Alarmins and c-Jun N-Terminal Kinase (JNK) Signaling in Neuroinflammation





Abstract
1. Introduction
2. JNK
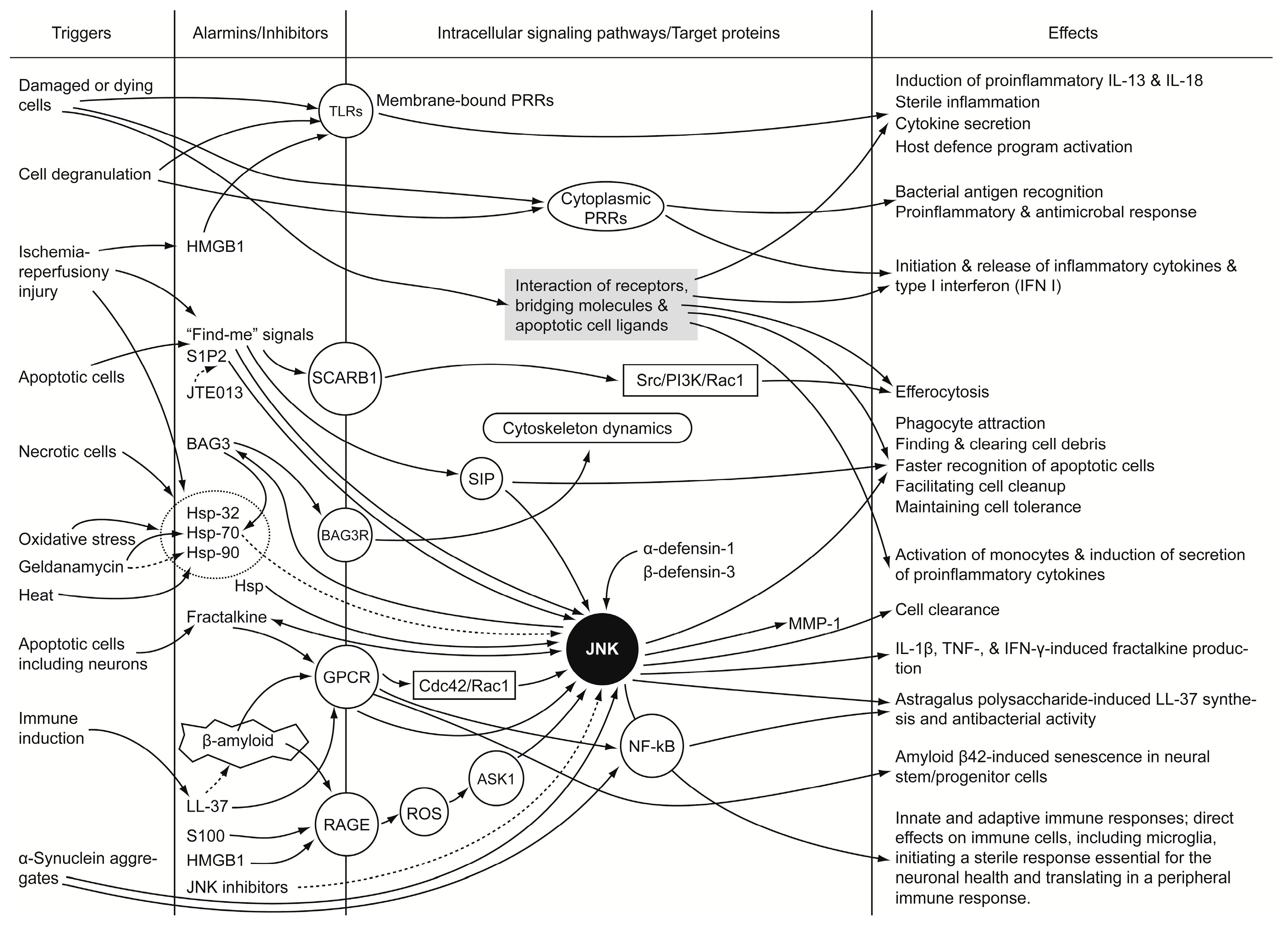
3. Alarmins and JNK-Signaling Cross-Talk
4. “Find-Me” Signals
5. Hsp
6. HMGB1
7. BAG3
8. S100 Calcium-Binding Protein B (S100B)
9. IL-33
10. β-Amyloid
11. Cathelicidin (LL-37)
12. Defensins
13. α-Synuclein
14. Mitochondrial DNA (mtDNA)
15. Anti-Alarmin Agents
16. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease; |
| ADP | adenosine diphosphate; |
| ASK1 | apoptosis-signal-regulated kinase 1; |
| BAG | Bcl-2-associated athanogene; |
| BAG3 | BAG family molecular chaperone regulator 3; |
| BEAS | pulmonary epithelial cell line |
| CHDPs | cationic host defense peptides |
| CNS | central nervous system |
| DAMP | damage-associated molecular patterns |
| DNA | deoxyribonucleic acid |
| ERK | extracellular signal-regulated kinase |
| FPR2 | N-formyl peptide receptor 2 |
| GPCR | G-protein-coupled receptors |
| HMGB1 | high mobility group box 1 |
| Hsp | heat shock proteins |
| HSPA1L | heat shock protein family A member 1 like |
| IFN | interferon |
| IL | interleukin |
| IRS-1 | insulin receptor substrate 1 |
| JNK | c-Jun N-terminal kinase |
| LC3-II | light chain 3 |
| LL-37 | cathelicidin |
| LPS | lipopolysaccharide |
| MAPK | mitogen-activated protein kinases |
| MerTK | Mer receptor tyrosine kinase |
| MFG-E8 | milk fat globule epidermal growth factor VIII |
| MMP | matrix metalloproteinase |
| MRP14 | myeloid-related protein 14 |
| mtDNA | mitochondrial deoxyribonucleic acid |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| NF-κB | nuclear factor κ light chain enhancer of activated B cells |
| NLRP3 | nucleotide-binding domain leucine-rich repeat (NLR) and pyrin domain containing receptor 3 |
| NO | nitric oxide |
| PAMP | pathogen-associated molecular patterns |
| PARP-1 | poly(ADP-ribose) polymerase-1 |
| PD | Parkinson’s disease |
| PI3K | phosphoinositide 3-kinase |
| Rac1 | Ras-related C3 botulinum toxin substrate 1 |
| RAGE | receptor for advanced glycation end products |
| RNA | ribonucleic acid |
| ROS | reactive oxygen species |
| S100B | S100 calcium-binding protein B |
| SBT2171 | exopolysaccharides produced by Lactobacillus helveticus |
| SCARB1 | scavenger receptor class B type I |
| SDF-1 | stromal cell-derived factor 1 |
| SIP | sphingosine 1-phosphate |
| Spz5 | protein spätzle |
| Src | proto-oncogene tyrosine-protein kinase |
| TLR | Toll-like receptor |
| TNF | tumor necrosis factor. |
References
- Banjara, M.; Ghosh, C. Sterile Neuroinflammation and Strategies for Therapeutic Intervention. Int. J. Inflam. 2017, 2017, 8385961. [Google Scholar] [CrossRef] [PubMed]
- Bustamante, A.; Simats, A.; Vilar-Bergua, A.; Garcia-Berrocoso, T.; Montaner, J. Blood/Brain Biomarkers of Inflammation After Stroke and Their Association With Outcome: From C-Reactive Protein to Damage-Associated Molecular Patterns. Neurotherapeutics 2016, 13, 671–684. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Mao, K.; Yu, H.; Chen, J. Neuroinflammatory Responses and Parkinson’ Disease: Pathogenic Mechanisms and Therapeutic Targets. J. Neuroimmune Pharmacol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Heiss, W.D. PET imaging in ischemic cerebrovascular disease: Current status and future directions. Neurosci. Bull. 2014, 30, 713–732. [Google Scholar] [CrossRef]
- Zhang, J. Mapping neuroinflammation in frontotemporal dementia with molecular PET imaging. J. Neuroinflammation 2015, 12, 108. [Google Scholar] [CrossRef]
- Galovic, M.; Koepp, M. Advances of Molecular Imaging in Epilepsy. Curr. Neurol. Neurosci. Rep. 2016, 16, 58. [Google Scholar] [CrossRef]
- Vivash, L.; O’Brien, T.J. Imaging Microglial Activation with TSPO PET: Lighting Up Neurologic Diseases? J. Nucl. Med. 2016, 57, 165–168. [Google Scholar] [CrossRef]
- Turkheimer, F.E.; Rizzo, G.; Bloomfield, P.S.; Howes, O.; Zanotti-Fregonara, P.; Bertoldo, A.; Veronese, M. The methodology of TSPO imaging with positron emission tomography. Biochem. Soc. Trans. 2015, 43, 586–592. [Google Scholar] [CrossRef]
- Gerhard, A. TSPO imaging in parkinsonian disorders. Clin. Transl. Imaging 2016, 4, 183–190. [Google Scholar] [CrossRef]
- Cunningham, C. Microglia and neurodegeneration: The role of systemic inflammation. Glia 2013, 61, 71–90. [Google Scholar] [CrossRef]
- Waisman, A.; Ginhoux, F.; Greter, M.; Bruttger, J. Homeostasis of Microglia in the Adult Brain: Review of Novel Microglia Depletion Systems. Trends Immunol. 2015, 36, 625–636. [Google Scholar] [CrossRef] [PubMed]
- De Schepper, S.; Crowley, G.; Hong, S. Understanding microglial diversity and implications for neuronal function in health and disease. Dev. Neurobiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Hanisch, U.K.; Kettenmann, H. Microglia: Active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 2007, 10, 1387–1394. [Google Scholar] [CrossRef] [PubMed]
- Poutiainen, P.; Jaronen, M.; Quintana, F.J.; Brownell, A.L. Precision Medicine in Multiple Sclerosis: Future of PET Imaging of Inflammation and Reactive Astrocytes. Front. Mol. Neurosci. 2016, 9, 85. [Google Scholar] [CrossRef]
- Nagy, E.E.; Frigy, A.; Szasz, J.A.; Horvath, E. Neuroinflammation and microglia/macrophage phenotype modulate the molecular background of post-stroke depression: A literature review. Exp. Ther. Med. 2020, 20, 2510–2523. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Masuda, T.; Sankowski, R.; Staszewski, O.; Prinz, M. Microglia Heterogeneity in the Single-Cell Era. Cell Rep. 2020, 30, 1271–1281. [Google Scholar] [CrossRef]
- Lawson, L.J.; Perry, V.H.; Dri, P.; Gordon, S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 1990, 39, 151–170. [Google Scholar] [CrossRef]
- Lawson, L.J.; Perry, V.H.; Gordon, S. Turnover of resident microglia in the normal adult mouse brain. Neuroscience 1992, 48, 405–415. [Google Scholar] [CrossRef]
- Filiano, A.J.; Gadani, S.P.; Kipnis, J. Interactions of innate and adaptive immunity in brain development and function. Brain Res. 2015, 1617, 18–27. [Google Scholar] [CrossRef]
- Kierdorf, K.; Prinz, M. Microglia in steady state. J. Clin. Investig. 2017, 127, 3201–3209. [Google Scholar] [CrossRef] [PubMed]
- Kettenmann, H.; Hanisch, U.K.; Noda, M.; Verkhratsky, A. Physiology of microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef] [PubMed]
- Svahn, A.J.; Becker, T.S.; Graeber, M.B. Emergent properties of microglia. Brain Pathol. 2014, 24, 665–670. [Google Scholar] [CrossRef] [PubMed]
- Doens, D.; Fernandez, P.L. Microglia receptors and their implications in the response to amyloid beta for Alzheimer’s disease pathogenesis. J. Neuroinflammation 2014, 11, 48. [Google Scholar] [CrossRef]
- Kreutzberg, G.W. Microglia: A sensor for pathological events in the CNS. Trends Neurosci. 1996, 19, 312–318. [Google Scholar] [CrossRef]
- Meda, L.; Cassatella, M.A.; Szendrei, G.I.; Otvos, L., Jr.; Baron, P.; Villalba, M.; Ferrari, D.; Rossi, F. Activation of microglial cells by b-amyloid protein and interferon-g. Nature 1995, 374, 647–650. [Google Scholar] [CrossRef]
- Dickson, D.W.; Lee, S.C.; Mattiace, L.A.; Yen, S.H.; Brosnan, C. Microglia and cytokines in neurological disease, with special reference to AIDS and Alzheimer’s disease. Glia 1993, 7, 75–83. [Google Scholar] [CrossRef]
- Merlo, S.; Spampinato, S.F.; Caruso, G.I.; Sortino, M.A. The ambiguous role of microglia in Abeta toxicity: Chances for therapeutic intervention. Curr. Neuropharmacol. 2020, 18, 446–455. [Google Scholar] [CrossRef]
- Liu, R.; Yang, J.; Liu, L.; Lu, Z.; Shi, Z.; Ji, W.; Shen, J.; Zhang, X. An “Amyloid-b Cleaner” for the Treatment of Alzheimer’s Disease by Normalizing Microglial Dysfunction. Adv. Sci. (Weinh.) 2020, 7, 1901555. [Google Scholar] [CrossRef]
- Ho, M.S. Microglia in Parkinson’s Disease. Adv. Exp. Med. Biol. 2019, 1175, 335–353. [Google Scholar] [CrossRef]
- Tajbakhsh, A.; Rezaee, M.; Barreto, G.E.; Moallem, S.A.; Henney, N.C.; Sahebkar, A. The role of nuclear factors as “Find-Me”/alarmin signals and immunostimulation in defective efferocytosis and related disorders. Int. Immunopharmacol. 2020, 80, 106134. [Google Scholar] [CrossRef] [PubMed]
- Roh, J.S.; Sohn, D.H. Damage-Associated Molecular Patterns in Inflammatory Diseases. Immune Netw. 2018, 18, e27. [Google Scholar] [CrossRef] [PubMed]
- Buzas, E.I.; Gyorgy, B.; Nagy, G.; Falus, A.; Gay, S. Emerging role of extracellular vesicles in inflammatory diseases. Nat. Rev. Rheumatol. 2014, 10, 356–364. [Google Scholar] [CrossRef] [PubMed]
- Collett, G.P.; Redman, C.W.; Sargent, I.L.; Vatish, M. Endoplasmic reticulum stress stimulates the release of extracellular vesicles carrying danger-associated molecular pattern (DAMP) molecules. Oncotarget 2018, 9, 6707–6717. [Google Scholar] [CrossRef]
- Armstrong, S.C. Protein kinase activation and myocardial ischemia/reperfusion injury. Cardiovasc. Res. 2004, 61, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Bode, A.M.; Dong, Z. The functional contrariety of JNK. Mol. Carcinog. 2007, 46, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Bogoyevitch, M.A.; Kobe, B. Uses for JNK: The many and varied substrates of the c-Jun N-terminal kinases. Microbiol. Mol. Biol. Rev. 2006, 70, 1061–1095. [Google Scholar] [CrossRef]
- Duplain, H. Salvage of ischemic myocardium: A focus on JNK. Adv. Exp. Med. Biol. 2006, 588, 157–164. [Google Scholar] [CrossRef]
- Knight, R.J.; Buxton, D.B. Stimulation of c-Jun kinase and mitogen-activated protein kinase by ischemia and reperfusion in the perfused rat heart. Biochem. Biophys. Res. Commun. 1996, 218, 83–88. [Google Scholar] [CrossRef]
- Gupta, S.; Barrett, T.; Whitmarsh, A.J.; Cavanagh, J.; Sluss, H.K.; Derijard, B.; Davis, R.J. Selective interaction of JNK protein kinase isoforms with transcription factors. EMBO J. 1996, 15, 2760–2770. [Google Scholar] [CrossRef]
- Javadov, S.; Jang, S.; Agostini, B. Crosstalk between mitogen-activated protein kinases and mitochondria in cardiac diseases: Therapeutic perspectives. Pharmacol. Ther. 2014, 144, 202–225. [Google Scholar] [CrossRef] [PubMed]
- Waetzig, V.; Herdegen, T. Context-specific inhibition of JNKs: Overcoming the dilemma of protection and damage. Trends Pharmacol. Sci. 2005, 26, 455–461. [Google Scholar] [CrossRef]
- Nijboer, C.H.; van der Kooij, M.A.; van Bel, F.; Ohl, F.; Heijnen, C.J.; Kavelaars, A. Inhibition of the JNK/AP-1 pathway reduces neuronal death and improves behavioral outcome after neonatal hypoxic-ischemic brain injury. Brain Behav. Immun. 2010, 24, 812–821. [Google Scholar] [CrossRef]
- Shvedova, M.; Anfinogenova, Y.; Atochina-Vasserman, E.N.; Schepetkin, I.A.; Atochin, D.N. c-Jun N-Terminal Kinases (JNKs) in Myocardial and Cerebral Ischemia/Reperfusion Injury. Front. Pharmacol. 2018, 9, 715. [Google Scholar] [CrossRef]
- Plotnikov, M.B.; Chernysheva, G.A.; Aliev, O.I.; Smol’iakova, V.I.; Fomina, T.I.; Osipenko, A.N.; Rydchenko, V.S.; Anfinogenova, Y.J.; Khlebnikov, A.I.; Schepetkin, I.A.; et al. Protective Effects of a New C-Jun N-terminal Kinase Inhibitor in the Model of Global Cerebral Ischemia in Rats. Molecules 2019, 24, 1722. [Google Scholar] [CrossRef]
- Plotnikov, M.B.; Aliev, O.I.; Shamanaev, A.Y.; Sidekhmenova, A.V.; Anishchenko, A.M.; Fomina, T.I.; Rydchenko, V.S.; Khlebnikov, A.I.; Anfinogenova, Y.J.; Schepetkin, I.A.; et al. Antihypertensive activity of a new c-Jun N-terminal kinase inhibitor in spontaneously hypertensive rats. Hypertens. Res. 2020. [Google Scholar] [CrossRef]
- Plotnikov, M.B.; Chernysheva, G.A.; Smolyakova, V.I.; Aliev, O.I.; Trofimova, E.S.; Sherstoboev, E.Y.; Osipenko, A.N.; Khlebnikov, A.I.; Anfinogenova, Y.J.; Schepetkin, I.A.; et al. Neuroprotective Effects of a Novel Inhibitor of c-Jun N-Terminal Kinase in the Rat Model of Transient Focal Cerebral Ischemia. Cells 2020, 9, 1860. [Google Scholar] [CrossRef] [PubMed]
- Schepetkin, I.A.; Khlebnikov, A.I.; Potapov, A.S.; Kovrizhina, A.R.; Matveevskaya, V.V.; Belyanin, M.L.; Atochin, D.N.; Zanoza, S.O.; Gaidarzhy, N.M.; Lyakhov, S.A.; et al. Synthesis, biological evaluation, and molecular modeling of 11H-indeno[1,2-b]quinoxalin-11-one derivatives and tryptanthrin-6-oxime as c-Jun N-terminal kinase inhibitors. Eur. J. Med. Chem. 2019, 161, 179–191. [Google Scholar] [CrossRef]
- Zeke, A.; Misheva, M.; Reményi, A.; Bogoyevitch, M.A. JNK Signaling: Regulation and Functions Based on Complex Protein-Protein Partnerships. Microbiol. Mol. Biol. Rev. 2016, 80, 793–835. [Google Scholar] [CrossRef] [PubMed]
- Ip, Y.T.; Davis, R.J. Signal transduction by the c-Jun N-terminal kinase (JNK)--from inflammation to development. Curr. Opin. Cell Biol. 1998, 10, 205–219. [Google Scholar] [CrossRef]
- Vlahopoulos, S.; Zoumpourlis, V.C. JNK: A key modulator of intracellular signaling. Biochemistry (Mosc.) 2004, 69, 844–854. [Google Scholar] [CrossRef]
- Shao, Z.; Bhattacharya, K.; Hsich, E.; Park, L.; Walters, B.; Germann, U.; Wang, Y.M.; Kyriakis, J.; Mohanlal, R.; Kuida, K.; et al. c-Jun N-terminal kinases mediate reactivation of Akt and cardiomyocyte survival after hypoxic injury in vitro and in vivo. Circ. Res. 2006, 98, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Chaudhury, H.; Zakkar, M.; Boyle, J.; Cuhlmann, S.; van der Heiden, K.; Luong le, A.; Davis, J.; Platt, A.; Mason, J.C.; Krams, R.; et al. c-Jun N-terminal kinase primes endothelial cells at atheroprone sites for apoptosis. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 546–553. [Google Scholar] [CrossRef]
- Wiltshire, C.; Gillespie, D.A.; May, G.H. Sab (SH3BP5), a novel mitochondria-localized JNK-interacting protein. Biochem. Soc. Trans. 2004, 32, 1075–1077. [Google Scholar] [CrossRef]
- Yang, D.; Rosenberg, H.F.; Chen, Q.; Dyer, K.D.; Kurosaka, K.; Oppenheim, J.J. Eosinophil-derived neurotoxin (EDN), an antimicrobial protein with chemotactic activities for dendritic cells. Blood 2003, 102, 3396–3403. [Google Scholar] [CrossRef]
- Di Salvo, E.; Casciaro, M.; Quartuccio, S.; Genovese, L.; Gangemi, S. Do Alarmins Have a Potential Role in Autism Spectrum Disorders Pathogenesis and Progression? Biomolecules 2018, 9, 2. [Google Scholar] [CrossRef]
- Uyangaa, E.; Choi, J.Y.; Patil, A.M.; Hossain, F.M.A.; Park, S.O.; Kim, B.; Kim, K.; Eo, S.K. Dual TLR2/9 Recognition of Herpes Simplex Virus Infection Is Required for Recruitment and Activation of Monocytes and NK Cells and Restriction of Viral Dissemination to the Central Nervous System. Front. Immunol. 2018, 9, 905. [Google Scholar] [CrossRef] [PubMed]
- Ferhat, M.; Robin, A.; Giraud, S.; Sena, S.; Goujon, J.M.; Touchard, G.; Hauet, T.; Girard, J.P.; Gombert, J.M.; Herbelin, A.; et al. Endogenous IL-33 Contributes to Kidney Ischemia-Reperfusion Injury as an Alarmin. J. Am. Soc. Nephrol. 2018, 29, 1272–1288. [Google Scholar] [CrossRef]
- Saez, P.J.; Vargas, P.; Shoji, K.F.; Harcha, P.A.; Lennon-Dumenil, A.M.; Saez, J.C. ATP promotes the fast migration of dendritic cells through the activity of pannexin 1 channels and P2X7 receptors. Sci. Signal. 2017, 10, aah7107. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Hu, Z.; Tamura, H.; Nagaoka, I. Release mechanism of high mobility group nucleosome binding domain 1 from lipopolysaccharide-stimulated macrophages. Mol. Med. Rep. 2016, 13, 3115–3120. [Google Scholar] [CrossRef]
- Oppenheim, J.J.; Yang, D. Alarmins: Chemotactic activators of immune responses. Curr. Opin. Immunol. 2005, 17, 359–365. [Google Scholar] [CrossRef]
- Gorgulu, B.; Bavbek, S. [Alarmins and anti-alarmin biologics in asthma]. Tuberk. Toraks 2018, 66, 166–175. [Google Scholar] [CrossRef]
- Leow-Dyke, S.; Allen, C.; Denes, A.; Nilsson, O.; Maysami, S.; Bowie, A.G.; Rothwell, N.J.; Pinteaux, E. Neuronal Toll-like receptor 4 signaling induces brain endothelial activation and neutrophil transmigration in vitro. J. Neuroinflammation 2012, 9, 230. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Yao, X.; Jiang, W.; Li, W.; Zhu, S.; Liao, C.; Zou, L.; Ding, R.; Chen, J. Advanced oxidation protein products induce microglia-mediated neuroinflammation via MAPKs-NF-kappaB signaling pathway and pyroptosis after secondary spinal cord injury. J. Neuroinflammation 2020, 17, 90. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, T.; Zulfiqar, A.; Arguelles, S.; Rasekhian, M.; Nabavi, S.F.; Silva, A.S.; Nabavi, S.M. Map kinase signaling as therapeutic target for neurodegeneration. Pharmacol. Res. 2020, 160, 105090. [Google Scholar] [CrossRef] [PubMed]
- Busquets, O.; Ettcheto, M.; Cano, A.; R Manzine, P.; Sanchez-Lopez, E.; Espinosa-Jimenez, T.; Verdaguer, E.; Dario Castro-Torres, R.; Beas-Zarate, C.; X Sureda, F.; et al. Role of c-Jun N-Terminal Kinases (JNKs) in Epilepsy and Metabolic Cognitive Impairment. Int. J. Mol. Sci. 2019, 21, 255. [Google Scholar] [CrossRef]
- Kaidanovich-Beilin, O.; Milman, A.; Weizman, A.; Pick, C.G.; Eldar-Finkelman, H. Rapid antidepressive-like activity of specific glycogen synthase kinase-3 inhibitor and its effect on beta-catenin in mouse hippocampus. Biol. Psychiatry 2004, 55, 781–784. [Google Scholar] [CrossRef]
- Elliott, M.R.; Chekeni, F.B.; Trampont, P.C.; Lazarowski, E.R.; Kadl, A.; Walk, S.F.; Park, D.; Woodson, R.I.; Ostankovich, M.; Sharma, P.; et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 2009, 461, 282–286. [Google Scholar] [CrossRef]
- Flannagan, R.S.; Jaumouille, V.; Grinstein, S. The cell biology of phagocytosis. Annu. Rev. Pathol. 2012, 7, 61–98. [Google Scholar] [CrossRef]
- Ravichandran, K.S. Find-me and eat-me signals in apoptotic cell clearance: Progress and conundrums. J. Exp. Med. 2010, 207, 1807–1817. [Google Scholar] [CrossRef]
- Segawa, K.; Nagata, S. An Apoptotic ‘Eat Me’ Signal: Phosphatidylserine Exposure. Trends Cell Biol. 2015, 25, 639–650. [Google Scholar] [CrossRef]
- McLaughlin, C.N.; Perry-Richardson, J.J.; Coutinho-Budd, J.C.; Broihier, H.T. Dying Neurons Utilize Innate Immune Signaling to Prime Glia for Phagocytosis during Development. Dev. Cell 2019, 48, 506–522 e506. [Google Scholar] [CrossRef]
- Elliott, M.R.; Ravichandran, K.S. The Dynamics of Apoptotic Cell Clearance. Dev. Cell 2016, 38, 147–160. [Google Scholar] [CrossRef]
- Linton, M.F.; Babaev, V.R.; Huang, J.; Linton, E.F.; Tao, H.; Yancey, P.G. Macrophage Apoptosis and Efferocytosis in the Pathogenesis of Atherosclerosis. Circ. J. 2016, 80, 2259–2268. [Google Scholar] [CrossRef]
- Kolb, J.P.; Martinez, J. Bon EPOtit! S1P-Mediated EPO Signaling Whets a Macrophage’s Appetite for Apoptotic Cells. Immunity 2016, 44, 209–211. [Google Scholar] [CrossRef]
- Luo, B.; Gan, W.; Liu, Z.; Shen, Z.; Wang, J.; Shi, R.; Liu, Y.; Liu, Y.; Jiang, M.; Zhang, Z.; et al. Erythropoeitin Signaling in Macrophages Promotes Dying Cell Clearance and Immune Tolerance. Immunity 2016, 44, 287–302. [Google Scholar] [CrossRef]
- Sapkota, A.; Gaire, B.P.; Kang, M.G.; Choi, J.W. S1P2 contributes to microglial activation and M1 polarization following cerebral ischemia through ERK1/2 and JNK. Sci. Rep. 2019, 9, 12106. [Google Scholar] [CrossRef]
- Sokolowski, J.D.; Chabanon-Hicks, C.N.; Han, C.Z.; Heffron, D.S.; Mandell, J.W. Fractalkine is a “find-me” signal released by neurons undergoing ethanol-induced apoptosis. Front. Cell. Neurosci. 2014, 8, 360. [Google Scholar] [CrossRef]
- Galan-Ganga, M.; Garcia-Yague, A.J.; Lastres-Becker, I. Role of MSK1 in the Induction of NF-kappaB by the Chemokine CX3CL1 in Microglial Cells. Cell. Mol. Neurobiol. 2019, 39, 331–340. [Google Scholar] [CrossRef]
- Atochin, D.N.; Schepetkin, I.A.; Khlebnikov, A.I.; Seledtsov, V.I.; Swanson, H.; Quinn, M.T.; Huang, P.L. A novel dual NO-donating oxime and c-Jun N-terminal kinase inhibitor protects against cerebral ischemia-reperfusion injury in mice. Neurosci. Lett. 2016, 618, 45–49. [Google Scholar] [CrossRef]
- Huang, S.J.; Chen, C.P.; Buchwalder, L.; Yu, Y.C.; Piao, L.; Huang, C.Y.; Schatz, F.; Lockwood, C.J. Regulation of CX3CL1 Expression in Human First-Trimester Decidual Cells: Implications for Preeclampsia. Reprod. Sci. 2019, 26, 1256–1265. [Google Scholar] [CrossRef] [PubMed]
- Sontheimer, R.D.; Racila, D.; Racila, E.; Eggleton, P.; Donnelly, S. Calreticulin’s role (s) in autoimmune disorders. In Calreticulin; Eggleton, P., Michalak, M., Eds.; Springer: Boston, MA, USA, 2003; pp. 180–192. [Google Scholar] [CrossRef]
- Rimaniol, A.C.; Till, S.J.; Garcia, G.; Capel, F.; Godot, V.; Balabanian, K.; Durand-Gasselin, I.; Varga, E.M.; Simonneau, G.; Emilie, D.; et al. The CX3C chemokine fractalkine in allergic asthma and rhinitis. J. Allergy Clin. Immunol. 2003, 112, 1139–1146. [Google Scholar] [CrossRef] [PubMed]
- Gerber, E.E.; Gallo, E.M.; Fontana, S.C.; Davis, E.C.; Wigley, F.M.; Huso, D.L.; Dietz, H.C. Integrin-modulating therapy prevents fibrosis and autoimmunity in mouse models of scleroderma. Nature 2013, 503, 126–130. [Google Scholar] [CrossRef]
- Abdolmaleki, F.; Farahani, N.; Gheibi Hayat, S.M.; Pirro, M.; Bianconi, V.; Barreto, G.E.; Sahebkar, A. The Role of Efferocytosis in Autoimmune Diseases. Front. Immunol. 2018, 9, 1645. [Google Scholar] [CrossRef] [PubMed]
- Gheibi Hayat, S.M.; Bianconi, V.; Pirro, M.; Sahebkar, A. Efferocytosis: Molecular mechanisms and pathophysiological perspectives. Immunol. Cell Biol. 2019, 97, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Tajbakhsh, A.; Gheibi Hayat, S.M.; Butler, A.E.; Sahebkar, A. Effect of soluble cleavage products of important receptors/ligands on efferocytosis: Their role in inflammatory, autoimmune and cardiovascular disease. Ageing Res. Rev. 2019, 50, 43–57. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, C.L.; Martínez-Cerdeño, V.; Noctor, S.C. Microglia regulate the number of neural precursor cells in the developing cerebral cortex. J. Neurosci. 2013, 33, 4216–4233. [Google Scholar] [CrossRef] [PubMed]
- VanRyzin, J.W.; Marquardt, A.E.; Argue, K.J.; Vecchiarelli, H.A.; Ashton, S.E.; Arambula, S.E.; Hill, M.N.; McCarthy, M.M. Microglial Phagocytosis of Newborn Cells Is Induced by Endocannabinoids and Sculpts Sex Differences in Juvenile Rat Social Play. Neuron 2019, 102, 435–449. [Google Scholar] [CrossRef]
- Sierra, A.; Encinas, J.M.; Deudero, J.J.; Chancey, J.H.; Enikolopov, G.; Overstreet-Wadiche, L.S.; Tsirka, S.E.; Maletic-Savatic, M. Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell 2010, 7, 483–495. [Google Scholar] [CrossRef]
- Fourgeaud, L.; Través, P.G.; Tufail, Y.; Leal-Bailey, H.; Lew, E.D.; Burrola, P.G.; Callaway, P.; Zagórska, A.; Rothlin, C.V.; Nimmerjahn, A.; et al. TAM receptors regulate multiple features of microglial physiology. Nature 2016, 532, 240–244. [Google Scholar] [CrossRef]
- Sierra, A.; Abiega, O.; Shahraz, A.; Neumann, H. Janus-faced microglia: Beneficial and detrimental consequences of microglial phagocytosis. Front. Cell. Neurosci. 2013, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Márquez-Ropero, M.; Benito, E.; Plaza-Zabala, A.; Sierra, A. Microglial Corpse Clearance: Lessons From Macrophages. Front. Immunol. 2020, 11, 506. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, E.; Gehrmann, M.; Brunet, M.; Multhoff, G.; Garrido, C. Intracellular and extracellular functions of heat shock proteins: Repercussions in cancer therapy. J. Leukoc. Biol. 2007, 81, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, M.E. DAMPs, PAMPs and alarmins: All we need to know about danger. J. Leukoc. Biol. 2007, 81, 1–5. [Google Scholar] [CrossRef]
- Wallin, R.P.; Lundqvist, A.; More, S.H.; von Bonin, A.; Kiessling, R.; Ljunggren, H.G. Heat-shock proteins as activators of the innate immune system. Trends Immunol. 2002, 23, 130–135. [Google Scholar] [CrossRef]
- Srivastava, P. Roles of heat-shock proteins in innate and adaptive immunity. Nat. Rev. Immunol. 2002, 2, 185–194. [Google Scholar] [CrossRef]
- Pockley, A.G. Heat shock proteins as regulators of the immune response. Lancet 2003, 362, 469–476. [Google Scholar] [CrossRef]
- Asea, A.; Kraeft, S.K.; Kurt-Jones, E.A.; Stevenson, M.A.; Chen, L.B.; Finberg, R.W.; Koo, G.C.; Calderwood, S.K. HSP70 stimulates cytokine production through a CD14-dependant pathway, demonstrating its dual role as a chaperone and cytokine. Nat. Med. 2000, 6, 435–442. [Google Scholar] [CrossRef]
- Vabulas, R.M.; Ahmad-Nejad, P.; Ghose, S.; Kirschning, C.J.; Issels, R.D.; Wagner, H. HSP70 as endogenous stimulus of the Toll/interleukin-1 receptor signal pathway. J. Biol. Chem. 2002, 277, 15107–15112. [Google Scholar] [CrossRef]
- Vabulas, R.M.; Wagner, H.; Schild, H. Heat shock proteins as ligands of toll-like receptors. Curr. Top. Microbiol. Immunol. 2002, 270, 169–184. [Google Scholar] [CrossRef]
- Min, H.; Youn, E.; Kawasaki, I.; Shim, Y.H. Caffeine-induced food-avoidance behavior is mediated by neuroendocrine signals in Caenorhabditis elegans. BMB Rep. 2017, 50, 31–36. [Google Scholar] [CrossRef]
- Kuta, R.; Larochelle, N.; Fernandez, M.; Pal, A.; Minotti, S.; Tibshirani, M.; St Louis, K.; Gentil, B.J.; Nalbantoglu, J.N.; Hermann, A.; et al. Depending on the stress, histone deacetylase inhibitors act as heat shock protein co-inducers in motor neurons and potentiate arimoclomol, exerting neuroprotection through multiple mechanisms in ALS models. Cell Stress Chaperones 2020, 25, 173–191. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.R.; Robinson, M.B.; Gifondorwa, D.J.; Tytell, M.; Milligan, C.E. Regulation of heat shock protein 70 release in astrocytes: Role of signaling kinases. Dev. Neurobiol. 2007, 67, 1815–1829. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.Y.; Yang, Y.; Ju, W.N.; Wang, X.; Zhang, H.L. Emerging Roles of Astrocytes in Neuro-Vascular Unit and the Tripartite Synapse With Emphasis on Reactive Gliosis in the Context of Alzheimer’s Disease. Front. Cell. Neurosci. 2018, 12, 193. [Google Scholar] [CrossRef]
- Lee, K.H.; Jeong, J.; Yoo, C.G. Positive feedback regulation of heat shock protein 70 (Hsp70) is mediated through Toll-like receptor 4-PI3K/Akt-glycogen synthase kinase-3beta pathway. Exp. Cell Res. 2013, 319, 88–95. [Google Scholar] [CrossRef]
- Shukla, A.K.; Pragya, P.; Chaouhan, H.S.; Tiwari, A.K.; Patel, D.K.; Abdin, M.Z.; Chowdhuri, D.K. Heat shock protein-70 (Hsp-70) suppresses paraquat-induced neurodegeneration by inhibiting JNK and caspase-3 activation in Drosophila model of Parkinson’s disease. PLoS ONE 2014, 9, e98886. [Google Scholar] [CrossRef]
- Porto, R.R.; Dutra, F.D.; Crestani, A.P.; Holsinger, R.M.D.; Quillfeldt, J.A.; Homem de Bittencourt, P.I., Jr.; de Oliveira Alvares, L. HSP70 Facilitates Memory Consolidation of Fear Conditioning through MAPK Pathway in the Hippocampus. Neuroscience 2018, 375, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Taran, A.S.; Shuvalova, L.D.; Lagarkova, M.A.; Alieva, I.B. Huntington’s Disease-An Outlook on the Interplay of the HTT Protein, Microtubules and Actin Cytoskeletal Components. Cells 2020, 9, 1514. [Google Scholar] [CrossRef]
- Chen, J.Y.; Parekh, M.; Seliman, H.; Bakshinskaya, D.; Dai, W.; Kwan, K.; Chen, K.Y.; Liu, A.Y.C. Heat shock promotes inclusion body formation of mutant huntingtin (mHtt) and alleviates mHtt-induced transcription factor dysfunction. J. Biol. Chem. 2018, 293, 15581–15593. [Google Scholar] [CrossRef]
- Choi, Y.J.; Kim, N.H.; Lim, M.S.; Lee, H.J.; Kim, S.S.; Chun, W. Geldanamycin attenuates 3nitropropionic acidinduced apoptosis and JNK activation through the expression of HSP 70 in striatal cells. Int. J. Mol. Med. 2014, 34, 24–34. [Google Scholar] [CrossRef][Green Version]
- Francis, S.P.; Kramarenko, I.I.; Brandon, C.S.; Lee, F.S.; Baker, T.G.; Cunningham, L.L. Celastrol inhibits aminoglycoside-induced ototoxicity via heat shock protein 32. Cell Death Dis. 2011, 2, e195. [Google Scholar] [CrossRef] [PubMed]
- Baker, T.G.; Roy, S.; Brandon, C.S.; Kramarenko, I.K.; Francis, S.P.; Taleb, M.; Marshall, K.M.; Schwendener, R.; Lee, F.S.; Cunningham, L.L. Heat shock protein-mediated protection against Cisplatin-induced hair cell death. J. Assoc. Res. Otolaryngol. 2015, 16, 67–80. [Google Scholar] [CrossRef]
- Biermann, J.; Lagreze, W.A.; Schallner, N.; Schwer, C.I.; Goebel, U. Inhalative preconditioning with hydrogen sulfide attenuated apoptosis after retinal ischemia/reperfusion injury. Mol. Vis. 2011, 17, 1275–1286. [Google Scholar] [PubMed]
- Anggayasti, W.L.; Ogino, K.; Yamamoto, E.; Helmerhorst, E.; Yasuoka, K.; Mancera, R.L. The acidic tail of HMGB1 regulates its secondary structure and conformational flexibility: A circular dichroism and molecular dynamics simulation study. Comput. Struct. Biotechnol. J. 2020, 18, 1160–1172. [Google Scholar] [CrossRef]
- Kang, R.; Chen, R.; Zhang, Q.; Hou, W.; Wu, S.; Cao, L.; Huang, J.; Yu, Y.; Fan, X.G.; Yan, Z.; et al. HMGB1 in health and disease. Mol. Aspects Med. 2014, 40, 1–116. [Google Scholar] [CrossRef]
- Yang, H.; Wang, H.; Ju, Z.; Ragab, A.A.; Lundback, P.; Long, W.; Valdes-Ferrer, S.I.; He, M.; Pribis, J.P.; Li, J.; et al. MD-2 is required for disulfide HMGB1-dependent TLR4 signaling. J. Exp. Med. 2015, 212, 5–14. [Google Scholar] [CrossRef]
- Hori, O.; Brett, J.; Slattery, T.; Cao, R.; Zhang, J.; Chen, J.X.; Nagashima, M.; Lundh, E.R.; Vijay, S.; Nitecki, D.; et al. The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin. Mediation of neurite outgrowth and co-expression of rage and amphoterin in the developing nervous system. J. Biol. Chem. 1995, 270, 25752–25761. [Google Scholar] [CrossRef]
- Sha, Y.; Zmijewski, J.; Xu, Z.; Abraham, E. HMGB1 develops enhanced proinflammatory activity by binding to cytokines. J. Immunol. 2008, 180, 2531–2537. [Google Scholar] [CrossRef] [PubMed]
- Urbonaviciute, V.; Furnrohr, B.G.; Meister, S.; Munoz, L.; Heyder, P.; De Marchis, F.; Bianchi, M.E.; Kirschning, C.; Wagner, H.; Manfredi, A.A.; et al. Induction of inflammatory and immune responses by HMGB1-nucleosome complexes: Implications for the pathogenesis of SLE. J. Exp. Med. 2008, 205, 3007–3018. [Google Scholar] [CrossRef] [PubMed]
- Hreggvidsdottir, H.S.; Ostberg, T.; Wahamaa, H.; Schierbeck, H.; Aveberger, A.C.; Klevenvall, L.; Palmblad, K.; Ottosson, L.; Andersson, U.; Harris, H.E. The alarmin HMGB1 acts in synergy with endogenous and exogenous danger signals to promote inflammation. J. Leukoc. Biol. 2009, 86, 655–662. [Google Scholar] [CrossRef]
- Youn, J.H.; Kwak, M.S.; Wu, J.; Kim, E.S.; Ji, Y.; Min, H.J.; Yoo, J.H.; Choi, J.E.; Cho, H.S.; Shin, J.S. Identification of lipopolysaccharide-binding peptide regions within HMGB1 and their effects on subclinical endotoxemia in a mouse model. Eur. J. Immunol. 2011, 41, 2753–2762. [Google Scholar] [CrossRef]
- Pisetsky, D.S. Mechanisms of Chromatin Remodeling and Repurposing During Extracellular Translocation. Adv. Protein Chem. Struct. Biol. 2017, 106, 113–137. [Google Scholar] [CrossRef]
- Andersson, U.; Yang, H.; Harris, H. Extracellular HMGB1 as a therapeutic target in inflammatory diseases. Expert Opin. Ther. Targets 2018, 22, 263–277. [Google Scholar] [CrossRef]
- Fritz, G. RAGE: A single receptor fits multiple ligands. Trends Biochem. Sci. 2011, 36, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Suchal, K.; Malik, S.; Khan, S.I.; Malhotra, R.K.; Goyal, S.N.; Bhatia, J.; Kumari, S.; Ojha, S.; Arya, D.S. Protective effect of mangiferin on myocardial ischemia-reperfusion injury in streptozotocin-induced diabetic rats: Role of AGE-RAGE/MAPK pathways. Sci. Rep. 2017, 7, 42027. [Google Scholar] [CrossRef] [PubMed]
- Wautier, M.P.; Chappey, O.; Corda, S.; Stern, D.M.; Schmidt, A.M.; Wautier, J.L. Activation of NADPH oxidase by AGE links oxidant stress to altered gene expression via RAGE. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E685–E694. [Google Scholar] [CrossRef]
- Qi, L.; Sun, X.; Li, F.E.; Zhu, B.S.; Braun, F.K.; Liu, Z.Q.; Tang, J.L.; Wu, C.; Xu, F.; Wang, H.H.; et al. HMGB1 Promotes Mitochondrial Dysfunction-Triggered Striatal Neurodegeneration via Autophagy and Apoptosis Activation. PLoS ONE 2015, 10, e0142901. [Google Scholar] [CrossRef]
- Zhu, L.; Huang, G.; Sheng, J.; Fu, Q.; Chen, A. High-mobility group box 1 induces neuron autophagy in a rat spinal root avulsion model. Neuroscience 2016, 315, 286–295. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Ryu, M.J.; Han, J.; Jang, Y.; Kim, J.; Lee, M.J.; Ryu, I.; Ju, X.; Oh, E.; Chung, W.; et al. Activation of the HMGB1-RAGE axis upregulates TH expression in dopaminergic neurons via JNK phosphorylation. Biochem. Biophys. Res. Commun. 2017, 493, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Ryu, M.J.; Han, J.; Jang, Y.; Lee, M.J.; Ju, X.; Ryu, I.; Lee, Y.L.; Oh, E.; Chung, W.; et al. Non-cell autonomous modulation of tyrosine hydroxylase by HMGB1 released from astrocytes in an acute MPTP-induced Parkinsonian mouse model. Lab. Investig. 2019, 99, 1389–1399. [Google Scholar] [CrossRef] [PubMed]
- Espinosa-Garcia, C.; Atif, F.; Yousuf, S.; Sayeed, I.; Neigh, G.N.; Stein, D.G. Progesterone Attenuates Stress-Induced NLRP3 Inflammasome Activation and Enhances Autophagy following Ischemic Brain Injury. Int. J. Mol. Sci. 2020, 21, 3740. [Google Scholar] [CrossRef]
- Qin, Y.; Chen, Z.; Han, X.; Wu, H.; Yu, Y.; Wu, J.; Liu, S.; Hou, Y. Progesterone attenuates Abeta(25-35)-induced neuronal toxicity via JNK inactivation and progesterone receptor membrane component 1-dependent inhibition of mitochondrial apoptotic pathway. J. Steroid Biochem. Mol. Biol. 2015, 154, 302–311. [Google Scholar] [CrossRef]
- Le, K.; Wu, S.; Chibaatar, E.; Ali, A.I.; Guo, Y. Alarmin HMGB1 Plays a Detrimental Role in Hippocampal Dysfunction Caused by Hypoxia-Ischemia Insult in Neonatal Mice: Evidence from the Application of the HMGB1 Inhibitor Glycyrrhizin. ACS Chem. Neurosci. 2020, 11, 979–993. [Google Scholar] [CrossRef]
- Qu, Y.; Zhan, Y.; Yang, S.; Ren, S.; Qiu, X.; Rehamn, Z.U.; Tan, L.; Sun, Y.; Meng, C.; Song, C.; et al. Newcastle disease virus infection triggers HMGB1 release to promote the inflammatory response. Virology 2018, 525, 19–31. [Google Scholar] [CrossRef]
- Klune, J.R.; Dhupar, R.; Cardinal, J.; Billiar, T.R.; Tsung, A. HMGB1: Endogenous danger signaling. Mol. Med. 2008, 14, 476–484. [Google Scholar] [CrossRef]
- Franceschelli, S.; Bruno, A.P.; Festa, M.; Falco, A.; Gionti, E.; d’Avenia, M.; De Marco, M.; Basile, A.; Iorio, V.; Marzullo, L.; et al. BAG3 Protein Is Involved in Endothelial Cell Response to Phenethyl Isothiocyanate. Oxid. Med. Cell. Longev. 2018, 2018, 5967890. [Google Scholar] [CrossRef]
- Meriin, A.B.; Narayanan, A.; Meng, L.; Alexandrov, I.; Varelas, X.; Cisse, I.I.; Sherman, M.Y. Hsp70-Bag3 complex is a hub for proteotoxicity-induced signaling that controls protein aggregation. Proc. Natl. Acad. Sci. USA 2018, 115, E7043–E7052. [Google Scholar] [CrossRef]
- Benz, A.P.; Niquet, J.; Wasterlain, C.G.; Rami, A. Status epilepticus in the immature rodent brain alters the dynamics of autophagy. Curr. Neurovasc. Res. 2014, 11, 125–135. [Google Scholar] [CrossRef]
- Muranova, L.K.; Ryzhavskaya, A.S.; Sudnitsyna, M.V.; Shatov, V.M.; Gusev, N.B. Small Heat Shock Proteins and Human Neurodegenerative Diseases. Biochemistry (Mosc) 2019, 84, 1256–1267. [Google Scholar] [CrossRef]
- Ji, C.; Tang, M.; Zeidler, C.; Hohfeld, J.; Johnson, G.V. BAG3 and SYNPO (synaptopodin) facilitate phospho-MAPT/Tau degradation via autophagy in neuronal processes. Autophagy 2019, 15, 1199–1213. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.Q.; Liu, B.Q.; Gao, Y.Y.; Meng, X.; Guan, Y.; Zhang, H.Y.; Du, Z.X. Inhibition of the JNK signalling pathway enhances proteasome inhibitor-induced apoptosis of kidney cancer cells by suppression of BAG3 expression. Br. J. Pharmacol. 2009, 158, 1405–1412. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Marzullo, L.; Turco, M.C.; De Marco, M. The multiple activities of BAG3 protein: Mechanisms. Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129628. [Google Scholar] [CrossRef] [PubMed]
- Muralidar, S.; Ambi, S.V.; Sekaran, S.; Thirumalai, D.; Palaniappan, B. Role of tau protein in Alzheimer’s disease: The prime pathological player. Int. J. Biol. Macromol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Kruger, U.; Wang, Y.; Kumar, S.; Mandelkow, E.M. Autophagic degradation of tau in primary neurons and its enhancement by trehalose. Neurobiol. Aging 2012, 33, 2291–2305. [Google Scholar] [CrossRef] [PubMed]
- Lei, Z.; Brizzee, C.; Johnson, G.V. BAG3 facilitates the clearance of endogenous tau in primary neurons. Neurobiol. Aging 2015, 36, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wang, H.; Won, S.J.; Basu, J.; Kapfhamer, D.; Swanson, R.A. Microglial activation induced by the alarmin S100B is regulated by poly(ADP-ribose) polymerase-1. Glia 2016, 64, 1869–1878. [Google Scholar] [CrossRef]
- Hagmeyer, S.; Romao, M.A.; Cristovao, J.S.; Vilella, A.; Zoli, M.; Gomes, C.M.; Grabrucker, A.M. Distribution and Relative Abundance of S100 Proteins in the Brain of the APP23 Alzheimer’s Disease Model Mice. Front. Neurosci. 2019, 13, 640. [Google Scholar] [CrossRef]
- Bellaver, B.; Souza, D.G.; Souza, D.O.; Quincozes-Santos, A. Hippocampal Astrocyte Cultures from Adult and Aged Rats Reproduce Changes in Glial Functionality Observed in the Aging Brain. Mol. Neurobiol. 2017, 54, 2969–2985. [Google Scholar] [CrossRef]
- Gomes, C.; Cunha, C.; Nascimento, F.; Ribeiro, J.A.; Vaz, A.R.; Brites, D. Cortical Neurotoxic Astrocytes with Early ALS Pathology and miR-146a Deficit Replicate Gliosis Markers of Symptomatic SOD1G93A Mouse Model. Mol. Neurobiol. 2019, 56, 2137–2158. [Google Scholar] [CrossRef]
- Fernandez-Lizarbe, S.; Civera-Tregon, A.; Cantarero, L.; Herrer, I.; Juarez, P.; Hoenicka, J.; Palau, F. Neuroinflammation in the pathogenesis of axonal Charcot-Marie-Tooth disease caused by lack of GDAP1. Exp. Neurol. 2019, 320, 113004. [Google Scholar] [CrossRef]
- Langeh, U.; Singh, S. Targetting S100b Protein as a Surrogate Biomarker and Its Role in Various Neurological Disorders. Curr. Neuropharmacol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Di Sante, G.; Amadio, S.; Sampaolese, B.; Clementi, M.E.; Valentini, M.; Volonte, C.; Casalbore, P.; Ria, F.; Michetti, F. The S100B Inhibitor Pentamidine Ameliorates Clinical Score and Neuropathology of Relapsing-Remitting Multiple Sclerosis Mouse Model. Cells 2020, 9, 748. [Google Scholar] [CrossRef] [PubMed]
- Valentim-Silva, J.R.; Macedo, S.R.A.; de Barros, N.B.; Dos Santos Ferreira, A.; da Silva, J.H.M.; de Figueiredo Nicolete, L.D.; Nicolete, R. Antileishmanial drugs activate inflammatory signaling pathways via toll-like receptors (docking approach) from Leishmania amazonensis-infected macrophages. Int. Immunopharmacol. 2020, 85, 106640. [Google Scholar] [CrossRef]
- Sung, H.Y.; Chen, W.Y.; Huang, H.T.; Wang, C.Y.; Chang, S.B.; Tzeng, S.F. Down-regulation of interleukin-33 expression in oligodendrocyte precursor cells impairs oligodendrocyte lineage progression. J. Neurochem. 2019, 150, 691–708. [Google Scholar] [CrossRef]
- Huang, H.T.; Tsai, S.F.; Wu, H.T.; Huang, H.Y.; Hsieh, H.H.; Kuo, Y.M.; Chen, P.S.; Yang, C.S.; Tzeng, S.F. Chronic exposure to high fat diet triggers myelin disruption and interleukin-33 upregulation in hypothalamus. BMC Neurosci. 2019, 20, 33. [Google Scholar] [CrossRef]
- Zarpelon, A.C.; Rodrigues, F.C.; Lopes, A.H.; Souza, G.R.; Carvalho, T.T.; Pinto, L.G.; Xu, D.; Ferreira, S.H.; Alves-Filho, J.C.; McInnes, I.B.; et al. Spinal cord oligodendrocyte-derived alarmin IL-33 mediates neuropathic pain. Faseb J. 2016, 30, 54–65. [Google Scholar] [CrossRef]
- Bamberger, M.E.; Harris, M.E.; McDonald, D.R.; Husemann, J.; Landreth, G.E. A cell surface receptor complex for fibrillar b-amyloid mediates microglial activation. J. Neurosci. 2003, 23, 2665–2674. [Google Scholar] [CrossRef]
- Paresce, D.M.; Ghosh, R.N.; Maxfield, F.R. Microglial cells internalize aggregates of the Alzheimer’s disease amyloid beta-protein via a scavenger receptor. Neuron 1996, 17, 553–565. [Google Scholar] [CrossRef]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.Y.; Landreth, G.E. The role of microglia in amyloid clearance from the AD brain. J. Neural. Transm. (Vienna) 2010, 117, 949–960. [Google Scholar] [CrossRef]
- Mawuenyega, K.G.; Sigurdson, W.; Ovod, V.; Munsell, L.; Kasten, T.; Morris, J.C.; Yarasheski, K.E.; Bateman, R.J. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science 2010, 330, 1774. [Google Scholar] [CrossRef] [PubMed]
- Hickman, S.E.; Allison, E.K.; El Khoury, J. Microglial dysfunction and defective b-amyloid clearance pathways in aging Alzheimer’s disease mice. J. Neurosci. 2008, 28, 8354–8360. [Google Scholar] [CrossRef] [PubMed]
- Giraldo, E.; Lloret, A.; Fuchsberger, T.; Vina, J. Abeta and tau toxicities in Alzheimer’s are linked via oxidative stress-induced p38 activation: Protective role of vitamin E. Redox. Biol. 2014, 2, 873–877. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.L.; Yang, F.; Rosario, E.R.; Ubeda, O.J.; Beech, W.; Gant, D.J.; Chen, P.P.; Hudspeth, B.; Chen, C.; Zhao, Y.; et al. Beta-amyloid oligomers induce phosphorylation of tau and inactivation of insulin receptor substrate via c-Jun N-terminal kinase signaling: Suppression by omega-3 fatty acids and curcumin. J. Neurosci. 2009, 29, 9078–9089. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Wu, X.; Ren, L.; Liu, G.; Li, L. Epigenetic mechanisms of amyloid-beta production in anisomycin-treated SH-SY5Y cells. Neuroscience 2011, 194, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Tokay, T.; Hachem, R.; Masmoudi-Kouki, O.; Gandolfo, P.; Desrues, L.; Leprince, J.; Castel, H.; Diallo, M.; Amri, M.; Vaudry, H.; et al. Beta-amyloid peptide stimulates endozepine release in cultured rat astrocytes through activation of N-formyl peptide receptors. Glia 2008, 56, 1380–1389. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ye, D. Molecular biology for formyl peptide receptors in human diseases. J. Mol. Med. (Berl.) 2013, 91, 781–789. [Google Scholar] [CrossRef]
- He, H.Q.; Ye, R.D. The Formyl Peptide Receptors: Diversity of Ligands and Mechanism for Recognition. Molecules 2017, 22, 455. [Google Scholar] [CrossRef]
- Trojan, E.; Bryniarska, N.; Leskiewicz, M.; Regulska, M.; Chamera, K.; Szuster, M.; Leopoldo, M.; Lacivita, E.; Basta-Kaim, A. The contribution of formyl peptide receptor dysfunction to the course of neuroinflammation: A potential role in the brain pathology. Curr. Neuropharmacol. 2019. [Google Scholar] [CrossRef]
- Cussell, P.J.G.; Gomez Escalada, M.; Milton, N.G.N.; Paterson, A.W.J. The N-formyl peptide receptors: Contemporary roles in neuronal function and dysfunction. Neural. Regen. Res. 2020, 15, 1191–1198. [Google Scholar] [CrossRef]
- He, N.; Jin, W.L.; Lok, K.H.; Wang, Y.; Yin, M.; Wang, Z.J. Amyloid-b1-42 oligomer accelerates senescence in adult hippocampal neural stem/progenitor cells via formylpeptide receptor 2. Cell Death Dis. 2013, 4, e924. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wang, D.; Gong, P.; Lin, A.; Zhang, Y.; Ye, R.D.; Yu, Y. Formyl Peptide Receptor 2 Deficiency Improves Cognition and Attenuates Tau Hyperphosphorylation and Astrogliosis in a Mouse Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2019, 67, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Wilmes, M.; Sahl, H.G. Defensin-based anti-infective strategies. Int. J. Med. Microbiol. 2014, 304, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Kountouras, J.; Deretzi, G.; Gavalas, E.; Zavos, C.; Polyzos, S.A.; Kazakos, E.; Giartza-Taxidou, E.; Vardaka, E.; Kountouras, C.; Katsinelos, P.; et al. A proposed role of human defensins in Helicobacter pylori-related neurodegenerative disorders. Med. Hypotheses 2014, 82, 368–373. [Google Scholar] [CrossRef]
- Hosoda, H.; Nakamura, K.; Hu, Z.; Tamura, H.; Reich, J.; Kuwahara-Arai, K.; Iba, T.; Tabe, Y.; Nagaoaka, I. Antimicrobial cathelicidin peptide LL37 induces NET formation and suppresses the inflammatory response in a mouse septic model. Mol. Med. Rep. 2017, 16, 5618–5626. [Google Scholar] [CrossRef]
- De Lorenzi, E.; Chiari, M.; Colombo, R.; Cretich, M.; Sola, L.; Vanna, R.; Gagni, P.; Bisceglia, F.; Morasso, C.; Lin, J.S.; et al. Evidence that the Human Innate Immune Peptide LL-37 may be a Binding Partner of Amyloid-beta and Inhibitor of Fibril Assembly. J. Alzheimers Dis. 2017, 59, 1213–1226. [Google Scholar] [CrossRef]
- Yu, X.; Quan, J.; Long, W.; Chen, H.; Wang, R.; Guo, J.; Lin, X.; Mai, S. LL-37 inhibits LPS-induced inflammation and stimulates the osteogenic differentiation of BMSCs via P2X7 receptor and MAPK signaling pathway. Exp. Cell Res. 2018, 372, 178–187. [Google Scholar] [CrossRef]
- Hemshekhar, M.; Choi, K.G.; Mookherjee, N. Host Defense Peptide LL-37-Mediated Chemoattractant Properties, but Not Anti-Inflammatory Cytokine IL-1RA Production, Is Selectively Controlled by Cdc42 Rho GTPase via G Protein-Coupled Receptors and JNK Mitogen-Activated Protein Kinase. Front. Immunol. 2018, 9, 1871. [Google Scholar] [CrossRef]
- Zhao, L.; Tan, S.; Zhang, H.; Liu, P.; Tan, Y.Z.; Li, J.C.; Jia, D.; Shen, X.F. Astragalus polysaccharides exerts anti-infective activity by inducing human cathelicidin antimicrobial peptide LL-37 in respiratory epithelial cells. Phytother. Res. 2018, 32, 1521–1529. [Google Scholar] [CrossRef]
- Zhou, W.; Gao, B.; Zhu, S. Did cis- and trans-defensins derive from a common ancestor? Immunogenetics 2019, 71, 61–69. [Google Scholar] [CrossRef]
- Zhao, Z.; Nelson, A.R.; Betsholtz, C.; Zlokovic, B.V. Establishment and Dysfunction of the Blood-Brain Barrier. Cell 2015, 163, 1064–1078. [Google Scholar] [CrossRef] [PubMed]
- Kazakos, E.I.; Kountouras, J.; Polyzos, S.A.; Deretzi, G. Novel aspects of defensins’ involvement in virus-induced autoimmunity in the central nervous system. Med. Hypotheses 2017, 102, 33–36. [Google Scholar] [CrossRef] [PubMed]
- Jafarzadeh, A.; Nemati, M.; Khorramdelazad, H.; Mirshafiey, A. The Toll-like Receptor 2 (TLR2)-related Immunopathological Responses in the Multiple Sclerosis and Experimental Autoimmune Encephalomyelitis. Iran J. Allergy Asthma Immunol. 2019, 18, 230–250. [Google Scholar] [CrossRef] [PubMed]
- Kobatake, E.; Kabuki, T. S-Layer Protein of Lactobacillus helveticus SBT2171 Promotes Human beta-Defensin 2 Expression via TLR2-JNK Signaling. Front. Microbiol. 2019, 10, 2414. [Google Scholar] [CrossRef] [PubMed]
- Lewis, S.B.; Prior, A.; Ellis, S.J.; Cook, V.; Chan, S.S.; Gelson, W.; Schuller, S. Flagellin Induces beta-Defensin 2 in Human Colonic Ex vivo Infection with Enterohemorrhagic Escherichia coli. Front. Cell. Infect. Microbiol. 2016, 6, 68. [Google Scholar] [CrossRef] [PubMed]
- Bian, T.; Li, H.; Zhou, Q.; Ni, C.; Zhang, Y.; Yan, F. Human beta-Defensin 3 Reduces TNF-alpha-Induced Inflammation and Monocyte Adhesion in Human Umbilical Vein Endothelial Cells. Mediat. Inflamm. 2017, 2017, 8529542. [Google Scholar] [CrossRef]
- Ahn, J.K.; Huang, B.; Bae, E.K.; Park, E.J.; Hwang, J.W.; Lee, J.; Koh, E.M.; Cha, H.S. The role of alpha-defensin-1 and related signal transduction mechanisms in the production of IL-6, IL-8 and MMPs in rheumatoid fibroblast-like synoviocytes. Rheumatology (Oxford) 2013, 52, 1368–1376. [Google Scholar] [CrossRef]
- Earls, R.H.; Menees, K.B.; Chung, J.; Barber, J.; Gutekunst, C.A.; Hazim, M.G.; Lee, J.K. Intrastriatal injection of preformed alpha-synuclein fibrils alters central and peripheral immune cell profiles in non-transgenic mice. J. Neuroinflammation 2019, 16, 250. [Google Scholar] [CrossRef]
- Ferreira, S.A.; Romero-Ramos, M. Microglia Response During Parkinson’s Disease: Alpha-Synuclein Intervention. Front. Cell. Neurosci. 2018, 12, 247. [Google Scholar] [CrossRef]
- Yu, W.W.; Cao, S.N.; Zang, C.X.; Wang, L.; Yang, H.Y.; Bao, X.Q.; Zhang, D. Heat shock protein 70 suppresses neuroinflammation induced by α-synuclein in astrocytes. Mol. Cell. Neurosci. 2018, 86, 58–64. [Google Scholar] [CrossRef]
- Christensen, D.P.; Ejlerskov, P.; Rasmussen, I.; Vilhardt, F. Reciprocal signals between microglia and neurons regulate α-synuclein secretion by exophagy through a neuronal cJUN-N-terminal kinase-signaling axis. J. Neuroinflammation 2016, 13, 59. [Google Scholar] [CrossRef]
- Lei, Z.; Cao, G.; Wei, G. A30P mutant α-synuclein impairs autophagic flux by inactivating JNK signaling to enhance ZKSCAN3 activity in midbrain dopaminergic neurons. Cell Death Dis. 2019, 10, 133. [Google Scholar] [CrossRef]
- Zhang, Y.; Wu, Q.; Zhang, L.; Wang, Q.; Yang, Z.; Liu, J.; Feng, L. Caffeic acid reduces A53T α-synuclein by activating JNK/Bcl-2-mediated autophagy in vitro and improves behaviour and protects dopaminergic neurons in a mouse model of Parkinson’s disease. Pharmacol. Res. 2019, 150, 104538. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Calvani, R.; Coelho-Junior, H.J.; Landi, F.; Bernabei, R.; Marzetti, E. Mitochondrial Dysfunction, Oxidative Stress, and Neuroinflammation: Intertwined Roads to Neurodegeneration. Antioxidants 2020, 9, 647. [Google Scholar] [CrossRef]
- Masouris, I.; Klein, M.; Dyckhoff, S.; Angele, B.; Pfister, H.W.; Koedel, U. Inhibition of DAMP signaling as an effective adjunctive treatment strategy in pneumococcal meningitis. J. Neuroinflammation 2017, 14, 214. [Google Scholar] [CrossRef]
- Finlay, W.J.; Cunningham, O.; Lambert, M.A.; Darmanin-Sheehan, A.; Liu, X.; Fennell, B.J.; Mahon, C.M.; Cummins, E.; Wade, J.M.; O’Sullivan, C.M.; et al. Affinity maturation of a humanized rat antibody for anti-RAGE therapy: Comprehensive mutagenesis reveals a high level of mutational plasticity both inside and outside the complementarity-determining regions. J. Mol. Biol. 2009, 388, 541–558. [Google Scholar] [CrossRef]


{kind=link}
| Alarmin | Origin | Receptor | Potential Biological Effects | Ref. |
|---|---|---|---|---|
| HMGB1 | Tissue damage | TLR4, RAGE | ROS-mediated JNK activation; NADPH-dependent ROS generation; oxidative stress signaling; autophagy; apoptosis; metabolic impairment | [119,120,121,122,123,124,125] |
| BAG3 | Stressful stimuli | Hsc70/Hsp70 ATPase domain | Chaperone-assisted selective autophagy; hsp70-dependent and independent functions; maintaining the intracellular levels of anti-apoptotic factors and other molecules; protein quality control; cytoskeleton dynamics; structural and functional roles in myocytes | [139,140,141] |
| S100 | Damaged cells | RAGE | ROS-mediated JNK activation; NADPH-dependent ROS generation | [125] |
| β-amyloid | AD pathogenesis | RAGE | ROS-mediated JNK activation; NADPH-dependent ROS generation | [125] |
| S1P | Activated platelets in the vasculature | Phagocytosis receptors, including MerTK and MFG-E8 on macrophages | Efficient phagocytosis; recruitment and priming of macrophages; | [75,76] |
| Spz5 | Cell damage or necrotic death | Toll-1 receptor | Prepares, or primes, glia for phagocytosis in the CNS; activates M1-relevant ERK1/2 and JNK in post-ischemic brain | [72,77] |
| Fractalkine | Apoptotic neurons | CX3CR1 | Activation of the proinflammatory pathway mediated by NF-κB as an early response in microglial cells | [78,79] |
| Hsp | Stressful conditions | TLR family | Inhibition of both aminoglycoside- and cisplatin-induced hair cell death in whole-organ cultures of utricles from adult mice | [112,113] |
| Hsp32 | Trauma; hemorrhage; H2S preconditioning | Neuroprotection; mediation of the protective effect of celastrol; inhibition of pro-apoptotic JNK activation and hair cell death | [112] | |
| Hsp70 | Necrotic cells; paraquat-induced oxidative stress; caspase-3-mediated dopaminergic neuronal cell death | c-Type lectin receptors (CLR) and scavenger receptors (SR) | Reduction of paraquat-induced oxidative stress, JNK- and caspase-3-mediated dopaminergic neuronal cell death; decrease in the activated forms of JNK and p38 in the hippocampus of a rat model of fear memory consolidation | [107] |
| HSP/c70 | Damaged astrocytes | TLR4 | Activation of JNK in macrophage RAW264.7 cells | [106] |
| Hsp90 | Stressful conditions | Glucocorticoid receptor | Neuroprotection | [114] |
| α-Synuclein | PD pathogenesis | Innate and adaptive immune responses; direct effects on immune cells, including microglia, initiating a sterile response essential for the neuronal health and translating in a peripheral immune response | [189,190] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anfinogenova, N.D.; Quinn, M.T.; Schepetkin, I.A.; Atochin, D.N. Alarmins and c-Jun N-Terminal Kinase (JNK) Signaling in Neuroinflammation. Cells 2020, 9, 2350. https://doi.org/10.3390/cells9112350
Anfinogenova ND, Quinn MT, Schepetkin IA, Atochin DN. Alarmins and c-Jun N-Terminal Kinase (JNK) Signaling in Neuroinflammation. Cells. 2020; 9(11):2350. https://doi.org/10.3390/cells9112350
Chicago/Turabian StyleAnfinogenova, Nina D., Mark T. Quinn, Igor A. Schepetkin, and Dmitriy N. Atochin. 2020. "Alarmins and c-Jun N-Terminal Kinase (JNK) Signaling in Neuroinflammation" Cells 9, no. 11: 2350. https://doi.org/10.3390/cells9112350
APA StyleAnfinogenova, N. D., Quinn, M. T., Schepetkin, I. A., & Atochin, D. N. (2020). Alarmins and c-Jun N-Terminal Kinase (JNK) Signaling in Neuroinflammation. Cells, 9(11), 2350. https://doi.org/10.3390/cells9112350