COVID-19-Associated Neurological Disorders: The Potential Route of CNS Invasion and Blood-Brain Barrier Relevance



Abstract
1. Introduction
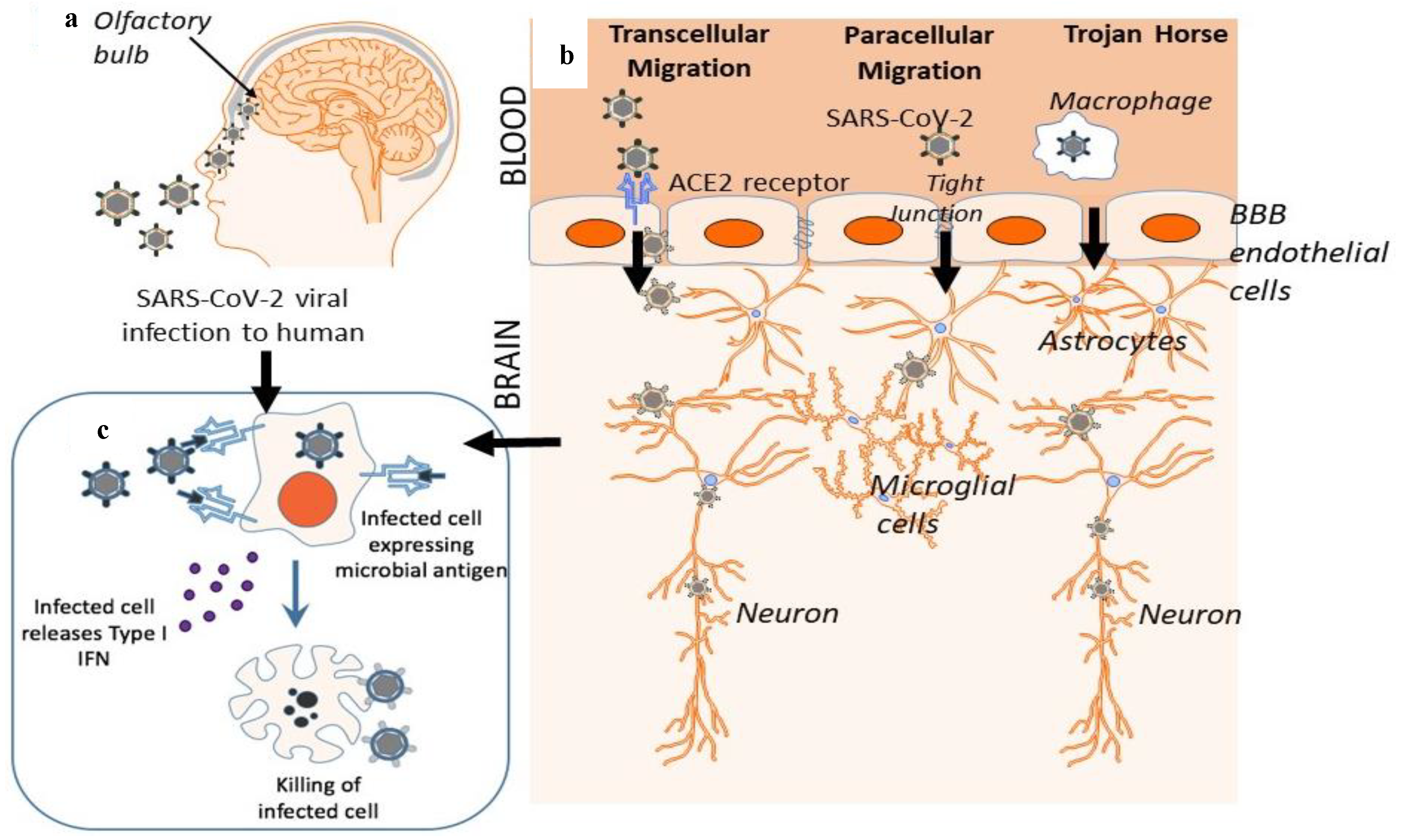
2. Gateway to the Brain: The Blood-Brain Barrier as a Possible Entry Route
3. Mechanism of Cellular Viral Invasion: ACE2 Receptor
4. Cytokine Storm on the Blood-Brain Barrier: A Malfunction of the Immune System

{kind=link}
| Cytokine | General Functions |
|---|---|
| Pro-inflammatory | |
| Tumor necrosis factor alpha (TNF-α) | Activation of neutrophils and platelets; enhances macrophage and natural killer cell effector function; anti-malignant cell cytotoxicity; necrosis and apoptosis [60]. |
| Interferon gamma (IFN-γ) | Th1 cell differentiation; activation of macrophages; upregulation of class I and II major histocompatibility complex and antigen presentation; specific cytotoxic immunity; induction of antiviral enzymes; cell growth inhibition [61]. |
| Interleukin-2 (IL-2) | T cell proliferation; long-term survival of T cells; development of T regulatory cells; NK cell growth factor; enhances NK cell cytotoxicity; antibody secretion; upregulation of B cell heavy and light chain gene expression [62]. |
| Interleukin-6 (IL-6) | T cell growth; CD8+ T cell proliferation; differentiation of macrophages, megakaryocytes, and osteoclasts; stimulation of B cells to produce immunoglobulins [63]. |
| Interleukin-8 (IL-8) | Recruitment and activation of neutrophils, increased expression of adhesion molecules; wound healing (stimulates migration and differentiation of fibroblasts); enhances metabolism of reactive oxygen species [64]. |
| Anti-inflammatory | |
| Interleukin-4 (IL-4) | Th2 cell differentiation; immunoglobulin class switch to IgG1 and IgE; activation of alternative macrophages [65]. |
| Interleukin-10 (IL-10) | Inhibition of pro-inflammatory cytokine production; downregulation of MHC molecules; B cell, mast cell, and thymocyte growth factor [66]. |
5. Neurological Manifestations: Case Studies
5.1. Ischaemic Stroke
5.2. Encephalitis
5.3. Other Encephalopathies
5.4. Epilepsy
5.5. Neurodegenerative Disease and Inflammatory-Mediated Neurological Disorder
5.5.1. Multiple Sclerosis
5.5.2. Parkinson’s Disease
5.5.3. Alzheimer’s Disease
6. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Pneumonia of unknown cause – China. Available online: https://www.who.int/csr/don/05-january-2020-pneumonia-of-unkown-cause-china/en/ (accessed on 3 June 2020).
- World map. Available online: https://www.cdc.gov/coronavirus/2019-ncov/global-covid-19/world-map.html (accessed on 3 June 2020).
- Cucinotta, D.; Vanelli, M. WHO Declares COVID-19 a Pandemic. Acta Biomed. 2020, 91, 157–160. [Google Scholar] [CrossRef]
- Weekly Operational Update on COVID-19. Available online: https://www.who.int/docs/default-source/coronaviruse/situation-reports/wou-4-september-2020-approved.pdf?sfvrsn=91215c78_2 (accessed on 4 September 2020).
- Ye, Z.W.; Yuan, S.; Yuen, K.S.; Fung, S.Y.; Chan, C.P.; Jin, D.Y. Zoonotic origins of human coronaviruses. Int. J. Biol. Sci. 2020, 16, 1686–1697. [Google Scholar] [CrossRef]
- Matoba, Y.; Abiko, C.; Ikeda, T.; Aoki, Y.; Suzuki, Y.; Yahagi, K.; Matsuzaki, Y.; Itagaki, T.; Katsushima, F.; Katsushima, Y.; et al. Detection of the human coronavirus 229E, HKU1, NL63, and OC43 between 2010 and 2013 in Yamagata, Japan. Jpn. J. Infect. Dis. 2015, 68, 138–141. [Google Scholar] [CrossRef]
- Tse, G.M.; To, K.F.; Chan, P.K.; Lo, A.W.; Ng, K.C.; Wu, A.; Lee, N.; Wong, H.C.; Mak, S.M.; Chan, K.F.; et al. Pulmonary pathological features in coronavirus associated severe acute respiratory syndrome (SARS). J. Clin. Pathol. 2004, 57, 260–265. [Google Scholar] [CrossRef]
- Widagdo, W.; Raj, V.S.; Schipper, D.; Kolijn, K.; van Leenders, G.; Bosch, B.J.; Bensaid, A.; Segales, J.; Baumgartner, W.; Osterhaus, A.; et al. Differential Expression of the Middle East Respiratory Syndrome Coronavirus Receptor in the Upper Respiratory Tracts of Humans and Dromedary Camels. J. Virol. 2016, 90, 4838–4842. [Google Scholar] [CrossRef]
- Wolfel, R.; Corman, V.M.; Guggemos, W.; Seilmaier, M.; Zange, S.; Muller, M.A.; Niemeyer, D.; Jones, T.C.; Vollmar, P.; Rothe, C.; et al. Virological assessment of hospitalized patients with COVID-2019. Nature 2020, 581, 465–469. [Google Scholar] [CrossRef]
- Symptoms of Coronavirus. Available online: https://www.cdc.gov/coronavirus/2019-ncov/symptoms-testing/symptoms.html (accessed on 29 May 2020).
- What Coronavirus does to the Lungs. Available online: https://www.hopkinsmedicine.org/health/conditions-and-diseases/coronavirus/what-coronavirus-does-to-the-lungs (accessed on 3 June 2020).
- People Who are at Increased Risk for Severe Illness. Available online: https://www.cdc.gov/coronavirus/2019-ncov/need-extra-precautions/people-at-increased-risk.html?CDC_AA_refVal=https%3A%2F%2Fwww.cdc.gov%2Fcoronavirus%2F2019-ncov%2Fneed-extra-precautions%2Fpeople-at-higher-risk.html (accessed on 25 June 2020).
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef]
- Zou, X.; Chen, K.; Zou, J.; Han, P.; Hao, J.; Han, Z. Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front. Med. 2020, 14, 185–192. [Google Scholar] [CrossRef]
- Mao, L.; Jin, H.; Wang, M.; Hu, Y.; Chen, S.; He, Q.; Chang, J.; Hong, C.; Zhou, Y.; Wang, D.; et al. Neurologic Manifestations of Hospitalized Patients With Coronavirus Disease 2019 in Wuhan, China. JAMA Neurol. 2020. [Google Scholar] [CrossRef]
- Varatharaj, A.; Thomas, N.; Ellul, M.A.; Davies, N.W.S.; Pollak, T.A.; Tenorio, E.L.; Sultan, M.; Easton, A.; Breen, G.; Zandi, M.; et al. Neurological and neuropsychiatric complications of COVID-19 in 153 patients: A UK-wide surveillance study. Lancet Psychiatry 2020. [Google Scholar] [CrossRef]
- Paterson, R.W.; Brown, R.L.; Benjamin, L.; Nortley, R.; Wiethoff, S.; Bharucha, T.; Jayaseelan, D.L.; Kumar, G.; Raftopoulos, R.E.; Zambreanu, L.; et al. The emerging spectrum of COVID-19 neurology: Clinical, radiological and laboratory findings. Brain 2020. [Google Scholar] [CrossRef]
- Tsivgoulis, G.; Palaiodimou, L.; Katsanos, A.H.; Caso, V.; Kohrmann, M.; Molina, C.; Cordonnier, C.; Fischer, U.; Kelly, P.; Sharma, V.K.; et al. Neurological manifestations and implications of COVID-19 pandemic. Adv. Neurol. Disord. 2020. [Google Scholar] [CrossRef]
- Helms, J.; Kremer, S.; Merdji, H.; Clere-Jehl, R.; Schenck, M.; Kummerlen, C.; Collange, O.; Boulay, C.; Fafi-Kremer, S.; Ohana, M.; et al. Neurologic Features in Severe SARS-CoV-2 Infection. N. Engl. J. Med. 2020, 382, 2268–2270. [Google Scholar] [CrossRef]
- Briguglio, M.; Bona, A.; Porta, M.; Dell’Osso, B.; Pregliasco, F.E.; Banfi, G. Disentangling the Hypothesis of Host Dysosmia and SARS-CoV-2: The Bait Symptom That Hides Neglected Neurophysiological Routes. Front. Physiol. 2020, 11. [Google Scholar] [CrossRef]
- Eliezer, M.; Hautefort, C.; Hamel, A.L.; Verillaud, B.; Herman, P.; Houdart, E.; Eloit, C. Sudden and Complete Olfactory Loss Function as a Possible Symptom of COVID-19. JAMA Otolaryngol. Head Neck Surg. 2020. [Google Scholar] [CrossRef]
- Klopfenstein, T.; Kadiane-Oussou, N.J.; Toko, L.; Royer, P.Y.; Lepiller, Q.; Gendrin, V.; Zayet, S. Features of anosmia in COVID-19. Med. Mal. Infect. 2020, 50, 436–439. [Google Scholar] [CrossRef]
- Meng, X.; Deng, Y.; Dai, Z.; Meng, Z. COVID-19 and anosmia: A review based on up-to-date knowledge. Am. J. Otolaryngol. 2020, 41. [Google Scholar] [CrossRef]
- Brann, D.H.; Tsukahara, T.; Weinreb, C.; Lipovsek, M.; Van den Berge, K.; Gong, B.; Chance, R.; Macaulay, I.C.; Chou, H.-J.; Fletcher, R.B.; et al. Non-neuronal expression of SARS-CoV-2 entry genes in the olfactory system suggests mechanisms underlying COVID-19-associated anosmia. Sci. Adv. 2020, 6. [Google Scholar] [CrossRef]
- Baig, A.M. Neurological manifestations in COVID-19 caused by SARS-CoV-2. Cns Neurosci. 2020, 26, 499–501. [Google Scholar] [CrossRef]
- Abbott, N.J. Astrocyte-endothelial interactions and blood-brain barrier permeability. J. Anat. 2002, 200, 629–638. [Google Scholar] [CrossRef]
- Abbott, N.J.; Ronnback, L.; Hansson, E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci 2006, 7, 41–53. [Google Scholar] [CrossRef]
- Daneman, R.; Prat, A. The blood-brain barrier. Cold Spring Harb Perspect Biol. 2015, 7. [Google Scholar] [CrossRef]
- Pardridge, W.M. The blood-brain barrier: Bottleneck in brain drug development. NeuroRx 2005, 2, 3–14. [Google Scholar] [CrossRef]
- Banjara, M.; Ghosh, C. Sterile Neuroinflammation and Strategies for Therapeutic Intervention. Int. J. Inflam 2017, 2017. [Google Scholar] [CrossRef]
- Williams, S.; Ghosh, C. Neurovascular glucocorticoid receptors and glucocorticoids: Implications in health, neurological disorders and drug therapy. Drug Discov. Today 2020, 25, 89–106. [Google Scholar] [CrossRef]
- Williams, S.; Hossain, M.; Mishra, S.; Gonzalez-Martinez, J.; Najm, I.; Ghosh, C. Expression and Functional Relevance of Death-Associated Protein Kinase in Human Drug-Resistant Epileptic Brain: Focusing on the Neurovascular Interface. Mol. Neurobiol. 2019, 56, 4904–4915. [Google Scholar] [CrossRef]
- Dahm, T.; Rudolph, H.; Schwerk, C.; Schroten, H.; Tenenbaum, T. Neuroinvasion and Inflammation in Viral Central Nervous System Infections. Mediat. Inflamm. 2016, 2016. [Google Scholar] [CrossRef]
- Robinson, C.P.; Busl, K.M. Neurologic Manifestations of Severe Respiratory Viral Contagions. Crit. Care Explor. 2020, 2. [Google Scholar] [CrossRef]
- Zhu, X.; Liu, Q.; Du, L.; Lu, L.; Jiang, S. Receptor-binding domain as a target for developing SARS vaccines. J. Thorac Dis. 2013, 5 (Suppl. 2), S142–S148. [Google Scholar] [CrossRef]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.Y.; et al. Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2. Cell 2020, 181, 894–904.e899. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e278. [Google Scholar] [CrossRef] [PubMed]
- Pellett, P.E.; Mitra, S.; Holland, T.C. Basics of virology. Handb Clin. Neurol. 2014, 123, 45–66. [Google Scholar] [CrossRef] [PubMed]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.M.; An, J. Cytokines, inflammation, and pain. Int. Anesth. Clin. 2007, 45, 27–37. [Google Scholar] [CrossRef]
- Mahallawi, W.H.; Khabour, O.F.; Zhang, Q.; Makhdoum, H.M.; Suliman, B.A. MERS-CoV infection in humans is associated with a pro-inflammatory Th1 and Th17 cytokine profile. Cytokine 2018, 104, 8–13. [Google Scholar] [CrossRef]
- Wong, C.K.; Lam, C.W.; Wu, A.K.; Ip, W.K.; Lee, N.L.; Chan, I.H.; Lit, L.C.; Hui, D.S.; Chan, M.H.; Chung, S.S.; et al. Plasma inflammatory cytokines and chemokines in severe acute respiratory syndrome. Clin. Exp. Immunol. 2004, 136, 95–103. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Han, H.; Ma, Q.; Li, C.; Liu, R.; Zhao, L.; Wang, W.; Zhang, P.; Liu, X.; Gao, G.; Liu, F.; et al. Profiling serum cytokines in COVID-19 patients reveals IL-6 and IL-10 are disease severity predictors. Emerg Microbes Infect. 2020, 9, 1123–1130. [Google Scholar] [CrossRef]
- Qin, C.; Zhou, L.; Hu, Z.; Zhang, S.; Yang, S.; Tao, Y.; Xie, C.; Ma, K.; Shang, K.; Wang, W.; et al. Dysregulation of Immune Response in Patients With Coronavirus 2019 (COVID-19) in Wuhan, China. Clin. Infect. Dis. 2020, 71, 762–768. [Google Scholar] [CrossRef]
- Liu, J.; Li, S.; Liu, J.; Liang, B.; Wang, X.; Wang, H.; Li, W.; Tong, Q.; Yi, J.; Zhao, L.; et al. Longitudinal characteristics of lymphocyte responses and cytokine profiles in the peripheral blood of SARS-CoV-2 infected patients. EBioMedicine 2020, 55. [Google Scholar] [CrossRef]
- Merad, M.; Martin, J.C. Pathological inflammation in patients with COVID-19: A key role for monocytes and macrophages. Nat. Rev. Immunol. 2020, 20, 355–362. [Google Scholar] [CrossRef]
- Del Valle, D.M.; Kim-Schulze, S.; Huang, H.H.; Beckmann, N.D.; Nirenberg, S.; Wang, B.; Lavin, Y.; Swartz, T.H.; Madduri, D.; Stock, A.; et al. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Mandel, M.; Harari, G.; Gurevich, M.; Achiron, A. Cytokine prediction of mortality in COVID19 patients. Cytokine 2020, 134. [Google Scholar] [CrossRef]
- Erta, M.; Quintana, A.; Hidalgo, J. Interleukin-6, a major cytokine in the central nervous system. Int. J. Biol. Sci. 2012, 8, 1254–1266. [Google Scholar] [CrossRef] [PubMed]
- Rochfort, K.D.; Collins, L.E.; Murphy, R.P.; Cummins, P.M. Downregulation of blood-brain barrier phenotype by proinflammatory cytokines involves NADPH oxidase-dependent ROS generation: Consequences for interendothelial adherens and tight junctions. PLoS ONE 2014, 9, e101815. [Google Scholar] [CrossRef]
- Zhang, J.; Sadowska, G.B.; Chen, X.; Park, S.Y.; Kim, J.E.; Bodge, C.A.; Cummings, E.; Lim, Y.P.; Makeyev, O.; Besio, W.G.; et al. Anti-IL-6 neutralizing antibody modulates blood-brain barrier function in the ovine fetus. FASEB J. 2015, 29, 1739–1753. [Google Scholar] [CrossRef]
- Benameur, K.; Agarwal, A.; Auld, S.C.; Butters, M.P.; Webster, A.S.; Ozturk, T.; Howell, J.C.; Bassit, L.C.; Velasquez, A.; Schinazi, R.F.; et al. Encephalopathy and Encephalitis Associated with Cerebrospinal Fluid Cytokine Alterations and Coronavirus Disease, Atlanta, Georgia, USA, 2020. Emerg. Infect. Dis. 2020, 26. [Google Scholar] [CrossRef]
- Ehrlich, L.C.; Hu, S.; Sheng, W.S.; Sutton, R.L.; Rockswold, G.L.; Peterson, P.K.; Chao, C.C. Cytokine regulation of human microglial cell IL-8 production. J. Immunol. 1998, 160, 1944–1948. [Google Scholar]
- Didangelos, A. COVID-19 Hyperinflammation: What about Neutrophils? mSphere 2020, 5. [Google Scholar] [CrossRef]
- Steardo, L.; Steardo, L., Jr.; Zorec, R.; Verkhratsky, A. Neuroinfection may contribute to pathophysiology and clinical manifestations of COVID-19. Acta Physiol. Oxf. 2020, 229, e13473. [Google Scholar] [CrossRef]
- Varatharaj, A.; Galea, I. The blood-brain barrier in systemic inflammation. Brain Behav. Immun. 2017, 60, 1–12. [Google Scholar] [CrossRef]
- Idriss, H.T.; Naismith, J.H. TNF alpha and the TNF receptor superfamily: Structure-function relationship(s). Microsc Res. Tech. 2000, 50, 184–195. [Google Scholar] [CrossRef]
- Schroder, K.; Hertzog, P.J.; Ravasi, T.; Hume, D.A. Interferon-gamma: An overview of signals, mechanisms and functions. J. Leukoc Biol. 2004, 75, 163–189. [Google Scholar] [CrossRef]
- Gaffen, S.L.; Liu, K.D. Overview of interleukin-2 function, production and clinical applications. Cytokine 2004, 28, 109–123. [Google Scholar] [CrossRef]
- Naka, T.; Nishimoto, N.; Kishimoto, T. The paradigm of IL-6: From basic science to medicine. Arthritis Res. 2002, 4 (Suppl. 3), S233–S242. [Google Scholar] [CrossRef]
- Qazi, B.S.; Tang, K.; Qazi, A. Recent advances in underlying pathologies provide insight into interleukin-8 expression-mediated inflammation and angiogenesis. Int. J. Inflam 2011, 2011. [Google Scholar] [CrossRef]
- Junttila, I.S. Tuning the Cytokine Responses: An Update on Interleukin (IL)-4 and IL-13 Receptor Complexes. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Walter, M.R. The molecular basis of IL-10 function: From receptor structure to the onset of signaling. Curr. Top. Microbiol. Immunol. 2014, 380, 191–212. [Google Scholar] [CrossRef]
- Sacco, R.L.; Kasner, S.E.; Broderick, J.P.; Caplan, L.R.; Connors, J.J.; Culebras, A.; Elkind, M.S.; George, M.G.; Hamdan, A.D.; Higashida, R.T.; et al. An updated definition of stroke for the 21st century: A statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2013, 44, 2064–2089. [Google Scholar] [CrossRef]
- Hui, C.; Tadi, P.; Patti, L. Ischemic Stroke. In StatPearls, Treasure Island (FL). 2020. Available online: https://www.ncbi.nlm.nih.gov/books/NBK499997/ (accessed on 27 October 2020).
- Zhai, P.; Ding, Y.; Li, Y. The impact of COVID-19 on ischemic stroke. Diagn Pathol. 2020, 15, 78. [Google Scholar] [CrossRef]
- Avula, A.; Nalleballe, K.; Narula, N.; Sapozhnikov, S.; Dandu, V.; Toom, S.; Glaser, A.; Elsayegh, D. COVID-19 presenting as stroke. Brain Behav. Immun. 2020, 87, 115–119. [Google Scholar] [CrossRef]
- Beyrouti, R.; Adams, M.E.; Benjamin, L.; Cohen, H.; Farmer, S.F.; Goh, Y.Y.; Humphries, F.; Jager, H.R.; Losseff, N.A.; Perry, R.J.; et al. Characteristics of ischaemic stroke associated with COVID-19. J. Neurol. Neurosurg Psychiatry 2020. [Google Scholar] [CrossRef]
- Oxley, T.J.; Mocco, J.; Majidi, S.; Kellner, C.P.; Shoirah, H.; Singh, I.P.; De Leacy, R.A.; Shigematsu, T.; Ladner, T.R.; Yaeger, K.A.; et al. Large-Vessel Stroke as a Presenting Feature of Covid-19 in the Young. N Engl. J. Med. 2020, 382, e60. [Google Scholar] [CrossRef]
- Jain, R.; Young, M.; Dogra, S.; Kennedy, H.; Nguyen, V.; Jones, S.; Bilaloglu, S.; Hochman, K.; Raz, E.; Galetta, S.; et al. COVID-19 related neuroimaging findings: A signal of thromboembolic complications and a strong prognostic marker of poor patient outcome. J. Neurol. Sci. 2020, 414. [Google Scholar] [CrossRef]
- Basi, S.; Hamdan, M.; Punekar, S. Clinical course of a 66-year-old man with an acute ischaemic stroke in the setting of a COVID-19 infection. Bmj Case Rep. 2020, 13. [Google Scholar] [CrossRef]
- Diaz-Segarra, N.; Edmond, A.; Kunac, A.; Yonclas, P. COVID-19 Ischemic Strokes as an Emerging Rehabilitation Population: A Case Series. Am. J. Phys. Med. Rehabil 2020. [Google Scholar] [CrossRef]
- Bhatia, R.; Srivastava, M.V.P. COVID-19 and Stroke: Incidental, Triggered or Causative. Ann. Indian Acad. Neurol. 2020, 23, 318–324. [Google Scholar] [CrossRef]
- Yaghi, S.; Ishida, K.; Torres, J.; Mac Grory, B.; Raz, E.; Humbert, K.; Henninger, N.; Trivedi, T.; Lillemoe, K.; Alam, S.; et al. SARS-CoV-2 and Stroke in a New York Healthcare System. Stroke 2020, 51, 2002–2011. [Google Scholar] [CrossRef]
- Spence, J.D.; de Freitas, G.R.; Pettigrew, L.C.; Ay, H.; Liebeskind, D.S.; Kase, C.S.; Del Brutto, O.H.; Hankey, G.J.; Venketasubramanian, N. Mechanisms of Stroke in COVID-19. Cereb. Dis. 2020, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Cao, J.; Wang, Q.; Shi, Q.; Liu, K.; Luo, Z.; Chen, X.; Chen, S.; Yu, K.; Huang, Z.; et al. D-dimer as a biomarker for disease severity and mortality in COVID-19 patients: A case control study. J. Intensive Care 2020, 8, 49. [Google Scholar] [CrossRef]
- Yu, H.H.; Qin, C.; Chen, M.; Wang, W.; Tian, D.S. D-dimer level is associated with the severity of COVID-19. Thromb Res. 2020, 195, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yan, X.; Fan, Q.; Liu, H.; Liu, X.; Liu, Z.; Zhang, Z. D-dimer levels on admission to predict in-hospital mortality in patients with Covid-19. J Thromb Haemost 2020, 18, 1324–1329. [Google Scholar] [CrossRef]
- Ellul, M.; Solomon, T. Acute encephalitis - diagnosis and management. Clin. Med. Lond. 2018, 18, 155–159. [Google Scholar] [CrossRef]
- Encephalitis. Available online: https://www.cancertherapyadvisor.com/home/decision-support-in-medicine/pediatrics/encephalitis-5/ (accessed on 15 October 2020).
- Moriguchi, T.; Harii, N.; Goto, J.; Harada, D.; Sugawara, H.; Takamino, J.; Ueno, M.; Sakata, H.; Kondo, K.; Myose, N.; et al. A first case of meningitis/encephalitis associated with SARS-Coronavirus-2. Int. J. Infect. Dis. 2020, 94, 55–58. [Google Scholar] [CrossRef] [PubMed]
- Ye, M.; Ren, Y.; Lv, T. Encephalitis as a clinical manifestation of COVID-19. Brain Behav. Immun. 2020. [Google Scholar] [CrossRef]
- Wong, P.F.; Craik, S.; Newman, P.; Makan, A.; Srinivasan, K.; Crawford, E.; Dev, D.; Moudgil, H.; Ahmad, N. Lessons of the month 1: A case of rhombencephalitis as a rare complication of acute COVID-19 infection. Clin. Med. Lond. 2020. [Google Scholar] [CrossRef]
- Zuhorn, F.; Omaimen, H.; Ruprecht, B.; Stellbrink, C.; Rauch, M.; Rogalewski, A.; Klingebiel, R.; Schabitz, W.R. Parainfectious encephalitis in COVID-19: “The Claustrum Sign”. J. Neurol. 2020. [Google Scholar] [CrossRef]
- Zambreanu, L.; Lightbody, S.; Bhandari, M.; Hoskote, C.; Kandil, H.; Houlihan, C.F.; Lunn, M.P. A case of limbic encephalitis associated with asymptomatic COVID-19 infection. J. Neurol. Neurosurg Psychiatry 2020. [Google Scholar] [CrossRef]
- Khoo, A.; McLoughlin, B.; Cheema, S.; Weil, R.S.; Lambert, C.; Manji, H.; Zandi, M.S.; Morrow, J.M. Postinfectious brainstem encephalitis associated with SARS-CoV-2. J. Neurol. Neurosurg Psychiatry 2020, 91, 1013–1014. [Google Scholar] [CrossRef]
- Pilotto, A.; Padovani, A.; Network, E.-B. Reply to the Letter “COVID-19-Associated Encephalopathy and Cytokine-Mediated Neuroinflammation”. Ann. Neurol. 2020. [Google Scholar] [CrossRef]
- Neumann, B.; Schmidbauer, M.L.; Dimitriadis, K.; Otto, S.; Knier, B.; Niesen, W.D.; Hosp, J.A.; Gunther, A.; Lindemann, S.; Nagy, G.; et al. Cerebrospinal fluid findings in COVID-19 patients with neurological symptoms. J. Neurol. Sci. 2020, 418. [Google Scholar] [CrossRef]
- Pilotto, A.; Masciocchi, S.; Volonghi, I.; del Zotto, E.; Magni, E.; De Giuli, V.; Caprioli, F.; Rifino, N.; Sessa, M.; Gennuso, M.; et al. The clinical spectrum of encephalitis in COVID-19 disease: The ENCOVID multicentre study. Preprint 2020. [Google Scholar] [CrossRef]
- Eden, A.; Kanberg, N.; Gostner, J.; Fuchs, D.; Hagberg, L.; Andersson, L.M.; Lindh, M.; Price, R.W.; Zetterberg, H.; Gisslen, M. CSF biomarkers in patients with COVID-19 and neurological symptoms: A case series. Neurology 2020. [Google Scholar] [CrossRef] [PubMed]
- Platt, M.P.; Agalliu, D.; Cutforth, T. Hello from the Other Side: How Autoantibodies Circumvent the Blood-Brain Barrier in Autoimmune Encephalitis. Front. Immunol. 2017, 8, 442. [Google Scholar] [CrossRef]
- Pilotto, A.; Odolini, S.; Masciocchi, S.; Comelli, A.; Volonghi, I.; Gazzina, S.; Nocivelli, S.; Pezzini, A.; Foca, E.; Caruso, A.; et al. Steroid-Responsive Encephalitis in Coronavirus Disease 2019. Ann. Neurol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Ruzek, D.; Salat, J.; Singh, S.K.; Kopecky, J. Breakdown of the blood-brain barrier during tick-borne encephalitis in mice is not dependent on CD8+ T-cells. PLoS ONE 2011, 6, e20472. [Google Scholar] [CrossRef]
- Li, F.; Wang, Y.; Yu, L.; Cao, S.; Wang, K.; Yuan, J.; Wang, C.; Wang, K.; Cui, M.; Fu, Z.F. Viral Infection of the Central Nervous System and Neuroinflammation Precede Blood-Brain Barrier Disruption during Japanese Encephalitis Virus Infection. J. Virol. 2015, 89, 5602–5614. [Google Scholar] [CrossRef]
- McCuddy, M.; Kelkar, P.; Zhao, Y.; Wicklund, D. Acute Demyelinating Encephalomyelitis (ADEM) in COVID-19 infection: A Case Series. Preprint 2020. [Google Scholar] [CrossRef]
- Novi, G.; Rossi, T.; Pedemonte, E.; Saitta, L.; Rolla, C.; Roccatagliata, L.; Inglese, M.; Farinini, D. Acute disseminated encephalomyelitis after SARS-CoV-2 infection. Neurol. Neuroimmunol. Neuroinflamm 2020, 7. [Google Scholar] [CrossRef]
- Utukuri, P.S.; Bautista, A.; Lignelli, A.; Moonis, G. Possible Acute Disseminated Encephalomyelitis Related to Severe Acute Respiratory Syndrome Coronavirus 2 Infection. Ajnr Am. J. Neuroradiol. 2020, 41, 82–83. [Google Scholar] [CrossRef]
- Palao, M.; Fernandez-Diaz, E.; Gracia-Gil, J.; Romero-Sanchez, C.M.; Diaz-Maroto, I.; Segura, T. Multiple sclerosis following SARS-CoV-2 infection. Mult. Scler. Relat. Disord. 2020, 45. [Google Scholar] [CrossRef]
- Perugula, M.L.; Lippmann, S. Encephalopathy or Psychosis? Innov. Clin. Neurosci. 2016, 13, 41–42. [Google Scholar]
- Poyiadji, N.; Shahin, G.; Noujaim, D.; Stone, M.; Patel, S.; Griffith, B. COVID-19-associated Acute Hemorrhagic Necrotizing Encephalopathy: Imaging Features. Radiology 2020, 296, 119–120. [Google Scholar] [CrossRef]
- Kihira, S.; Delman, B.N.; Belani, P.; Stein, L.; Aggarwal, A.; Rigney, B.; Schefflein, J.; Doshi, A.H.; Pawha, P.S. Imaging Features of Acute Encephalopathy in Patients with COVID-19: A Case Series. Ajnr Am. J. Neuroradiol. 2020, 41, 1804–1808. [Google Scholar] [CrossRef]
- Al Mazrouei, S.S.; Saeed, G.A.; Al Helali, A.A.; Ahmed, M. COVID-19-associated encephalopathy: Neurological manifestation of COVID-19. Radiol. Case Rep. 2020, 15, 1646–1649. [Google Scholar] [CrossRef]
- Krett, J.D.; Jewett, G.A.E.; Elton-Lacasse, C.; Fonseca, K.; Hahn, C.; Au, S.; Koch, M.W. Hemorrhagic encephalopathy associated with COVID-19. J. Neuroimmunol. 2020, 346. [Google Scholar] [CrossRef]
- Umapathi, T.; Jason, Q.W.M.; Min, Y.J.; Wai, K.H.S.; Yuan, M.Y.; Yee, J.C.C.; Min, L.L.; Wai-Yung, Y. Encephalopathy in COVID-19 patients; viral, parainfectious, or both? eNeurologicalSci 2020. [Google Scholar] [CrossRef]
- Farhadian, S.; Glick, L.R.; Vogels, C.B.F.; Thomas, J.; Chiarella, J.; Casanovas-Massana, A.; Zhou, J.; Odio, C.; Vijayakumar, P.; Geng, B.; et al. Acute encephalopathy with elevated CSF inflammatory markers as the initial presentation of COVID-19. BMC Neurol. 2020, 20, 248. [Google Scholar] [CrossRef]
- Stafstrom, C.E.; Carmant, L. Seizures and epilepsy: An overview for neuroscientists. Cold Spring Harb. Perspect Med. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Scharfman, H.E. The neurobiology of epilepsy. Curr. Neurol. Neurosci. Rep. 2007, 7, 348–354. [Google Scholar] [CrossRef]
- Sirven, J.I. Epilepsy: A Spectrum Disorder. Cold Spring Harb. Perspect. Med. 2015, 5, a022848. [Google Scholar] [CrossRef]
- Vollono, C.; Rollo, E.; Romozzi, M.; Frisullo, G.; Servidei, S.; Borghetti, A.; Calabresi, P. Focal status epilepticus as unique clinical feature of COVID-19: A case report. Seizure 2020, 78, 109–112. [Google Scholar] [CrossRef]
- Karimi, N.; Razavi, A.S.; Rouhani, N. Frequent Convulsive Seizures in an Adult Patient with COVID-19: A Case Report. Iran. Crescent Med J. 2020, 22. [Google Scholar] [CrossRef]
- Hepburn, M.; Mullaguri, N.; George, P.; Hantus, S.; Punia, V.; Bhimraj, A.; Newey, C.R. Acute Symptomatic Seizures in Critically Ill Patients with COVID-19: Is There an Association? Neurocrit Care 2020. [Google Scholar] [CrossRef]
- Lyons, S.; O’Kelly, B.; Woods, S.; Rowan, C.; Brady, D.; Sheehan, G.; Smyth, S. Seizure with CSF lymphocytosis as a presenting feature of COVID-19 in an otherwise healthy young man. Seizure 2020, 80, 113–114. [Google Scholar] [CrossRef]
- Kadono, Y.; Nakamura, Y.; Ogawa, Y.; Yamamoto, S.; Kajikawa, R.; Nakajima, Y.; Matsumoto, M.; Kishima, H. A case of COVID-19 infection presenting with a seizure following severe brain edema. Seizure 2020, 80, 53–55. [Google Scholar] [CrossRef]
- Anand, P.; Al-Faraj, A.; Sader, E.; Dashkoff, J.; Abdennadher, M.; Murugesan, R.; Cervantes-Arslanian, A.M.; Daneshmand, A. Seizure as the presenting symptom of COVID-19: A retrospective case series. Epilepsy Behav. 2020, 112. [Google Scholar] [CrossRef]
- Asadi-Pooya, A.A. Seizures associated with coronavirus infections. Seizure 2020, 79, 49–52. [Google Scholar] [CrossRef] [PubMed]
- Obermeier, B.; Daneman, R.; Ransohoff, R.M. Development, maintenance and disruption of the blood-brain barrier. Nat. Med. 2013, 19, 1584–1596. [Google Scholar] [CrossRef]
- Fieschi, C.; Lenzi, G.L.; Zanette, E.; Orzi, F.; Passero, S. Effects on EEG of the osmotic opening of the blood-brain barrier in rats. Life Sci. 1980, 27, 239–243. [Google Scholar] [CrossRef]
- Marchi, N.; Angelov, L.; Masaryk, T.; Fazio, V.; Granata, T.; Hernandez, N.; Hallene, K.; Diglaw, T.; Franic, L.; Najm, I.; et al. Seizure-promoting effect of blood-brain barrier disruption. Epilepsia 2007, 48, 732–742. [Google Scholar] [CrossRef]
- Loscher, W.; Friedman, A. Structural, Molecular, and Functional Alterations of the Blood-Brain Barrier during Epileptogenesis and Epilepsy: A Cause, Consequence, or Both? Int. J. Mol. Sci. 2020, 21, 591. [Google Scholar] [CrossRef]
- Abate, G.; Memo, M.; Uberti, D. Impact of COVID-19 on Alzheimer’s Disease Risk: Viewpoint for Research Action. Healthc. Basel 2020, 8, 286. [Google Scholar] [CrossRef]
- Tafti, D.; Ehsan, M.; Xixis, K.L. Multiple Sclerosis. In StatPearls, Treasure Island (FL). 2020. Available online: https://www.ncbi.nlm.nih.gov/books/NBK499849/ (accessed on 27 October 2020).
- Willis, M.D.; Robertson, N.P. Multiple sclerosis and the risk of infection: Considerations in the threat of the novel coronavirus, COVID-19/SARS-CoV-2. J. Neurol. 2020, 267, 1567–1569. [Google Scholar] [CrossRef]
- Winkelmann, A.; Loebermann, M.; Reisinger, E.C.; Hartung, H.P.; Zettl, U.K. Disease-modifying therapies and infectious risks in multiple sclerosis. Nat. Rev. Neurol. 2016, 12, 217–233. [Google Scholar] [CrossRef]
- Ghajarzadeh, M.; Bonavita, S. Are patients with multiple sclerosis (MS) at higher risk of COVID-19 infection? Neurol. Sci. 2020, 41, 2315–2316. [Google Scholar] [CrossRef]
- Crescenzo, F.; Marastoni, D.; Bovo, C.; Calabrese, M. Frequency and severity of COVID-19 in multiple sclerosis: A short single-site report from northern Italy. Mult. Scler. Relat. Disord. 2020, 44. [Google Scholar] [CrossRef]
- Fan, M.; Qiu, W.; Bu, B.; Xu, Y.; Yang, H.; Huang, D.; Lau, A.Y.; Guo, J.; Zhang, M.N.; Zhang, X.; et al. Risk of COVID-19 infection in MS and neuromyelitis optica spectrum disorders. Neurol. Neuroimmunol. Neuroinflamm 2020, 7. [Google Scholar] [CrossRef]
- Louapre, C.; Collongues, N.; Stankoff, B.; Giannesini, C.; Papeix, C.; Bensa, C.; Deschamps, R.; Creange, A.; Wahab, A.; Pelletier, J.; et al. Clinical Characteristics and Outcomes in Patients With Coronavirus Disease 2019 and Multiple Sclerosis. JAMA Neurol. 2020. [Google Scholar] [CrossRef]
- Ferini-Strambi, L.; Salsone, M. COVID-19 and neurological disorders: Are neurodegenerative or neuroimmunological diseases more vulnerable? J. Neurol. 2020. [Google Scholar] [CrossRef]
- Sadeghmousavi, S.; Rezaei, N. COVID-19 and Multiple Sclerosis: Predisposition and Precautions in Treatment. SN Compr. Clin. Med. 2020, 1–6. [Google Scholar] [CrossRef]
- Buljevac, D.; Flach, H.Z.; Hop, W.C.; Hijdra, D.; Laman, J.D.; Savelkoul, H.F.; van Der Meche, F.G.; van Doorn, P.A.; Hintzen, R.Q. Prospective study on the relationship between infections and multiple sclerosis exacerbations. Brain 2002, 125, 952–960. [Google Scholar] [CrossRef]
- Correale, J.; Fiol, M.; Gilmore, W. The risk of relapses in multiple sclerosis during systemic infections. Neurology 2006, 67, 652–659. [Google Scholar] [CrossRef]
- Parkinson Disease. Available online: https://www.ncbi.nlm.nih.gov/books/NBK470193/ (accessed on 27 October 2020).
- Del Prete, E.; Francesconi, A.; Palermo, G.; Mazzucchi, S.; Frosini, D.; Morganti, R.; Coleschi, P.; Raglione, L.M.; Vanni, P.; Ramat, S.; et al. Prevalence and impact of COVID-19 in Parkinson’s disease: Evidence from a multi-center survey in Tuscany region. J. Neurol. 2020. [Google Scholar] [CrossRef]
- Fasano, A.; Elia, A.E.; Dallocchio, C.; Canesi, M.; Alimonti, D.; Sorbera, C.; Alonso-Canovas, A.; Pezzoli, G. Predictors of COVID-19 outcome in Parkinson’s disease. Parkinsonism Relat. Disord. 2020, 78, 134–137. [Google Scholar] [CrossRef]
- Fasano, A.; Cereda, E.; Barichella, M.; Cassani, E.; Ferri, V.; Zecchinelli, A.L.; Pezzoli, G. COVID-19 in Parkinson’s Disease Patients Living in Lombardy, Italy. Mov. Disord. 2020, 35, 1089–1093. [Google Scholar] [CrossRef]
- Papa, S.M.; Brundin, P.; Fung, V.S.C.; Kang, U.J.; Burn, D.J.; Colosimo, C.; Chiang, H.L.; Alcalay, R.N.; Trenkwalder, C.; Committee, M.D.-S.I. Impact of the COVID-19 Pandemic on Parkinson’s Disease and Movement Disorders. Mov. Disord. 2020, 35, 711–715. [Google Scholar] [CrossRef]
- Naughton, S.X.; Raval, U.; Pasinetti, G.M. Potential Novel Role of COVID-19 in Alzheimer’s Disease and Preventative Mitigation Strategies. J. Alzheimers Dis. 2020, 76, 21–25. [Google Scholar] [CrossRef]
- Alzheimer Disease. Available online: https://www.ncbi.nlm.nih.gov/books/NBK499922/ (accessed on 27 October 2020).
- Bianchetti, A.; Rozzini, R.; Guerini, F.; Boffelli, S.; Ranieri, P.; Minelli, G.; Bianchetti, L.; Trabucchi, M. Clinical Presentation of COVID19 in Dementia Patients. J. Nutr. Health Aging 2020, 24, 560–562. [Google Scholar] [CrossRef] [PubMed]
- Roy, E.R.; Wang, B.; Wan, Y.W.; Chiu, G.; Cole, A.; Yin, Z.; Propson, N.E.; Xu, Y.; Jankowsky, J.L.; Liu, Z.; et al. Type I interferon response drives neuroinflammation and synapse loss in Alzheimer disease. J. Clin. Investig. 2020, 130, 1912–1930. [Google Scholar] [CrossRef]
- Garvin, M.R.; Alvarez, C.; Miller, J.I.; Prates, E.T.; Walker, A.M.; Amos, B.K.; Mast, A.E.; Justice, A.; Aronow, B.; Jacobson, D. A mechanistic model and therapeutic interventions for COVID-19 involving a RAS-mediated bradykinin storm. Elife 2020, 9. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Achar, A.; Ghosh, C. COVID-19-Associated Neurological Disorders: The Potential Route of CNS Invasion and Blood-Brain Barrier Relevance. Cells 2020, 9, 2360. https://doi.org/10.3390/cells9112360
Achar A, Ghosh C. COVID-19-Associated Neurological Disorders: The Potential Route of CNS Invasion and Blood-Brain Barrier Relevance. Cells. 2020; 9(11):2360. https://doi.org/10.3390/cells9112360
Chicago/Turabian StyleAchar, Aneesha, and Chaitali Ghosh. 2020. "COVID-19-Associated Neurological Disorders: The Potential Route of CNS Invasion and Blood-Brain Barrier Relevance" Cells 9, no. 11: 2360. https://doi.org/10.3390/cells9112360
APA StyleAchar, A., & Ghosh, C. (2020). COVID-19-Associated Neurological Disorders: The Potential Route of CNS Invasion and Blood-Brain Barrier Relevance. Cells, 9(11), 2360. https://doi.org/10.3390/cells9112360