Functional Characterization of the N-Acetylmuramyl-l-Alanine Amidase, Ami1, from Mycobacterium abscessus

 ,
,  , , , and
, , , and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Generation of Unmarked Deletion Mutant
2.2. Cloning for Recombinant Protein Expression
2.3. Mycobacterial Culture Conditions
2.4. Single-Cell Preparation for M. abscessus
2.5. Protein Expression and Purification
2.6. Crystallization, Data Collection, and Structure Refinement
2.7. In Silico Docking
2.8. Drug Susceptibility Testing and MIC Determination
2.9. Macrophage Experiments
2.10. Synthesis of Peptidoglycan Dipeptide, l-Alanyl-d-Isoglutamine (DP)
2.11. Chemical Library
2.12. General Procedures Relative to the Chemistry of Ami1Mab Inhibitors
 4-chlorofuro [3,2-c]pyridine-3-carboxylic acid (inhibitor 1) 1H NMR (300 MHz, DMSO-d6) δ 8.81 (s, 1H), 8.38 (d, J = 5.7 Hz, 1H), 7.85 (d, J = 5.7 Hz, 1H), LRMS (ESI-MS) m/z = 198.0 + 200.0 [M+H]+.
4-chlorofuro [3,2-c]pyridine-3-carboxylic acid (inhibitor 1) 1H NMR (300 MHz, DMSO-d6) δ 8.81 (s, 1H), 8.38 (d, J = 5.7 Hz, 1H), 7.85 (d, J = 5.7 Hz, 1H), LRMS (ESI-MS) m/z = 198.0 + 200.0 [M+H]+. 2-chloropyridine-3,4-dicarboxylic acid (inhibitor 2) 1H NMR (300 MHz, DMSO-d6) δ 14.07 (s, 2H), 8.62 (d, J = 5.0 Hz, 1H), 7.82 (d, J = 5.0 Hz, 1H). LRMS (ESI-MS) m/z = 202.0 + 204.0 [M+H]+.
2-chloropyridine-3,4-dicarboxylic acid (inhibitor 2) 1H NMR (300 MHz, DMSO-d6) δ 14.07 (s, 2H), 8.62 (d, J = 5.0 Hz, 1H), 7.82 (d, J = 5.0 Hz, 1H). LRMS (ESI-MS) m/z = 202.0 + 204.0 [M+H]+. 2-(1H-tetrazol-5-yl)aniline (inhibitor 3) 1H NMR (300 MHz, DMSO-d6) δ 9.09 (s, 3H), 7.84 (dd, J = 7.9, 1.2 Hz, 1H), 7.37 – 7.18 (m, 1H), 7.00 (d, J = 8.0 Hz, 1H), 6.80 (t, J = 7.2 Hz, 1H), LRMS (ESI-MS) m/z = 162.2 [M+H]+.
2-(1H-tetrazol-5-yl)aniline (inhibitor 3) 1H NMR (300 MHz, DMSO-d6) δ 9.09 (s, 3H), 7.84 (dd, J = 7.9, 1.2 Hz, 1H), 7.37 – 7.18 (m, 1H), 7.00 (d, J = 8.0 Hz, 1H), 6.80 (t, J = 7.2 Hz, 1H), LRMS (ESI-MS) m/z = 162.2 [M+H]+.



2.13. Transmission Electron Microscopy
2.14. Zebrafish Infection Experiments
2.15. Zebrafish Breeding and Ethics
2.16. Statistical Analyses
2.17. Muramyl-Dipeptide Hydrolysis Assay
2.18. Compounds Screening by Thermal-Shift Assay
3. Results
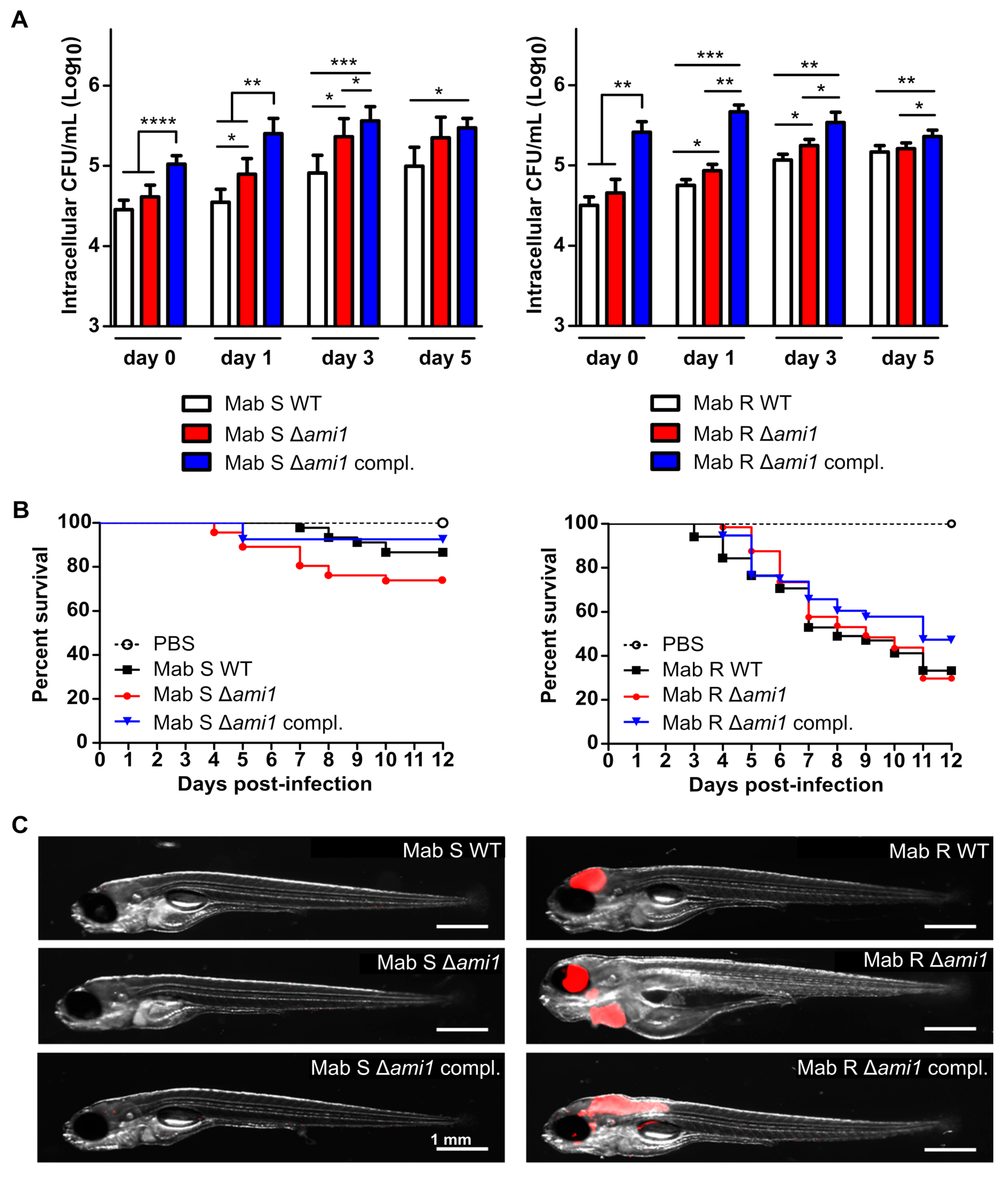
3.1. Phenotypic Analysis of ami1 Deletion Mutant in the R and S Morphotypes
3.1.1. ami1Mab Is Dispensable for M. abscessus Growth in Vitro
3.1.2. ami1Mab Deletion Mutants Do Not Present Division Defects
3.1.3. Δami1Mab Is More Sensitive to β-Lactam Antibiotics and Detergent
3.2. Analysis of the Virulence Phenotype of the M. abscessus ami1 Deletion Mutant
3.2.1. Δami1Mab Is Not Impaired in Uptake and Persistence in Human Macrophages
3.2.2. Virulence of Δami1Mab Is Not Affected in the Zebrafish Model of Infection
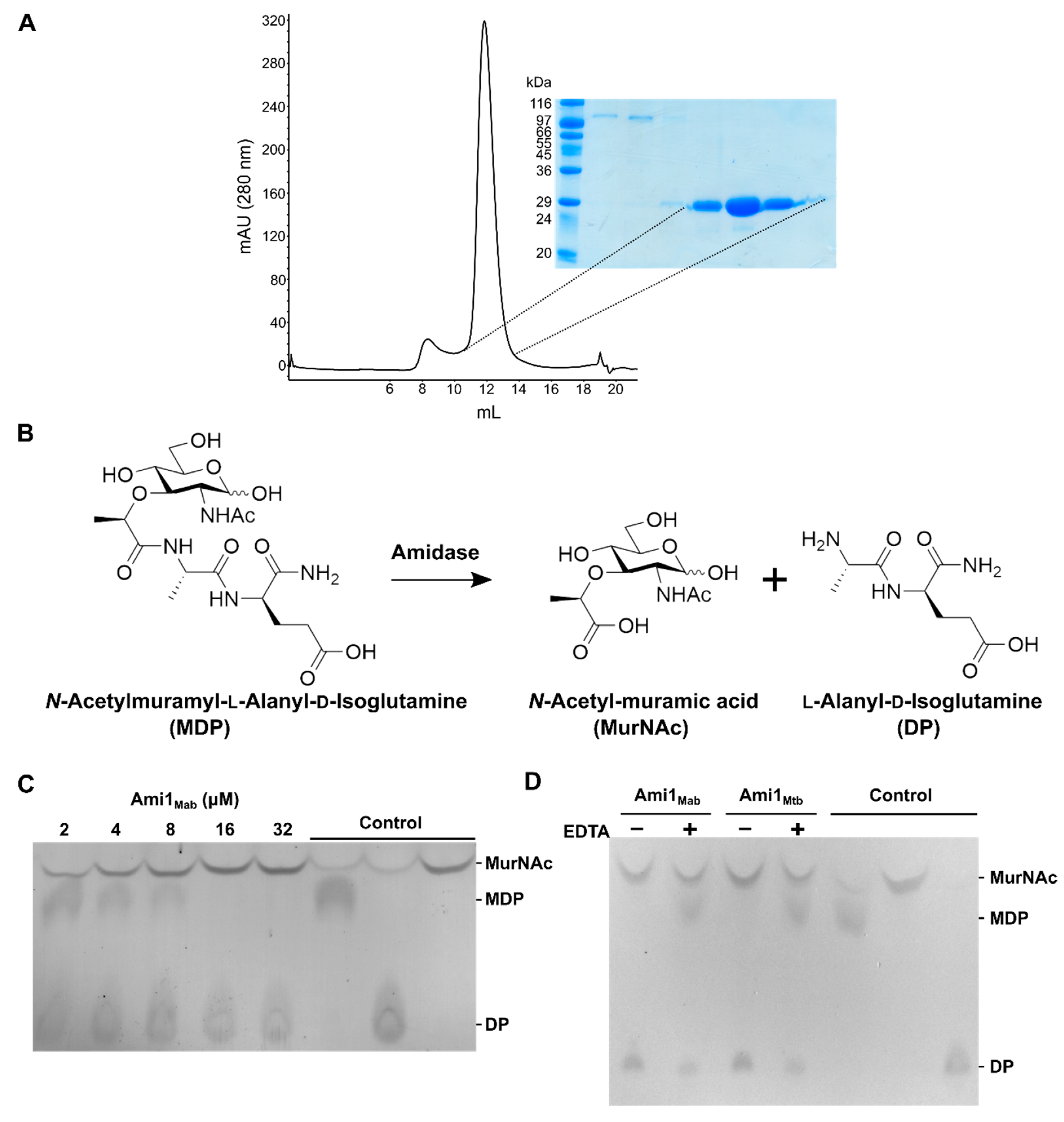
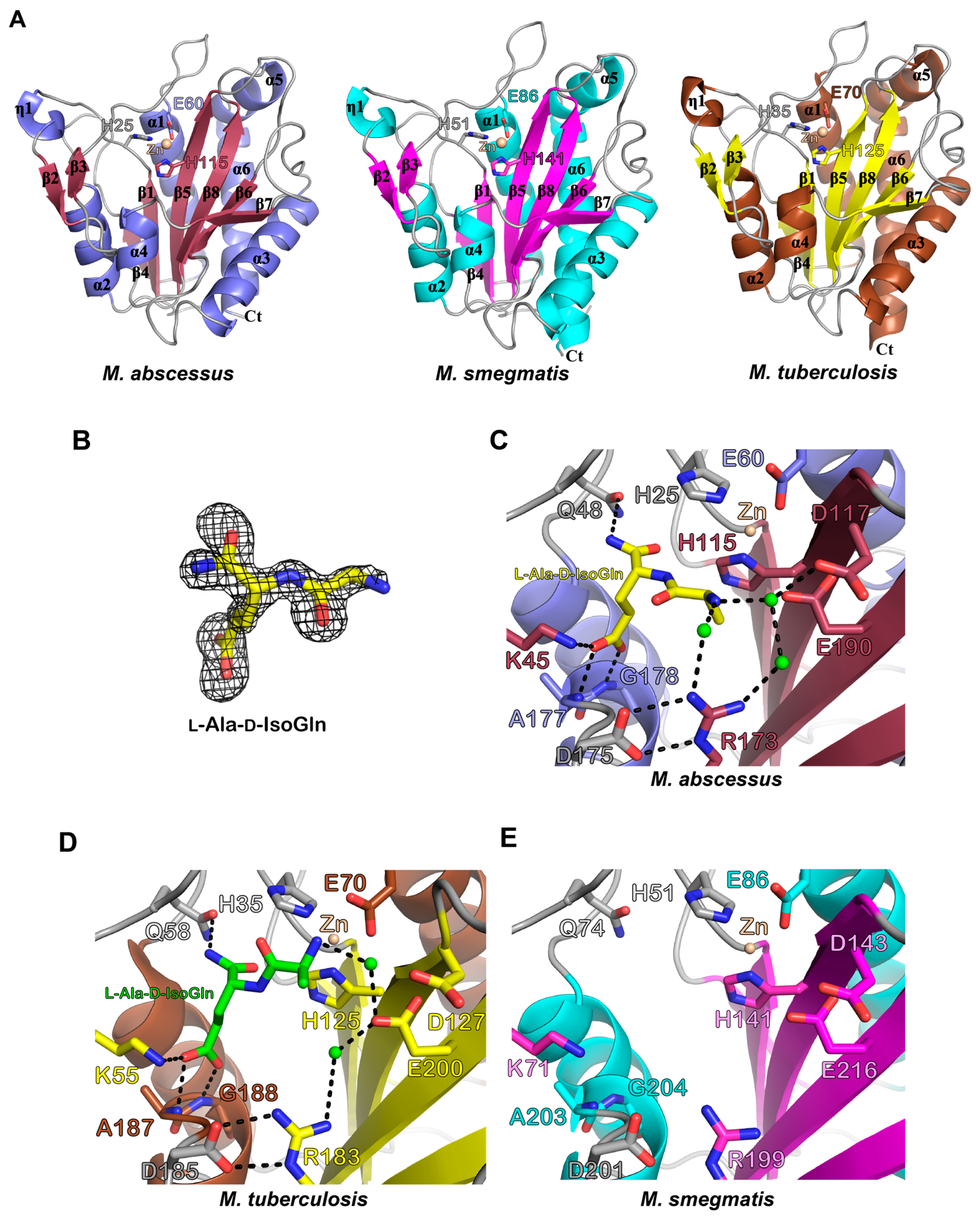
3.3. Biochemical and Structural Characterization of Ami1Mab
3.3.1. Ami1Mab Is an Active PG Hydrolase
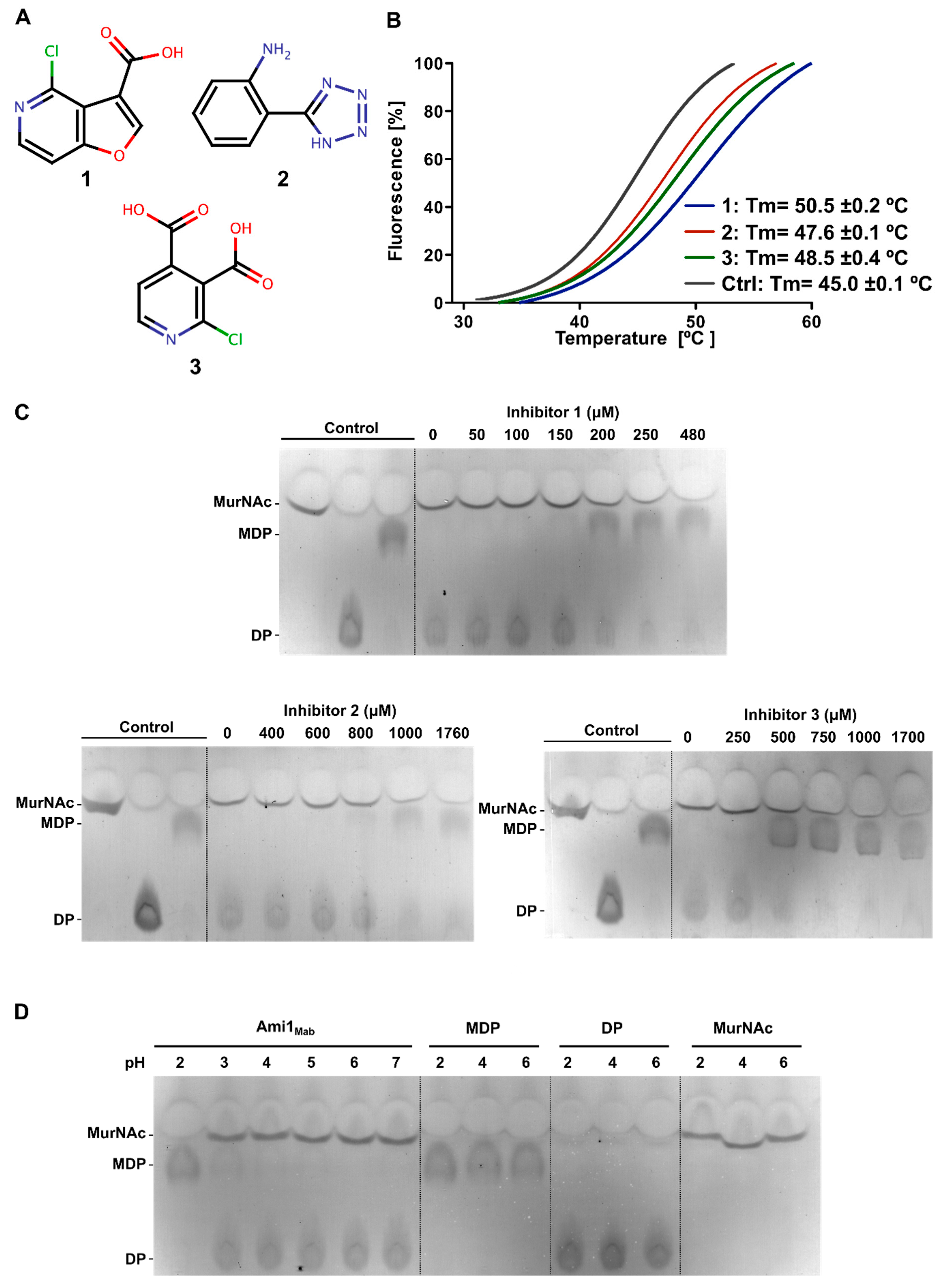
3.3.2. Thermal-Shift-Based Drug Screening Identified Three Potent Inhibitors of Ami1Mab
3.3.3. Biochemical Characterization of Ami1Mab Inhibitors
3.3.4. Biochemical Characterization of Ami1Mab Inhibitors
3.3.5. In Silico Docking Allows Predicting That Ami1Mab Inhibitors May Compete with Substrate Binding
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Meroueh, S.O.; Bencze, K.Z.; Hesek, D.; Lee, M.; Fisher, J.F.; Stemmler, T.L.; Mobashery, S. Three-dimensional structure of the bacterial cell wall peptidoglycan. Proc. Natl. Acad. Sci. USA 2006, 103, 4404–4409. [Google Scholar] [CrossRef]
- Typas, A.; Banzhaf, M.; Gross, C.A.; Vollmer, W. From the regulation of peptidoglycan synthesis to bacterial growth and morphology. Nat. Rev. Genet. 2012, 10, 123–136. [Google Scholar] [CrossRef]
- Egan, A.J.F.; Vollmer, W. The physiology of bacterial cell division. Ann. N. Y. Acad. Sci. 2012, 1277, 8–28. [Google Scholar] [CrossRef]
- Vollmer, W. Bacterial growth does require peptidoglycan hydrolases. Mol. Microbiol. 2012, 86, 1031–1035. [Google Scholar] [CrossRef]
- Daffé, M.; Marrakchi, H. Unraveling the Structure of the Mycobacterial Envelope. Microbiol. Spectr. 2019, 7, 7. [Google Scholar] [CrossRef]
- Anantharaman, V.; Aravind, L. Evolutionary history, structural features and biochemical diversity of the NlpC/P60 superfamily of enzymes. Genome Biol. 2003, 4, R11. [Google Scholar] [CrossRef] [PubMed]
- Hett, E.C.; Chao, M.C.; Rubin, E.J. Interaction and Modulation of Two Antagonistic Cell Wall Enzymes of Mycobacteria. PLoS Pathog. 2010, 6, e1001020. [Google Scholar] [CrossRef] [PubMed]
- Botella, H.; Vaubourgeix, J.; Lee, M.H.; Song, N.; Xu, W.; Makinoshima, H.; Glickman, M.S.; Ehrt, S. Mycobacterium tuberculosis protease MarP activates a peptidoglycan hydrolase during acid stress. Embo J. 2017, 36, 536–548. [Google Scholar] [CrossRef] [PubMed]
- Boutte, C.C.; Baer, C.E.; Papavinasasundaram, K.; Liu, W.; Chase, M.R.; Meniche, X.; Fortune, S.M.; Sassetti, C.M.; Ioerger, T.R.; Rubin, E.J. A cytoplasmic peptidoglycan amidase homologue controls mycobacterial cell wall synthesis. eLife 2016, 5, e14590. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.J.; Boutte, C.C.; Ioerger, T.R.; Rubin, E.J. Mycobacterium smegmatis HtrA Blocks the Toxic Activity of a Putative Cell Wall Amidase. Cell Rep. 2019, 27, 2468–2479.e3. [Google Scholar] [CrossRef] [PubMed]
- Senzani, S.; Li, D.; Bhaskar, A.; Ealand, C.; Chang, J.; Rimal, B.; Liu, C.; Kim, S.J.; Dhar, N.; Kana, B. An Amidase_3 domain-containing N-acetylmuramyl-l-alanine amidase is required for mycobacterial cell division. Sci. Rep. 2017, 7, 1–16. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Fu, W.; Zhao, S.; Zhang, C.; Sun, T.; Jiang, T. Lack of MSMEG_6281, a peptidoglycan amidase, affects cell wall integrity and virulence of Mycobacterium smegmatis. Microb. Pathog. 2019, 128, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; He, J.; Fu, W.; Cao, P.; Zhang, S.; Jiang, T. Effect of Mycobacterium tuberculosis Rv3717 on cell division and cell adhesion. Microb. Pathog. 2018, 117, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Miao, J.; Liu, H.; Qu, Y.; Fu, W.; Qi, K.; Zang, S.; He, J.; Zhao, S.; Chen, S.; Jiang, T. Effect of peptidoglycan amidase MSMEG_6281 on fatty acid metabolism in Mycobacterium smegmatis. Microb. Pathog. 2020, 140, 103939. [Google Scholar] [CrossRef]
- Prigozhin, D.M.; Mavrici, D.; Huizar, J.P.; Vansell, H.J.; Alber, T. Structural and Biochemical Analyses of Mycobacterium tuberculosis N-Acetylmuramyl-l-alanine Amidase Rv3717 Point to a Role in Peptidoglycan Fragment Recycling. J. Biol. Chem. 2013, 288, 31549–31555. [Google Scholar] [CrossRef]
- Kumar, A.; Kumar, S.; Kumar, D.; Mishra, A.; Dewangan, R.P.; Shrivastava, P.; Ramachandran, S.; Taneja, B. The structure of Rv3717 reveals a novel amidase from Mycobacterium tuberculosis. Acta Crystallogr. Sect. D Biol. Crystallogr. 2013, 69, 2543–2554. [Google Scholar] [CrossRef]
- Heidrich, C.; Templin, M.F.; Ursinus, A.; Merdanovic, M.; Berger, J.; Schwarz, H.; De Pedro, M.A.; Höltje, J.-V. Involvement of N-acetylmuramyl-l-alanine amidases in cell separation and antibiotic-induced autolysis of Escherichia coli. Mol. Microbiol. 2001, 41, 167–178. [Google Scholar] [CrossRef]
- Heidrich, C.; Ursinus, A.; Berger, J.; Schwarz, H.; Höltje, J.-V. Effects of Multiple Deletions of Murein Hydrolases on Viability, Septum Cleavage, and Sensitivity to Large Toxic Molecules in Escherichia coli. J. Bacteriol. 2002, 184, 6093–6099. [Google Scholar] [CrossRef]
- Van Heijenoort, J. Peptidoglycan Hydrolases of Escherichia coli. Microbiol. Mol. Biol. Rev. 2011, 75, 636–663. [Google Scholar] [CrossRef]
- Healy, C.; Gouzy, A.; Ehrt, S. Peptidoglycan Hydrolases RipA and Ami1 Are Critical for Replication and Persistence of Mycobacterium tuberculosis in the Host. mBio 2020, 11. [Google Scholar] [CrossRef]
- Johansen, M.D.; Herrmann, J.-L.; Kremer, L. Non-tuberculous mycobacteria and the rise of Mycobacterium abscessus. Nat. Rev. Genet. 2020, 18, 392–407. [Google Scholar] [CrossRef]
- Bernut, A.; Herrmann, J.-L.; Kissa, K.; Dubremetz, J.-F.; Gaillard, J.-L.; Lutfalla, G.; Kremer, L. Mycobacterium abscessuscording prevents phagocytosis and promotes abscess formation. Proc. Natl. Acad. Sci. USA 2014, 111, E943–E952. [Google Scholar] [CrossRef]
- Bernut, A.; Herrmann, J.-L.; Ordway, D.; Kremer, L. The Diverse Cellular and Animal Models to Decipher the Physiopathological Traits of Mycobacterium abscessus Infection. Front. Cell. Infect. Microbiol. 2017, 7, 100. [Google Scholar] [CrossRef]
- Gutiérrez, A.V.; Viljoen, A.; Ghigo, E.; Herrmann, J.-L.; Kremer, L. Glycopeptidolipids, a Double-Edged Sword of the Mycobacterium abscessus Complex. Front. Microbiol. 2018, 9, 1145. [Google Scholar] [CrossRef]
- Li, B.; Ye, M.; Zhao, L.; Guo, Q.; Chen, J.; Xu, B.; Zhan, M.; Zhang, Y.; Zhang, Z.; Chu, H. Glycopeptidolipid Genotype Correlates With the Severity of Mycobacterium abscessus Lung Disease. J. Infect. Dis. 2020, 221, S257–S262. [Google Scholar] [CrossRef]
- Richard, M.; Gutiérrez, A.V.; Viljoen, A.; Rodriguez-Rincon, D.; Roquet-Baneres, F.; Blaise, M.; Everall, I.; Parkhill, J.; Floto, R.A.; Kremer, L. Mutations in the MAB_2299c TetR Regulator Confer Cross-Resistance to Clofazimine and Bedaquiline in Mycobacterium abscessus. Antimicrob. Agents Chemother. 2018, 63. [Google Scholar] [CrossRef]
- Woods, G.L.; Brown-Elliott, B.A.; Conville, P.S.; Desmond, E.P.; Hall, G.S.; Lin, G.; Pfyffer, G.E.; Ridderhof, J.C.; Siddiqi, S.H.; Wallace, R.J.; et al. Susceptibility Testing of Mycobacteria, Nocardiae, and Other Aerobic Actinomycetes, 2nd ed.; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2011; ISBN 978-1-56238-746-4. [Google Scholar]
- Adams, P.D.; Afonine, P.V.; Bunkóczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.-W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef]
- Dallakyan, S.; Olson, A.J. Small-Molecule Library Screening by Docking with PyRx. Methods Mol. Biol. 2015, 1263, 243–250. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef]
- Lamason, R.L.; Mohideen, M.-A.P.; Mest, J.R.; Wong, A.C.; Norton, H.L.; Aros, M.C.; Jurynec, M.J.; Mao, X.; Humphreville, V.R.; Humbert, J.E.; et al. SLC24A5, a Putative Cation Exchanger, Affects Pigmentation in Zebrafish and Humans. Science 2005, 310, 1782–1786. [Google Scholar] [CrossRef]
- Niesen, F.H.; Berglund, H.; Vedadi, M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc. 2007, 2, 2212–2221. [Google Scholar] [CrossRef]
- Stover, C.K.; De La Cruz, V.F.; Fuerst, T.R.; Burlein, J.E.; Benson, L.A.; Bennett, L.T.; Bansal, G.P.; Young, J.F.; Lee, M.H.; Hatfull, G.F.; et al. New use of BCG for recombinant vaccines. Nat. Cell Biol. 1991, 351, 456–460. [Google Scholar] [CrossRef]
- Chaput, C.; Ecobichon, C.; Pouradier, N.; Rousselle, J.-C.; Namane, A.; Boneca, I.G. Role of theN-Acetylmuramoyl-l-Alanyl Amidase, AmiA, ofHelicobacter pyloriin Peptidoglycan Metabolism, Daughter Cell Separation, and Virulence. Microb. Drug Resist. 2016, 22, 477–486. [Google Scholar] [CrossRef]
- Halloum, I.; Carrère-Kremer, S.; Blaise, M.; Viljoen, A.; Bernut, A.; Le Moigne, V.; Vilchèze, C.; Guérardel, Y.; Lutfalla, G.; Herrmann, J.-L.; et al. Deletion of a dehydratase important for intracellular growth and cording renders rough Mycobacterium abscessus avirulent. Proc. Natl. Acad. Sci. USA 2016, 113, E4228–E4237. [Google Scholar] [CrossRef]
- Bernut, A.; Viljoen, A.; Dupont, C.; Sapriel, G.; Blaise, M.; Bouchier, C.; Brosch, R.; De Chastellier, C.; Herrmann, J.L.; Kremer, L. Insights into the smooth-to-rough transitioning in Mycobacterium bolletii unravels a functional Tyr residue conserved in all mycobacterial MmpL family members. Mol. Microbiol. 2015, 99, 866–883. [Google Scholar] [CrossRef]
- Pantoliano, M.W.; Petrella, E.C.; Kwasnoski, J.D.; Lobanov, V.S.; Myslik, J.; Graf, E.; Carver, T.; Asel, E.; Springer, B.A.; Lane, P.; et al. High-Density Miniaturized Thermal Shift Assays as a General Strategy for Drug Discovery. J. Biomol. Screen. 2001, 6, 429–440. [Google Scholar] [CrossRef]
- Lo, M.-C.; Aulabaugh, A.; Jin, G.; Cowling, R.; Bard, J.; Malamas, M.; Ellestad, G. Evaluation of fluorescence-based thermal shift assays for hit identification in drug discovery. Anal. Biochem. 2004, 332, 153–159. [Google Scholar] [CrossRef]
- Tian, W.; Chen, C.; Lei, X.; Zhao, J.; Liang, J. CASTp 3.0: Computed atlas of surface topography of proteins. Nucleic Acids Res. 2018, 46, W363–W367. [Google Scholar] [CrossRef]
- Hugonnet, J.-E.; Tremblay, L.W.; Boshoff, H.I.; Barry, C.E.; Blanchard, J.S. Meropenem-Clavulanate Is Effective Against Extensively Drug-Resistant Mycobacterium tuberculosis. Science 2009, 323, 1215–1218. [Google Scholar] [CrossRef] [PubMed]
- Dubée, V.; Bernut, A.; Cortes, M.; Lesne, T.; Dorchene, D.; Lefebvre, A.-L.; Hugonnet, J.-E.; Gutmann, L.; Mainardi, J.-L.; Herrmann, J.-L.; et al. -Lactamase inhibition by avibactam in Mycobacterium abscessus. J. Antimicrob. Chemother. 2015, 70, 1051–1058. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, A.-L.; Le Moigne, V.; Bernut, A.; Veckerlé, C.; Compain, F.; Herrmann, J.-L.; Kremer, L.; Arthur, M.; Mainardi, J.-L. Inhibition of the β-Lactamase BlaMab by Avibactam Improves the In Vitro and In Vivo Efficacy of Imipenem against Mycobacterium abscessus. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [PubMed]
- Hett, E.C.; Rubin, E.J. Bacterial Growth and Cell Division: A Mycobacterial Perspective. Microbiol. Mol. Biol. Rev. 2008, 72, 126–156. [Google Scholar] [CrossRef]
- Böth, D.; Schneider, G.; Schnell, R. Peptidoglycan Remodeling in Mycobacterium tuberculosis: Comparison of Structures and Catalytic Activities of RipA and RipB. J. Mol. Biol. 2011, 413, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Ruggiero, A.; Marasco, D.; Squeglia, F.; Soldini, S.; Pedone, E.M.; Pedone, C.; Berisio, R. Structure and Functional Regulation of RipA, a Mycobacterial Enzyme Essential for Daughter Cell Separation. Structure 2010, 18, 1184–1190. [Google Scholar] [CrossRef]
- Mavrici, D.; Marakalala, M.J.; Holton, J.M.; Prigozhin, D.M.; Gee, C.L.; Zhang, Y.J.; Rubin, E.J.; Alber, T. Mycobacterium tuberculosis FtsX extracellular domain activates the peptidoglycan hydrolase, RipC. Proc. Natl. Acad. Sci. USA 2014, 111, 8037–8042. [Google Scholar] [CrossRef]
- Böth, D.; Steiner, E.-M.; Izumi, A.; Schneider, G.; Schnell, R. RipD (Rv1566c) from Mycobacterium tuberculosis: Adaptation of an NlpC/p60 domain to a non-catalytic peptidoglycan-binding function. Biochem. J. 2013, 457, 33–41. [Google Scholar] [CrossRef]
- Hett, E.C.; Chao, M.C.; Deng, L.; Rubin, E.J. A Mycobacterial Enzyme Essential for Cell Division Synergizes with Resuscitation-Promoting Factor. PLoS Pathog. 2008, 4, e1000001. [Google Scholar] [CrossRef]
- Martinelli, D.J.; Pavelka, J.M.S. The RipA and RipB Peptidoglycan Endopeptidases Are Individually Nonessential to Mycobacterium smegmatis. J. Bacteriol. 2016, 198, 1464–1475. [Google Scholar] [CrossRef]
- Lavollay, M.; Fourgeaud, M.; Herrmann, J.L.; Dubost, L.; Marie, A.; Gutmann, L.; Arthur, M.; Mainardi, J.-L. The Peptidoglycan of Mycobacterium abscessus Is Predominantly Cross-Linked by l,d-Transpeptidases. J. Bacteriol. 2010, 193, 778–782. [Google Scholar] [CrossRef]
- Botella, H.; Yang, G.; Ouerfelli, O.; Ehrt, S.; Nathan, C.F.; Vaubourgeix, J. Distinct Spatiotemporal Dynamics of Peptidoglycan Synthesis between Mycobacterium smegmatis and Mycobacterium tuberculosis. mBio 2017, 8. [Google Scholar] [CrossRef]
- Dubois, V.; Pawlik, A.; Bories, A.; Le Moigne, V.; Sismeiro, O.; Legendre, R.; Varet, H.; Rodríguez-Ordóñez, M.D.P.; Gaillard, J.-L.; Coppée, J.-Y.; et al. Mycobacterium abscessus virulence traits unraveled by transcriptomic profiling in amoeba and macrophages. PLoS Pathog. 2019, 15, e1008069. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| M. abscessus S WT | M. abscessus S Δami1 | M. abscessus S Δami1 Compl. | |
|---|---|---|---|
| Imipenem | 4 | 2 | 4 |
| Cefoxitine | 16 | 16 | 16 |
| Cefuroxime | 187–375 | 93 | 187 |
| Cefamandole | 2000 | 500–1000 | 2000 |
| Amikacin | 16 | 16 | 16 |
| Clofazimine | 0.78 | 0.78 | 0.78 |
| Tigecycline | 4 | 4 | 4 |
| Ami1Mab | Ami1Mab l-Ala-d-IsoGln | Ami1Msm | |
|---|---|---|---|
| PDB accession | 7AGL | 7AGO | 7AGM |
| Beamlime | ESRF-ID30B | SLS-PXIII-X06DA | SLS-PXIII-S06DA |
| Wavelength | 0.979 | 1 | 0.979 |
| Resolution range | 38.4–1.6 (1.65–1.6) | 42.8–1.7 (1.76–1.7) | 46.7–1.35 (1.39–1.35) |
| Space group | P 41 21 2 | P 41 21 2 | P 1 21 1 |
| Unit cell | 85.92 85.92 73.66 90 90 90 | 85.74 85.74 75.63 90 90 90 | 41.85 68.84 63.72 90 91.866 90 |
| Total reflections | 228,475 (22,850) | 824,584 (80,940) | 526,861 (52,052) |
| Multiplicity | 6.4 (6.3) | 26.1 (26.1) | 6.7 (6.6) |
| Completeness (%) | 96.7 (99.2) | 99.8 (100) | 99.9 (99.9) |
| Mean I/sigma(I) | 11.1 (1.3) | 18.8 (2.4) | 16.0 (1.3) |
| Wilson B-factor | 22.4 | 20.9 | 15.4 |
| R-meas | 0.094 (1.25) | 0.131 (1.29) | 0.069 (1.47) |
| CC1/2 | 0.99 (0.48) | 0.99 (0.85) | 0.99 (0.61) |
| Reflections used in refinement | 35,744 (3601) | 31,542 (3107) | 79,191 (7884) |
| Reflections used for R-free | 2000 (201) | 2000 (197) | 3960 (394) |
| R-work | 0.168 (0.294) | 0.162 (0.256) | 0.155 (0.285) |
| R-free | 0.195 (0.329) | 0.186 (0.307) | 0.180 (0.319) |
| Number of non-hydrogen atoms | 1831 | 1857 | 3672 |
| Macromolecules | 1598 | 1628 | 3218 |
| Ligands | 1 | 15 | 2 |
| Solvent | 232 | 242 | 452 |
| Protein residues | 212 | 214 | 434 |
| RMS (bonds, Å) | 0.007 | 0.006 | 0.005 |
| RMS (angles, °) | 0.89 | 0.79 | 0.77 |
| Ramachandran favored (%) | 97.14 | 97.17 | 97.91 |
| Ramachandran allowed (%) | 2.86 | 2.83 | 2.09 |
| Ramachandran outliers (%) | 0.00 | 0.00 | 0.00 |
| Rotamer outliers (%) | 1.18 | 1.18 | 0.30 |
| Clashscore | 0.63 | 1.55 | 1.72 |
| Average B-factor (Å2) | 27.6 | 22.6 | 23.2 |
| Macromolecules | 25.8 | 20.5 | 21.6 |
| Ligands | 20.4 | 33.4 | 18.3 |
| Solvent | 40.1 | 35.8 | 34.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Küssau, T.; Van Wyk, N.; Johansen, M.D.; Alsarraf, H.M.A.B.; Neyret, A.; Hamela, C.; Sørensen, K.K.; Thygesen, M.B.; Beauvineau, C.; Kremer, L.; et al. Functional Characterization of the N-Acetylmuramyl-l-Alanine Amidase, Ami1, from Mycobacterium abscessus. Cells 2020, 9, 2410. https://doi.org/10.3390/cells9112410
Küssau T, Van Wyk N, Johansen MD, Alsarraf HMAB, Neyret A, Hamela C, Sørensen KK, Thygesen MB, Beauvineau C, Kremer L, et al. Functional Characterization of the N-Acetylmuramyl-l-Alanine Amidase, Ami1, from Mycobacterium abscessus. Cells. 2020; 9(11):2410. https://doi.org/10.3390/cells9112410
Chicago/Turabian StyleKüssau, Tanja, Niël Van Wyk, Matt D. Johansen, Husam M. A. B. Alsarraf, Aymeric Neyret, Claire Hamela, Kasper K. Sørensen, Mikkel B. Thygesen, Claire Beauvineau, Laurent Kremer, and et al. 2020. "Functional Characterization of the N-Acetylmuramyl-l-Alanine Amidase, Ami1, from Mycobacterium abscessus" Cells 9, no. 11: 2410. https://doi.org/10.3390/cells9112410
APA StyleKüssau, T., Van Wyk, N., Johansen, M. D., Alsarraf, H. M. A. B., Neyret, A., Hamela, C., Sørensen, K. K., Thygesen, M. B., Beauvineau, C., Kremer, L., & Blaise, M. (2020). Functional Characterization of the N-Acetylmuramyl-l-Alanine Amidase, Ami1, from Mycobacterium abscessus. Cells, 9(11), 2410. https://doi.org/10.3390/cells9112410