez-Bonilla, M.; Delrow, J.J.; et al. Germline ETV6 mutations in familial thrombocytopenia and hematologic malignancy. Nat. Genet. 2015, 47, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Feurstein, S.; Godley, L.A. Germline ETV6 mutations and predisposition to hematological malignancies. Int. J. Hematol. 2017, 106, 189–195. [Google Scholar] [CrossRef]
- Sébert, M.; Passet, M.; Raimbault, A.; Rahmé, R.; Raffoux, E.; Sicre de Fontbrune, F.; Cerrano, M.; Quentin, S.; Vasquez, N.; Da Costa, M.; et al. Germline DDX41 mutations define a significant entity within adult MDS/AML patients. Blood 2019, 134, 1441–1444. [Google Scholar] [CrossRef]
- Polprasert, C.; Schulze, I.; Sekeres, M.A.; Makishima, H.; Przychodzen, B.; Hosono, N.; Singh, J.; Padgett, R.A.; Gu, X.; Phillips, J.G.; et al. Inherited and Somatic Defects in DDX41 in Myeloid Neoplasms. Cancer Cell 2015, 27, 658–670. [Google Scholar] [CrossRef]
- Douglas Tremblay, M.D. Implications of Mutation Profiling in Myeloid Malignancies—PART 1: Myelodysplastic Syndromes and Acute Myeloid Leukemia. Available online: https://www.cancernetwork.com/article/implications-mutation-profiling-myeloid-malignancies (accessed on 26 March 2020).
- Wen, X.; Hu, J.; Yang, J.; Qian, W.; Yao, D.; Deng, Z.; Zhang, Y.; Zhu, X.; Guo, H.; Lin, J.; et al. CEBPA methylation and mutation in myelodysplastic syndrome. Med. Oncol. 2015, 32, 192. [Google Scholar] [CrossRef]
- Kennedy, A.L.; Shimamura, A. Genetic predisposition to MDS: Clinical features and clonal evolution. Blood 2019, 133, 1071–1085. [Google Scholar] [CrossRef]
- Steensma, D.P. Clinical consequences of clonal hematopoiesis of indeterminate potential. Blood Adv. 2018, 2, 3404–3410. [Google Scholar] [CrossRef]
- Malcovati, L.; Gallì, A.; Travaglino, E.; Ambaglio, I.; Rizzo, E.; Molteni, E.; Elena, C.; Ferretti, V.V.; Catricalà, S.; Bono, E.; et al. Clinical significance of somatic mutation in unexplained blood cytopenia. Blood 2017, 129, 3371–3378. [Google Scholar] [CrossRef]
- Steensma, D.P.; Bejar, R.; Jaiswal, S.; Lindsley, R.C.; Sekeres, M.A.; Hasserjian, R.P.; Ebert, B.L. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015, 126, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Valent, P.; Orazi, A.; Steensma, D.P.; Ebert, B.L.; Haase, D.; Malcovati, L.; van de Loosdrecht, A.A.; Haferlach, T.; Westers, T.M.; Wells, D.A.; et al. Proposed minimal diagnostic criteria for myelodysplastic syndromes (MDS) and potential pre-MDS conditions. Oncotarget 2017, 8, 73483–73500. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Lu, C.; Wang, J.; McLellan, M.D.; Johnson, K.J.; Wendl, M.C.; McMichael, J.F.; Schmidt, H.K.; Yellapantula, V.; Miller, C.A.; et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med. 2014, 20, 1472–1478. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef] [PubMed]
- Kwok, B.; Hall, J.M.; Witte, J.S.; Xu, Y.; Reddy, P.; Lin, K.; Flamholz, R.; Dabbas, B.; Yung, A.; Al-Hafidh, J.; et al. MDS-associated somatic mutations and clonal hematopoiesis are common in idiopathic cytopenias of undetermined significance. Blood 2015, 126, 2355–2361. [Google Scholar] [CrossRef]
- Genovese, G.; Kähler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef]
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; Gibson, C.J.; Bick, A.G.; Shvartz, E.; McConkey, M.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 111–121. [Google Scholar] [CrossRef]
- Frick, M.; Chan, W.; Arends, C.M.; Hablesreiter, R.; Halik, A.; Heuser, M.; Michonneau, D.; Blau, O.; Hoyer, K.; Christen, F.; et al. Role of Donor Clonal Hematopoiesis in Allogeneic Hematopoietic Stem-Cell Transplantation. J. Clin. Oncol. 2019, 37, 375–385. [Google Scholar] [CrossRef]
- Gibson, C.J.; Lindsley, R.C.; Tchekmedyian, V.; Mar, B.G.; Shi, J.; Jaiswal, S.; Bosworth, A.; Francisco, L.; He, J.; Bansal, A.; et al. Clonal Hematopoiesis Associated With Adverse Outcomes After Autologous Stem-Cell Transplantation for Lymphoma. J. Clin. Oncol. 2017, 35, 1598–1605. [Google Scholar] [CrossRef]
- Pellagatti, A.; Cazzola, M.; Giagounidis, A.; Perry, J.; Malcovati, L.; Della Porta, M.G.; Jädersten, M.; Killick, S.; Verma, A.; Norbury, C.J.; et al. Deregulated gene expression pathways in myelodysplastic syndrome hematopoietic stem cells. Leukemia 2010, 24, 756–764. [Google Scholar] [CrossRef]
- Shiozawa, Y.; Malcovati, L.; Gallì, A.; Pellagatti, A.; Karimi, M.; Sato-Otsubo, A.; Sato, Y.; Suzuki, H.; Yoshizato, T.; Yoshida, K.; et al. Gene expression and risk of leukemic transformation in myelodysplasia. Blood 2017, 130, 2642–2653. [Google Scholar] [CrossRef] [PubMed]
- Visconte, V.; Avishai, N.; Mahfouz, R.; Tabarroki, A.; Cowen, J.; Sharghi-Moshtaghin, R.; Hitomi, M.; Rogers, H.J.; Hasrouni, E.; Phillips, J.; et al. Distinct iron architecture in SF3B1-mutant myelodysplastic syndrome patients is linked to an SLC25A37 splice variant with a retained intron. Leukemia 2015, 29, 188–195. [Google Scholar] [CrossRef] [PubMed]
- del Rey, M.; Benito, R.; Fontanillo, C.; Campos-Laborie, F.J.; Janusz, K.; Velasco-Hernández, T.; Abáigar, M.; Hernández, M.; Cuello, R.; Borrego, D.; et al. Deregulation of Genes Related to Iron and Mitochondrial Metabolism in Refractory Anemia with Ring Sideroblasts. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Dolatshad, H.; Pellagatti, A.; Liberante, F.G.; Llorian, M.; Repapi, E.; Steeples, V.; Roy, S.; Scifo, L.; Armstrong, R.N.; Shaw, J.; et al. Cryptic splicing events in the iron transporter ABCB7 and other key target genes in SF3B1-mutant myelodysplastic syndromes. Leukemia 2016, 30, 2322–2331. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Ahmed, D.; Dolatshad, H.; Tatwavedi, D.; Schulze, U.; Sanchi, A.; Ryley, S.; Dhir, A.; Carpenter, L.; Watt, S.M.; et al. SF3B1 mutations induce R-loop accumulation and DNA damage in MDS and leukemia cells with therapeutic implications. Leukemia 2020, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Pellagatti, A.; Armstrong, R.N.; Steeples, V.; Sharma, E.; Repapi, E.; Singh, S.; Sanchi, A.; Radujkovic, A.; Horn, P.; Dolatshad, H.; et al. Impact of spliceosome mutations on RNA splicing in myelodysplasia: Dysregulated genes/pathways and clinical associations. Blood 2018, 132, 1225–1240. [Google Scholar] [CrossRef]
- Follo, M.Y.; Pellagatti, A.; Ratti, S.; Ramazzotti, G.; Faenza, I.; Fiume, R.; Mongiorgi, S.; Suh, P.-G.; McCubrey, J.A.; Manzoli, L.; et al. Recent advances in MDS mutation landscape: Splicing and signalling. Adv. Biol. Regul. 2020, 75, 100673. [Google Scholar] [CrossRef]
- Follo, M.Y.; Pellagatti, A.; Armstrong, R.N.; Ratti, S.; Mongiorgi, S.; De Fanti, S.; Bochicchio, M.T.; Russo, D.; Gobbi, M.; Miglino, M.; et al. Response of high-risk MDS to azacitidine and lenalidomide is impacted by baseline and acquired mutations in a cluster of three inositide-specific genes. Leukemia 2019, 33, 2276–2290. [Google Scholar] [CrossRef]
- Pellagatti, A.; Boultwood, J. Splicing factor mutant myelodysplastic syndromes: Recent advances. Adv. Biol. Regul. 2020, 75, 100655. [Google Scholar] [CrossRef]
- Wong, J.J.-L.; Lau, K.A.; Pinello, N.; Rasko, J.E.J. Epigenetic modifications of splicing factor genes in myelodysplastic syndromes and acute myeloid leukemia. Cancer Sci. 2014, 105, 1457–1463. [Google Scholar] [CrossRef]
- Fong, J.Y.; Pignata, L.; Goy, P.-A.; Kawabata, K.C.; Lee, S.C.-W.; Koh, C.M.; Musiani, D.; Massignani, E.; Kotini, A.G.; Penson, A.; et al. Therapeutic Targeting of RNA Splicing Catalysis through Inhibition of Protein Arginine Methylation. Cancer Cell 2019, 36, 194–209.e9. [Google Scholar] [CrossRef] [PubMed]
- Grignano, E.; Jachiet, V.; Fenaux, P.; Ades, L.; Fain, O.; Mekinian, A. Autoimmune manifestations associated with myelodysplastic syndromes. Ann. Hematol. 2018, 97, 2015–2023. [Google Scholar] [CrossRef] [PubMed]
- Enright, H.; Miller, W. Autoimmune phenomena in patients with myelodysplastic syndromes. Leuk. Lymphoma 1997, 24, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Enright, H.; Jacob, H.S.; Vercellotti, G.; Howe, R.; Belzer, M.; Miller, W. Paraneoplastic autoimmune phenomena in patients with myelodysplastic syndromes: Response to immunosuppressive therapy. Br. J. Haematol. 1995, 91, 403–408. [Google Scholar] [CrossRef]
- Rochet, N.M.; Chavan, R.N.; Cappel, M.A.; Wada, D.A.; Gibson, L.E. Sweet syndrome: Clinical presentation, associations, and response to treatment in 77 patients. J. Am. Acad. Dermatol. 2013, 69, 557–564. [Google Scholar] [CrossRef]
- Ten Oever, J.; Kuijper, P.H.M.; Kuijpers, A.L.A.; Dercksen, M.W.; Vreugdenhil, G. Complete remission of MDS RAEB following immunosuppressive treatment in a patient with Sweet’s syndrome. Neth. J. Med. 2009, 67, 347–350. [Google Scholar]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Greenberg, P.L.; Tuechler, H.; Schanz, J.; Sanz, G.; Garcia-Manero, G.; Solé, F.; Bennett, J.M.; Bowen, D.; Fenaux, P.; Dreyfus, F.; et al. Revised International Prognostic Scoring System for Myelodysplastic Syndromes. Blood 2012, 120, 2454–2465. [Google Scholar] [CrossRef]
- Steensma, D.P.; Wermke, M.; Klimek, V.M.; Greenberg, P.L.; Font, P.; Komrokji, R.S.; Yang, J.; Brunner, A.M.; Carraway, H.E.; Ades, L.; et al. Results of a Clinical Trial of H3B-8800, a Splicing Modulator, in Patients with Myelodysplastic Syndromes (MDS), Acute Myeloid Leukemia (AML) or Chronic Myelomonocytic Leukemia (CMML). Blood 2019, 134, 673. [Google Scholar] [CrossRef]
- Fenaux, P.; Platzbecker, U.; Mufti, G.J.; Garcia-Manero, G.; Buckstein, R.; Santini, V.; Díez-Campelo, M.; Finelli, C.; Cazzola, M.; Ilhan, O.; et al. Luspatercept in Patients with Lower-Risk Myelodysplastic Syndromes. N. Engl. J. Med. 2020, 382, 140–151. [Google Scholar] [CrossRef]
- Han, T.; Goralski, M.; Gaskill, N.; Capota, E.; Kim, J.; Ting, T.C.; Xie, Y.; Williams, N.S.; Nijhawan, D. Anticancer sulfonamides target splicing by inducing RBM39 degradation via recruitment to DCAF15. Science 2017, 356. [Google Scholar] [CrossRef] [PubMed]
- Cimmino, L.; Dolgalev, I.; Wang, Y.; Yoshimi, A.; Martin, G.H.; Wang, J.; Ng, V.; Xia, B.; Witkowski, M.T.; Mitchell-Flack, M.; et al. Restoration of TET2 Function Blocks Aberrant Self-Renewal and Leukemia Progression. Cell 2017, 170, 1079–1095.e20. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.S.; Fathi, A.T. Targeting isocitrate dehydrogenase mutations (IDH) in AML: Wielding the double-edged sword of differentiation. Curr. Cancer Drug Targets 2020. [Google Scholar] [CrossRef] [PubMed]
- Kaur, R.P.; Vasudeva, K.; Kumar, R.; Munshi, A. Role of p53 Gene in Breast Cancer: Focus on Mutation Spectrum and Therapeutic Strategies. Curr. Pharm. Des. 2018, 24, 3566–3575. [Google Scholar] [CrossRef]
- Maslah, N.; Salomao, N.; Drevon, L.; Verger, E.; Partouche, N.; Ly, P.; Aubin, P.; Naoui, N.; Schlageter, M.-H.; Bally, C.; et al. Synergistic effects of PRIMA-1Met (APR-246) and Azacitidine in TP53-mutated myelodysplastic syndromes and acute myeloid leukemia. Haematologica 2019. [Google Scholar] [CrossRef]
- Kröger, N.; Brand, R.; van Biezen, A.; Zander, A.; Dierlamm, J.; Niederwieser, D.; Devergie, A.; Ruutu, T.; Cornish, J.; Ljungman, P.; et al. Risk factors for therapy-related myelodysplastic syndrome and acute myeloid leukemia treated with allogeneic stem cell transplantation. Haematologica 2009, 94, 542–549. [Google Scholar] [CrossRef]
- Yeung, C.C.S.; Gerds, A.T.; Fang, M.; Scott, B.L.; Flowers, M.E.D.; Gooley, T.; Deeg, H.J. Relapse after Allogeneic Hematopoietic Cell Transplantation for Myelodysplastic Syndromes: Analysis of Late Relapse Using Comparative Karyotype and Chromosome Genome Array Testing. Biol. Blood Marrow Transpl. 2015, 21, 1565–1575. [Google Scholar] [CrossRef]
- Dietz, A.C.; DeFor, T.E.; Brunstein, C.G.; Wagner, J.E. Donor-derived myelodysplastic syndrome and acute leukaemia after allogeneic haematopoietic stem cell transplantation: Incidence, natural history and treatment response. Br. J. Haematol. 2014, 166, 209–212. [Google Scholar] [CrossRef]
- Komrokji, R.; Ifthikharuddin, J.J.; Felgar, R.E.; Abboud, C.N.; Wedow, L.A.; Connaughton, A.; Bennett, J.M. Donor cell myelodysplastic syndrome after allogeneic stem cell transplantation responding to donor lymphocyte infusion: Case report and literature review. Am. J. Hematol. 2004, 76, 389–394. [Google Scholar] [CrossRef]
- Engel, N.; Rovo, A.; Badoglio, M.; Labopin, M.; Basak, G.W.; Beguin, Y.; Guyotat, D.; Ljungman, P.; Nagler, A.; Schattenberg, A.; et al. European experience and risk factor analysis of donor cell-derived leukaemias/MDS following haematopoietic cell transplantation. Leukemia 2019, 33, 508–517. [Google Scholar] [CrossRef]
- Farina, M.; Bernardi, S.; Gandolfi, L.; Zanaglio, C.; Morello, E.; Turra, A.; Zollner, T.; Gramegna, D.; Rambaldi, B.; Cattina, F.; et al. Case Report: Late Onset of Myelodysplastic Syndrome From Donor Progenitor Cells After Allogeneic Stem Cell Transplantation. Which Lessons Can We Draw From the Reported Case? Front. Oncol. 2020, 10, 564521. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Hutchinson, C.B.; Huang, Q.; Lu, C.M.; Crow, J.; Wang, F.F.; Sebastian, S.; Rehder, C.; Lagoo, A.; Horwitz, M.; et al. Donor cell-derived leukemias/myelodysplastic neoplasms in allogeneic hematopoietic stem cell transplant recipients: A clinicopathologic study of 10 cases and a comprehensive review of the literature. Am. J. Clin. Pathol. 2011, 135, 525–540. [Google Scholar] [CrossRef] [PubMed]
- West, R.R.; Stafford, D.A.; White, A.D.; Bowen, D.T.; Padua, R.A. Cytogenetic abnormalities in the myelodysplastic syndromes and occupational or environmental exposure. Blood 2000, 95, 2093–2097. [Google Scholar] [CrossRef] [PubMed]
- Farquhar, M.J.; Bowen, D.T. Oxidative stress and the myelodysplastic syndromes. Int. J. Hematol. 2003, 77, 342–350. [Google Scholar] [CrossRef]
- Yin, S.N.; Hayes, R.B.; Linet, M.S.; Li, G.L.; Dosemeci, M.; Travis, L.B.; Li, C.Y.; Zhang, Z.N.; Li, D.G.; Chow, W.H.; et al. A cohort study of cancer among benzene-exposed workers in China: Overall results. Am. J. Ind. Med. 1996, 29, 227–235. [Google Scholar] [CrossRef]
- Aksoy, M.; Dinçol, K.; Erdem, S.; Dinçol, G. Acute leukemia due to chronic exposure to benzene. Am. J. Med. 1972, 52, 160–166. [Google Scholar] [CrossRef]
- Aul, C.; Bowen, D.T.; Yoshida, Y. Pathogenesis, etiology and epidemiology of myelodysplastic syndromes. Haematologica 1998, 83, 71–86. [Google Scholar]
- Rothman, N.; Haas, R.; Hayes, R.B.; Li, G.L.; Wiemels, J.; Campleman, S.; Quintana, P.J.; Xi, L.J.; Dosemeci, M.; Titenko-Holland, N. Benzene induces gene-duplicating but not gene-inactivating mutations at the glycophorin A locus in exposed humans. Proc. Natl. Acad. Sci. USA 1995, 92, 4069–4073. [Google Scholar] [CrossRef]
- Brugnone, F.; Perbellini, L.; Maranelli, G.; Romeo, L.; Alexopoulos, C.; Gobbi, M. Effects of cigarette smoking on blood and alveolar air levels of benzene. Med. Lav. 1990, 81, 101–106. [Google Scholar]
- Tong, H.; Hu, C.; Yin, X.; Yu, M.; Yang, J.; Jin, J. A Meta-Analysis of the Relationship between Cigarette Smoking and Incidence of Myelodysplastic Syndromes. PLoS ONE 2013, 8, e67537. [Google Scholar] [CrossRef]


{kind=link}
| Idiopathic Cytopenia of Undetermined Significance (ICUS) | Clonal Hematopoiesis of Indeterminate Potential (CHIP) | Clonal Cytopenia of Undetermined Significance (CCUS) | |
|---|---|---|---|
| Incidence | NA/unknown | >10% in age ≥70 years | 40% of patients with ICUS |
| Dysplasia | <10% | <10% | <10% |
| Cytopenia # | Present | Absent | Present |
| Clonality | Absent | Present | Present |
| Bone marrow blast % | <5% | <5% | <5% |
| Mutation in the leukemia-associated gene | Absent | Present | Present |
| Variant allele frequency % | <2% | ≥2% | ≥2% |
| Common mutated somatic genes | U2AF1, SRSF2, ASXL1, TP53, TET2, DNMT3A | DNMT3A, TET2, ASXL1, TP53, JAK2 | TET2, DNMT3A, ASXL1, TP53, U2AF1, ZRSR2, SRSF2, JAK2, RUNX1 |
| Risk of progression to MDS | Low | Very low | High risk |
| Risk of progression to AML | Very low | Very low | Low |
| Rate of progression to MDS/AML | Variable | 0.5–1% per year | Variable |
| Risk of cardiovascular disease | Unknown | Increased risk | Unknown |
| Outcome/Mortality $ | Unknown | Increased mortality | Unknown |
| Genetic Abnormality | Mutational Frequency | Clinical Characteristics | Therapeutics or Potential Targeted Therapy | Prognosis | Additional Comments |
| del(5q) | 10–15% | Anemia with or without neutropenia/thrombocytosis | Lenalidomide | Good | Isolated del(5q) is the only cytogenetic abnormality identified as a specific subtype in the WHO classification of MDS |
| del(7q) | 50% (t-MDS), 10% (de novo MDS) | Severe cytopenias, increased risk of infection | Hypomethylating agents | Intermediate | |
| trisomy 8 | 5–10% | Cytopenias, autoimmune manifestations | Immunosuppressive agents | Intermediate | |
| del(20q) | 2% | Thrombocytopenia | NA | Good | |
| del(17p) | 1% | Associated with t-MDS | NA | Very poor | |
| -Y | NA | Benign clinical course | NA | Very good | Age-related phenomenon |
| -X | Rare | Usually benign course | NA | Intermediate | Age-related phenomenon |
| Complex karyotype with 3 chromosomal abnormalities | 10% | Cytopenias, increased resistance to chemotherapeutic agents | NA | Poor | Complex karyotype with >3 chromosomal abnormalities confers very poor prognosis |
| TP53 mutation | 5–20% | Commonly found in t-MDS and AML | Hypomethylating agents, APR-246 | Poor | TP53 is an independent marker for prognosis and relapse even after allogeneic BMT |
| TP53 mutation and del(5q) | 20% | Aggressive disease with increased risk for transformation to AML | Hypomethylating agents | Poor | No response to lenalidomide |
| TP53 mutation and complex karyotype | High bone marrow blast proportion, severe anemia, thrombocytopenia | Hypomethylating agents | Very poor |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Awada, H.; Thapa, B.; Visconte, V. The Genomics of Myelodysplastic Syndromes: Origins of Disease Evolution, Biological Pathways, and Prognostic Implications. Cells 2020, 9, 2512. https://doi.org/10.3390/cells9112512
Awada H, Thapa B, Visconte V. The Genomics of Myelodysplastic Syndromes: Origins of Disease Evolution, Biological Pathways, and Prognostic Implications. Cells. 2020; 9(11):2512. https://doi.org/10.3390/cells9112512
Chicago/Turabian StyleAwada, Hassan, Bicky Thapa, and Valeria Visconte. 2020. "The Genomics of Myelodysplastic Syndromes: Origins of Disease Evolution, Biological Pathways, and Prognostic Implications" Cells 9, no. 11: 2512. https://doi.org/10.3390/cells9112512
APA StyleAwada, H., Thapa, B., & Visconte, V. (2020). The Genomics of Myelodysplastic Syndromes: Origins of Disease Evolution, Biological Pathways, and Prognostic Implications. Cells, 9(11), 2512. https://doi.org/10.3390/cells9112512