Ribosomal Protein L10: From Function to Dysfunction



Abstract
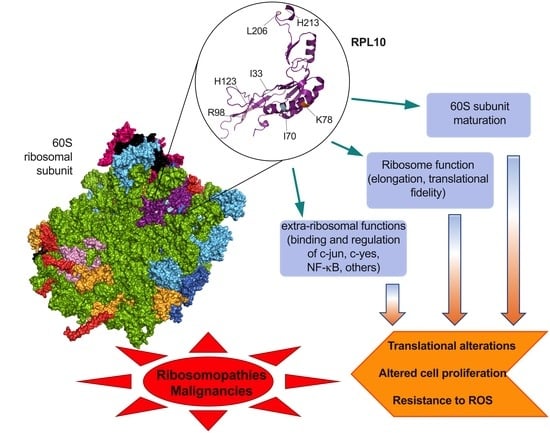
1. Introduction
2. RPL10 Gene and Protein

3. RPL10 Ribosomal Functions
4. RPL10 Extra-Ribosomal Functions
5. RPL10: A Player in Inherited Ribosomopathies
6. RPL10: A Player in Cancer
7. Summary and Outlook
Funding
Acknowledgments
Conflicts of Interest
References
- Baßler, J.; Hurt, E. Eukaryotic Ribosome Assembly. Annu. Rev. Biochem. 2019, 88, 281–306. [Google Scholar] [CrossRef]
- Robledo, S.; Idol, R.A.; Crimmins, D.L.; Ladenson, J.H.; Mason, P.J.; Bessler, M. The role of human ribosomal proteins in the maturation of rRNA and ribosome production. RNA 2008, 14, 1918–1929. [Google Scholar] [CrossRef]
- Yamashita, R.; Suzuki, Y.; Takeuchi, N.; Wakaguri, H.; Ueda, T.; Sugano, S.; Nakai, K. Comprehensive detection of human terminal oligo-pyrimidine (TOP) genes and analysis of their characteristics. Nucleic Acids Res. 2008, 36, 3707–3715. [Google Scholar] [CrossRef]
- De la Cruz, J.; Karbstein, K.; Woolford, J.L. Functions of Ribosomal Proteins in Assembly of Eukaryotic Ribosomes In Vivo. Annu Rev. Biochem. 2015, 84, 93–129. [Google Scholar] [CrossRef]
- Peña, C.; Hurt, E.; Panse, V.G. Eukaryotic ribosome assembly, transport and quality control. Nat. Struct. Mol. Biol. 2017, 24, 689–699. [Google Scholar] [CrossRef]
- Genuth, N.R.; Barna, M. The Discovery of Ribosome Heterogeneity and Its Implications for Gene Regulation and Organismal Life. Mol. Cell 2018, 71, 364–374. [Google Scholar] [CrossRef] [PubMed]
- Venezia, N.D.; Vincent, A.; Marcel, V.; Catez, F.; Diaz, J.J. Emerging role of eukaryote ribosomes in translational control. Int. J. Mol. Sci. 2019, 20, 1226. [Google Scholar] [CrossRef]
- Farley-Barnes, K.I.; Ogawa, L.M.; Baserga, S.J. Ribosomopathies: Old Concepts, New Controversies. Trends Genet. 2019, 35, 754–767. [Google Scholar] [CrossRef] [PubMed]
- Ban, N.; Beckmann, R.; Cate, J.H.D.; Dinman, J.D.; Dragon, F.; Ellis, S.R.; Lafontaine, D.L.J.; Lindahl, L.; Liljas, A.; Lipton, J.M.; et al. A new system for naming ribosomal proteins. Curr. Opin. Struct. Biol. 2014, 24, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.S.; Kwon, H.; Sun, S.K.; Yang, C.H. QM, a putative tumor suppressor, regulates proto-oncogene c-Yes. J. Biol. Chem. 2002, 277, 36489–36498. [Google Scholar] [CrossRef]
- Farmer, A.A.; Loftus, T.M.; Mills, A.A.; Sato, K.Y.; Neill, J.D.; Tron, T.; Yang, M.; Trumpower, B.L.; Stanbridge, E.J. Extreme evolutionary conservation of QM, a novel c-Jun associated transcription factor. Hum. Mol. Genet. 1994, 3, 23–28. [Google Scholar]
- Shi, J.; Zhang, L.; Zhou, D.; Zhang, J.; Lin, Q.; Guan, W.; Zhang, J.; Ren, W.; Xu, G. Biological function of ribosomal protein L10 on cell behavior in human epithelial ovarian cancer. J. Cancer 2018, 9, 45–56. [Google Scholar] [CrossRef]
- Klauck, S.M.; Felder, B.; Kolb-Kokocinski, A.; Schuster, C.; Chiocchetti, A.; Schupp, I.; Wellenreuther, R.; Schmötzer, G.; Poustka, F.; Breitenbach-Koller, L.; et al. Mutations in the ribosomal protein gene RPL10 suggest a novel modulating disease mechanism for autism. Mol. Psychiatry 2006, 11, 1073–1084. [Google Scholar] [CrossRef]
- Del Campo, E.M.; Casano, L.M.; Barreno, E. Evolutionary implications of intron-exon distribution and the properties and sequences of the RPL10A gene in eukaryotes. Mol. Phylogenet. Evol. 2013, 66, 857–867. [Google Scholar] [CrossRef]
- Uechi, T.; Maeda, N.; Tanaka, T.; Kenmochi, N. Functional second genes generated by retrotransposition of the X-linked ribosomal protein genes. Nucleic Acids Res. 2002, 30, 5369–5375. [Google Scholar]
- Jiang, L.; Li, T.; Zhang, X.; Zhang, B.; Yu, C.; Li, Y.; Fan, S.; Jiang, X.; Khan, T.; Hao, Q.; et al. RPL10L is Required for Male Meiotic Division by Compensating for RPL10 during Meiotic Sex Chromosome Inactivation in Mice. Curr. Biol. 2017, 27, 1498–1505.e6. [Google Scholar] [CrossRef]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2-A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef]
- Natchiar, S.K.; Myasnikov, A.G.; Kratzat, H.; Hazemann, I.; Klaholz, B.P. Visualization of chemical modifications in the human 80S ribosome structure. Nature 2017, 551, 472–477. [Google Scholar] [CrossRef]
- Zanni, G.; Kalscheuer, V.M.; Friedrich, A.; Barresi, S.; Alfieri, P.; Di Capua, M.; Haas, S.A.; Piccini, G.; Karl, T.; Klauck, S.M.; et al. A Novel Mutation in RPL10 (Ribosomal Protein L10) Causes X-Linked Intellectual Disability, Cerebellar Hypoplasia, and Spondylo-Epiphyseal Dysplasia. Hum. Mutat. 2015, 36, 1155–1158. [Google Scholar] [CrossRef]
- Klinge, S.; Woolford, J.L. Ribosome assembly coming into focus. Nat. Rev. Mol. Cell Biol. 2019, 20, 116–131. [Google Scholar] [CrossRef]
- Spahn, C.M.T.; Beckmann, R.; Eswar, N.; Penczek, P.A.; Sali, A.; Blobel, G.; Frank, J. Structure of the 80S ribosome from Saccharomyces cerevisiae—tRNA-ribosome and subunit-subunit interactions. Cell 2001, 107, 373–386. [Google Scholar] [CrossRef] [PubMed]
- Hofer, A.; Bussiere, C.; Johnson, A.W. Mutational analysis of the ribosomal protein Rpl10 from yeast. J. Biol. Chem. 2007, 282, 32630–32639. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, L.; Lambert, N.J.; Maklan, E.J.; Horan, L.H.; Noller, H.F. The sarcin-ricin loop of 23S rRNA is essential for assembly of the functional core of the 50S ribosomal subunit. RNA 2008, 14, 1999–2012. [Google Scholar] [CrossRef]
- Sulima, S.O.; Patchett, S.; Advani, V.M.; De Keersmaecker, K.; Johnson, A.W.; Dinman, J.D. Bypass of the pre-60S ribosomal quality control as a pathway to oncogenesis. Proc. Natl. Acad. Sci. USA 2014, 111, 5640–5645. [Google Scholar] [CrossRef]
- Schmidt, C.; Becker, T.; Heuer, A.; Braunger, K.; Shanmuganathan, V.; Pech, M.; Berninghausen, O.; Wilson, D.N.; Beckmann, R. Structure of the hypusinylated eukaryotic translation factor eIF-5A bound to the ribosome. Nucleic Acids Res. 2015, 44, 1944–1951. [Google Scholar] [CrossRef] [PubMed]
- Weis, F.; Giudice, E.; Churcher, M.; Jin, L.; Hilcenko, C.; Wong, C.C.; Traynor, D.; Kay, R.R.; Warren, A.J. Mechanism of eIF6 release from the nascent 60S ribosomal subunit. Nat. Struct. Mol. Biol. 2015, 22, 914–919. [Google Scholar] [CrossRef] [PubMed]
- Oender, K.; Loeffler, M.; Doppler, E.; Eder, M.; Lach, S.; Heinrich, F.; Karl, T.; Moesl, R.; Hundsberger, H.; Klade, T.; et al. Translational regulator RpL10p/Grc5p interacts physically and functionally with Sed1p, a dynamic component of the yeast cell surface. Yeast 2003, 20, 281–294. [Google Scholar] [CrossRef]
- Chiocchetti, A.G.; Haslinger, D.; Boesch, M.; Karl, T.; Wiemann, S.; Freitag, C.M.; Poustka, F.; Scheibe, B.; Bauer, J.W.; Hintner, H.; et al. Protein signatures of oxidative stress response in a patient specific cell line model for autism. Mol. Autism 2014, 5. [Google Scholar] [CrossRef]
- West, M.; Hedges, J.B.; Chen, A.; Johnson, A.W. Defining the Order in Which Nmd3p and Rpl10p Load onto Nascent 60S Ribosomal Subunits. Mol. Cell. Biol. 2005, 25, 3802–3813. [Google Scholar] [CrossRef]
- Pachler, K.; Karl, T.; Kolmann, K.; Mehlmer, N.; Eder, M.; Loeffler, M.; Oender, K.; Hochleitner, E.O.; Lottspeich, F.; Bresgen, N.; et al. Functional interaction in establishment of ribosomal integrity between small subunit protein rpS6 and translational regulator rpL10/Grc5p. FEMS Yeast Res. 2004, 5, 71–80. [Google Scholar] [CrossRef]
- Zemp, I.; Kutay, U. Nuclear export and cytoplasmic maturation of ribosomal subunits. FEBS Lett. 2007, 581, 2783–2793. [Google Scholar] [CrossRef] [PubMed]
- Panse, V.G.; Johnson, A.W. Maturation of eukaryotic ribosomes: Acquisition of functionality. Trends Biochem. Sci. 2010, 35, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Karbstein, K. Quality control mechanisms during ribosome maturation. Trends Cell Biol. 2013, 23, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Kargas, V.; Castro-Hartmann, P.; Escudero-Urquijo, N.; Dent, K.; Hilcenko, C.; Sailer, C.; Zisser, G.; Marques-Carvalho, M.J.; Pellegrino, S.; Wawiórka, L.; et al. Mechanism of completion of peptidyltransferase centre assembly in eukaryotes. eLife 2019, 8. [Google Scholar] [CrossRef]
- Zhou, Y.; Musalgaonkar, S.; Johnson, A.W.; Taylor, D.W. Tightly-orchestrated rearrangements govern catalytic center assembly of the ribosome. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef]
- Liang, X.; Zuo, M.Q.; Zhang, Y.; Li, N.; Ma, C.; Dong, M.Q.; Gao, N. Structural snapshots of human pre-60S ribosomal particles before and after nuclear export. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef]
- Patchett, S.; Musalgaonkar, S.; Malyutin, A.G.; Johnson, A.W. The T-cell leukemia related rpl10-R98S mutant traps the 60S export adapter Nmd3 in the ribosomal P site in yeast. PLOS Genet. 2017, 13, e1006894. [Google Scholar] [CrossRef]
- Russell, D.W.; Spremulli, L.L. Mechanism of action of the wheat germ ribosome dissociation factor: Interaction with the 60 S subunit. Arch. Biochem. Biophys 1980, 201, 518–526. [Google Scholar] [CrossRef]
- Gartmann, M.; Blau, M.; Armache, J.P.; Mielke, T.; Topf, M.; Beckmann, R. Mechanism of eIF6-mediated inhibition of ribosomal subunit joining. J. Biol. Chem. 2010, 285, 14848–14851. [Google Scholar] [CrossRef]
- Ceci, M.; Gaviraghi, C.; Gorrini, C.; Sala, L.A.; Offenhäuser, N.; Marchisio, P.C.; Biffo, S. Release of eIF6 (p27BBP) from the 60S subunit allows 80S ribosome assembly. Nature 2003, 426, 579–584. [Google Scholar] [CrossRef]
- Raychaudhuri, P.; Stringer, E.; Valenzuela, D.; Maitra, U. Ribosomal subunit antiassociation activity in rabbit reticulocyte lysates. Evidence for a low molecular weight ribosomal subunit antiassociation protein factor (Mr = 25,000). J. Biol. Chem. 1984, 259, 11930–11935. [Google Scholar] [PubMed]
- Boocock, G.R.B.; Morrison, J.A.; Popovic, M.; Richards, N.; Ellis, L.; Durie, P.R.; Rommens, J.M. Mutations in SBDS are associated with Shwachman-Diamond syndrome. Nat. Genet. 2003, 33, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Bezzerri, V.; Cipolli, M. Shwachman-Diamond Syndrome: Molecular Mechanisms and Current Perspectives. Mol. Diagn. Ther. 2019, 23, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Petrov, A.N.; Meskauskas, A.; Roshwalb, S.C.; Dinman, J.D. Yeast ribosomal protein L10 helps coordinate tRNA movement through the large subunit. Nucleic Acids Res. 2008, 36, 6187–6198. [Google Scholar] [CrossRef]
- Bussiere, C.; Hashem, Y.; Arora, S.; Frank, J.; Johnson, A.W. Integrity of the P-site is probed during maturation of the 60S ribosomal subunit. J. Cell Biol. 2012, 197, 747–759. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Hwang, J.S.; Goo, T.W.; Yun, E.Y.; Lee, J.H.; Kang, S.W.; Kim, K.Y.; Kwon, O.Y. Tissue-/stage-dependent expression of a cloned Bombyx mandarina QM homologue. Biomol. Eng. 2000, 16, 211–215. [Google Scholar] [CrossRef]
- Rooks, S.S.; Wall, A.L.; Golzio, C.; Reid, D.W.; Kondyles, A.; Willer, J.R.; Botti, C.; Nicchitta, C.V.; Katsanis, N.; Davis, E.E. A novel ribosomopathy caused by dysfunction of RPL10 disrupts neurodevelopment and causes X-linked microcephaly in humans. Genetics 2014, 198, 723–733. [Google Scholar] [CrossRef]
- Imafuku, I.; Masaki, T.; Waragai, M.; Takeuchi, S.; Kawabata, M.; Hirai, S.I.; Ohno, S.; Nee, L.E.; Lippa, C.F.; Kanazawa, I.; et al. Presenilin 1 suppresses the function of c-Jun homodimers via interaction with QM/Jif-1. J. Cell Biol. 1999, 147, 121–133. [Google Scholar] [CrossRef]
- Yang, J.; Chen, Z.; Liu, N.; Chen, Y. Ribosomal protein L10 in mitochondria serves as a regulator for ROS level in pancreatic cancer cells. Redox Biol. 2018, 19, 158–165. [Google Scholar] [CrossRef]
- Monteclaro, F.S.; Vogt, P.K. A Jun-binding protein related to a putative tumor suppressor. Proc. Natl. Acad. Sci. USA 1993, 90, 6726–6730. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.L.; Diaz, J.J.; Denoroy, L.; Madjar, J.J.; Wool, I.G. The primary structure of rat ribosomal protein L10: Relationship to a Jun-binding protein and to a putative Wilms’ tumor suppressor. Biochem. Biophys. Res. Commun. 1996, 225, 952–956. [Google Scholar] [CrossRef]
- Kampen, K.R.; Sulima, S.O.; Verbelen, B.; Girardi, T.; Vereecke, S.; Rinaldi, G.; Verbeeck, J.; Op de Beeck, J.; Uyttebroeck, A.; Meijerink, J.P.P.; et al. The ribosomal RPL10 R98S mutation drives IRES-dependent BCL-2 translation in T-ALL. Leukemia 2019, 33, 319–332. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Wang, Y.; Guo, Y.; Chen, Y.; Liu, N. Cooperative down-regulation of ribosomal protein L10 and NF-κB signaling pathway is responsible for the anti-proliferative effects by DMAPT in pancreatic cancer cells. Oncotarget 2017, 8, 5009–5018. [Google Scholar] [CrossRef]
- Rocha, C.S.; Santos, A.A.; Machado, J.P.B.; Fontes, E.P.B. The ribosomal protein L10/QM-like protein is a component of the NIK-mediated antiviral signaling. Virology 2008, 380, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Ferreyra, M.L.F.; Biarc, J.; Burlingame, A.L.; Casati, P. Arabidopsis l10 ribosomal proteins in UV-B responses. Plant Signal. Behav. 2010, 5, 222–225. [Google Scholar] [CrossRef]
- Chen, C.; Wanduragala, S.; Becker, D.F.; Dickman, M.B. Tomato QM-like protein protects Saccharomyces cerevisiae cells against oxidative stress by regulating intracellular proline levels. Appl. Environ. Microbiol. 2006, 72, 4001–4006. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.W.; Green, R. Ribosomopathies: There’s strength in numbers. Science 2017, 358, eaan2755. [Google Scholar] [CrossRef]
- Nakhoul, H.; Ke, J.; Zhou, X.; Liao, W.; Zeng, S.X.; Lu, H. Ribosomopathies: Mechanisms of disease. Clin. Med. Insights Blood Disord. 2014, 7, 7–16. [Google Scholar] [CrossRef]
- De Keersmaecker, K.; Sulima, S.O.; Dinman, J.D. Ribosomopathies and the paradox of cellular hypo- to hyperproliferation. Blood 2015, 125, 1377–1382. [Google Scholar] [CrossRef]
- Da Costa, L.; O’donohue, M.-F.; Van Dooijeweert, B.; Albrecht, K.; Unal, S.; Ramenghi, U.; Leblanc, T.; Dianzani, I.; Tamary, H.; Bartels, M.; et al. Molecular approaches to diagnose Diamond-Blackfan anemia: The EuroDBA experience. Articleinfo 2017. [Google Scholar] [CrossRef]
- Khajuria, R.K.; Munschauer, M.; Ulirsch, J.C.; Fiorini, C.; Ludwig, L.S.; McFarland, S.K.; Abdulhay, N.J.; Specht, H.; Keshishian, H.; Mani, D.R.; et al. Ribosome Levels Selectively Regulate Translation and Lineage Commitment in Human Hematopoiesis. Cell 2018, 173, 90–103.e19. [Google Scholar] [CrossRef] [PubMed]
- Bursac, S.; Brdovcak, M.C.; Donati, G.; Volarevic, S. Activation of the tumor suppressor p53 upon impairment of ribosome biogenesis. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 817–830. [Google Scholar] [CrossRef]
- Xue, S.; Barna, M. Specialized ribosomes: A new frontier in gene regulation and organismal biology. Nat. Rev. Mol. Cell Biol. 2012, 13, 355–369. [Google Scholar] [CrossRef] [PubMed]
- Ferretti, M.B.; Karbstein, K. Does functional specialization of ribosomes really exist? RNA 2019, 25, 521–538. [Google Scholar] [CrossRef] [PubMed]
- Lezzerini, M.; Penzo, M.; O’Donohue, M.F.; Dos Santos Vieira, C.M.; Saby, M.; Elfrink, H.L.; Diets, I.J.; Hesse, A.M.; Couté, Y.; Gastou, M.; et al. Ribosomal protein gene RPL9 variants can differentially impair ribosome function and cellular metabolism. Nucleic Acids Res. 2020, 48, 770–787. [Google Scholar] [CrossRef]
- Chiocchetti, A.; Pakalapati, G.; Duketis, E.; Wiemann, S.; Poustka, A.; Poustka, F.; Klauck, S.M. Mutation and expression analyses of the ribosomal protein gene RPL10 in an extended German sample of patients with autism spectrum disorder. Am. J. Med. Genet. Part A 2011, 155, 1472–1475. [Google Scholar] [CrossRef]
- Bourque, D.K.; Hartley, T.; Nikkel, S.M.; Pohl, D.; Tétreault, M.; Kernohan, K.D.; Dyment, D.A. A de novo mutation in RPL10 causes a rare X-linked ribosomopathy characterized by syndromic intellectual disability and epilepsy: A new case and review of the literature. Eur. J. Med. Genet. 2018, 61, 89–93. [Google Scholar] [CrossRef]
- Thevenon, J.; Michot, C.; Bole, C.; Nitschke, P.; Nizon, M.; Faivre, L.; Munnich, A.; Lyonnet, S.; Bonnefont, J.P.; Des Portes, V.; et al. RPL10 mutation segregating in a family with X-linked syndromic Intellectual Disability. Am. J. Med. Genet. Part A 2015, 167, 1908–1912. [Google Scholar] [CrossRef]
- Goudarzi, K.M.; Lindström, M.S. Role of ribosomal protein mutations in tumor development (Review). Int. J. Oncol. 2016, 48, 1313–1324. [Google Scholar] [CrossRef]
- Vlachos, A. Acquired ribosomopathies in leukemia and solid tumors. Hematology 2017, 2017, 716–719. [Google Scholar] [CrossRef] [PubMed]
- Penzo, M.; Clima, R.; Trerè, D.; Montanaro, L. Separated Siamese Twins: Intronic Small Nucleolar RNAs and Matched Host Genes May be Altered in Conjunction or Separately in Multiple Cancer Types. Cells 2020, 9, 87. [Google Scholar] [CrossRef]
- Ajore, R.; Raiser, D.; McConkey, M.; Jöud, M.; Boidol, B.; Mar, B.; Saksena, G.; Weinstock, D.M.; Armstrong, S.; Ellis, S.R.; et al. Deletion of ribosomal protein genes is a common vulnerability in human cancer, especially in concert with TP53 mutations. EMBO Mol. Med. 2017, 9, 98–507. [Google Scholar] [CrossRef]
- Dolezal, J.M.; Dash, A.P.; Prochownik, E.V. Diagnostic and prognostic implications of ribosomal protein transcript expression patterns in human cancers. BMC Cancer 2018, 18, 275. [Google Scholar] [CrossRef]
- Dowdy, S.F.; Lai, K.; Weissman, B.E.; Matsui, Y.; Hogan, B.L.M.; Stanbridge, E.J. The isolation and characterization of a novel cDNA demonstrating an altered mRNA level in nontumorigenic wilms’ microcell hybrid cells. Nucleic Acids Res. 1991, 19, 5763–5769. [Google Scholar] [CrossRef]
- De Keersmaecker, K.; Atak, Z.K.; Li, N.; Vicente, C.; Patchett, S.; Girardi, T.; Gianfelici, V.; Geerdens, E.; Clappier, E.; Porcu, M.; et al. Exome sequencing identifies mutation in CNOT3 and ribosomal genes RPL5 and RPL10 in T-cell acute lymphoblastic leukemia. Nat. Genet. 2012, 45, 186–190. [Google Scholar] [CrossRef] [PubMed]
- Ben-Shem, A.; Garreau de Loubresse, N.; Melnikov, S.; Jenner, L.; Yusupova, G.; Yusupov, M. The Structure of the Eukaryotic Ribosome at 3.0 A Resolution. Science 2011, 334, 1524–1529. [Google Scholar] [CrossRef]
- Girardi, T.; Vereecke, S.; Sulima, S.O.; Khan, Y.; Fancello, L.; Briggs, J.W.; Schwab, C.; De Beeck, J.O.; Verbeeck, J.; Royaert, J.; et al. The T-cell leukemia-associated ribosomal RPL10 R98S mutation enhances JAK-STAT signaling. Leukemia 2018, 32, 809–819. [Google Scholar] [CrossRef]
- Kampen, K.R.; Fancello, L.; Girardi, T.; Rinaldi, G.; Planque, M.; Sulima, S.O.; Loayza-Puch, F.; Verbelen, B.; Vereecke, S.; Verbeeck, J.; et al. Translatome analysis reveals altered serine and glycine metabolism in T-cell acute lymphoblastic leukemia cells. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef]
- Hofman, I.J.F.; Patchett, S.; Van Duin, M.; Geerdens, E.; Verbeeck, J.; Michaux, L.; Delforge, M.; Sonneveld, P.; Johnson, A.W.; De Keersmaecker, K. Low frequency mutations in ribosomal proteins RPL10 and RPL5 in multiple myeloma. Haematologica 2017, 102, e317–e320. [Google Scholar] [CrossRef]
- Altinok, G.; Powell, I.J.; Che, M.; Hormont, K.; Sarkar, F.H.; Sakr, W.A.; Grignon, D.; Liao, D.J. Reduction of QM protein expression correlates with tumor grade in prostatic adenocarcinoma. Prostate Cancer Prostatic. Dis. 2006, 9, 7–82. [Google Scholar] [CrossRef][Green Version]
- Shen, X.J.; Ali-Fehmi, R.; Weng, C.R.; Sarkar, F.H.; Grignon, D.; Liao, D.J. Loss of heterozygosity and microsatellite instability at the Xq28 and the A/G heterozygosity of the QM gene are associated with ovarian cancer. Cancer Biol. 2006, 5, 23–28. [Google Scholar] [CrossRef]
- Ren, Y.; Tao, C.; Wang, X.; Ju, Y. Identification of RPL5 and RPL10 as novel diagnostic biomarkers of Atypical teratoid/rhabdoid tumors. Cancer Cell Int. 2018, 18. [Google Scholar] [CrossRef]
- Ford, D. Ribosomal heterogeneity—A new inroad for pharmacological innovation. Biochem. Pharmacol. 2020, 175. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CDS Position | aa Substitution | Pathology | Reference |
|---|---|---|---|
| c.191C > T | A64V | intellectual disability, cerebellar hypoplasia, skeletal abnormalities | [13] |
| c.232A > G | K78E | mental retardation, seizures and microcephaly, epilepsy | [37,56] |
| c.481G > A | G161S | syndromic intellectual disability | [69] |
| c.605G > A | S202N | intellectual disability | [9] |
| c.616C > A | L206M | autism | [9,55] |
| c.639C > G | H213Q | autism, pancreatic cancer | [9,20,40,44] |
| c.97A > G | I33V | multiple myeloma | [68] |
| c.197A > G | E66G | multiple myeloma | [68] |
| c.208A > C | I70L | multiple myeloma | [68] |
| c.210T > G | I70M | multiple myeloma | [68] |
| c.292C > A | R98S | infant T-ALL | [64] |
| c.292C > T | R98C | infant T-ALL | [64] |
| c.368A > C | Q123P | infant T-ALL | [64] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pollutri, D.; Penzo, M. Ribosomal Protein L10: From Function to Dysfunction. Cells 2020, 9, 2503. https://doi.org/10.3390/cells9112503
Pollutri D, Penzo M. Ribosomal Protein L10: From Function to Dysfunction. Cells. 2020; 9(11):2503. https://doi.org/10.3390/cells9112503
Chicago/Turabian StylePollutri, Daniela, and Marianna Penzo. 2020. "Ribosomal Protein L10: From Function to Dysfunction" Cells 9, no. 11: 2503. https://doi.org/10.3390/cells9112503
APA StylePollutri, D., & Penzo, M. (2020). Ribosomal Protein L10: From Function to Dysfunction. Cells, 9(11), 2503. https://doi.org/10.3390/cells9112503