Tafazzin Mutation Affecting Cardiolipin Leads to Increased Mitochondrial Superoxide Anions and Mitophagy Inhibition in Barth Syndrome



Abstract
:
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Transfections
2.2. Lipid Analysis
2.3. Analysis of HeLa Cell Proliferation Using xCELLigence
2.4. Cellular Bioenergetic Assays of Mitochondrial Functions
2.5. AMP/ADP/ATP Determination by HPLC and ATP by Luminometry
2.6. Confocal Microscopy
2.7. Microspectrofluorimetry
2.8. Acridine Orange Staining of the Acidic Compartment
2.9. Determination of Lipid Peroxidation and Protein Carbonylation
2.10. Oxidative Stress, Production of Reactive Oxygen Species, and Mitochondrial Membrane Potential
2.11. NAD(P)H and NADH Determination
2.12. Caspase 3/7 Activation
2.13. Mitochondrial DNA Assay
2.14. Specific Lysosomal Staining
2.15. Detection of Lysosomes and Autophagolysosomes with Monodansylcadaverine (MDC)
2.16. Transmission Electron Microscopy
2.17. Immunoblotting
2.18. Semiquantitative and Real-Time RT-PCR
2.19. 3[H]-Leucine Incorporation Assay
2.20. Transfection and Luciferase Assay
2.21. Statistical Analysis
3. Results
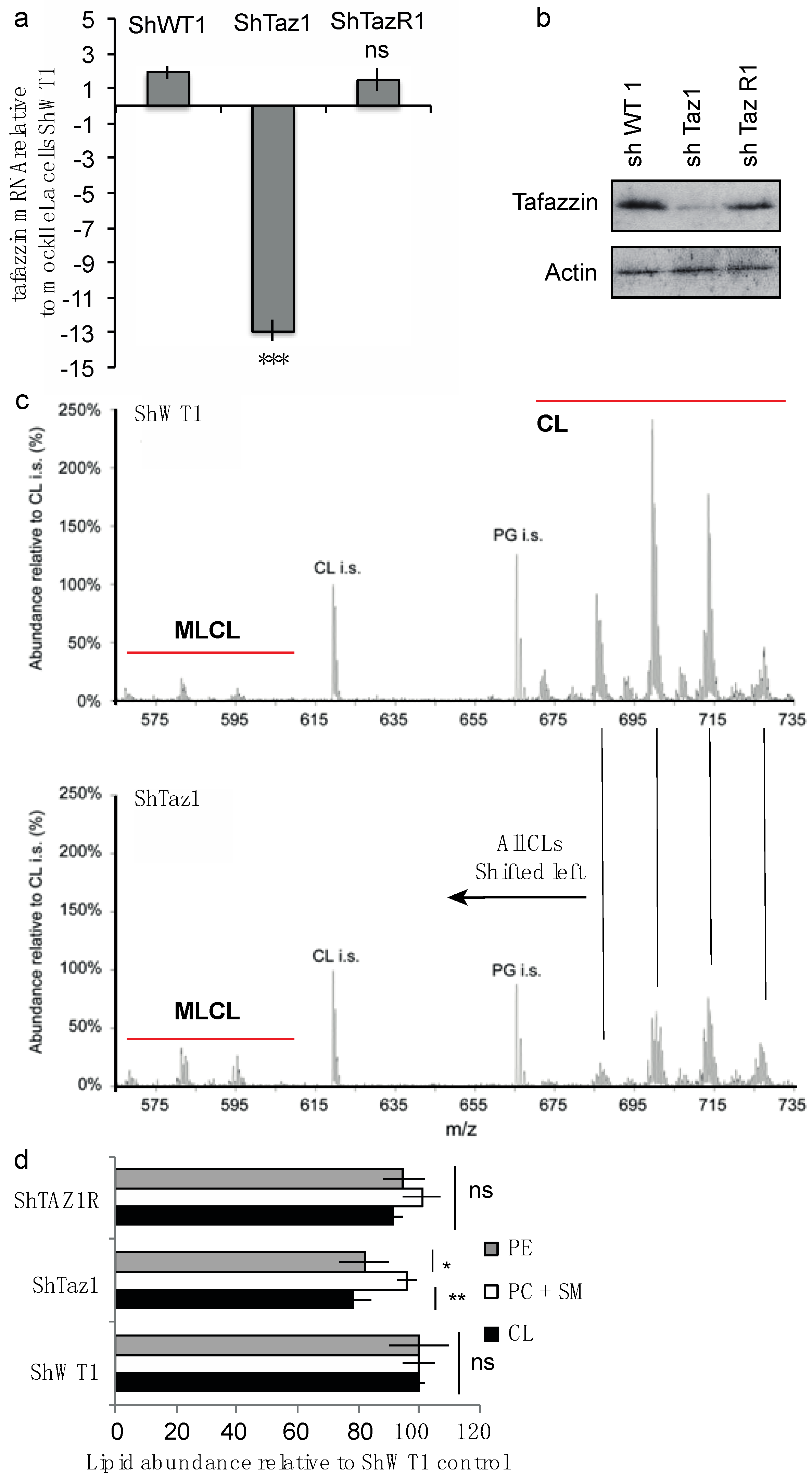
3.1. Biochemical Features of ShTaz1 and ShTaz1R Cells Versus ShWT1 HeLa Cells
3.1.1. Cardiolipin Modifications
3.1.2. Early Apoptosis-Associated Events are also Inhibited
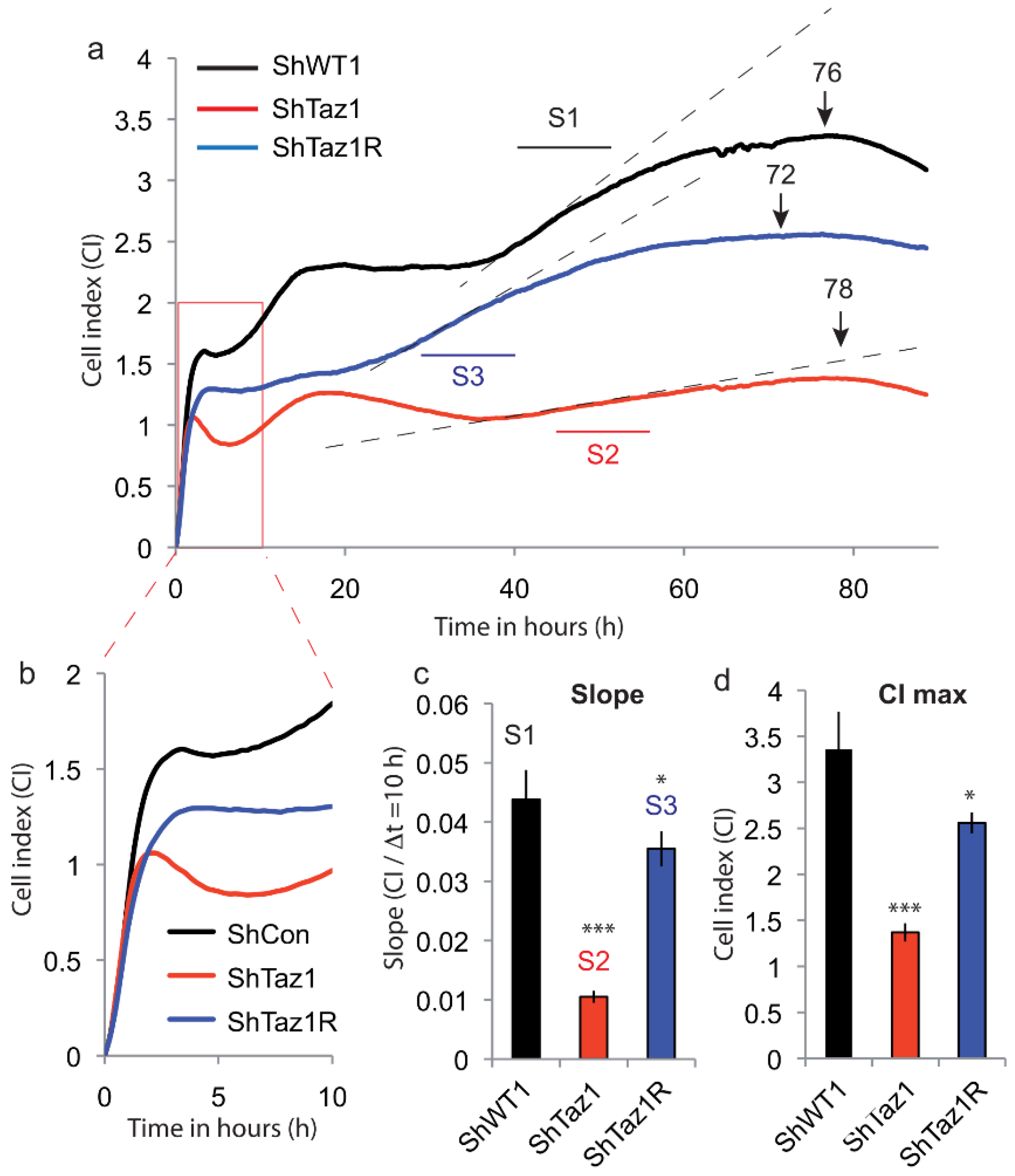
3.2. Cell Adhesion and Proliferation
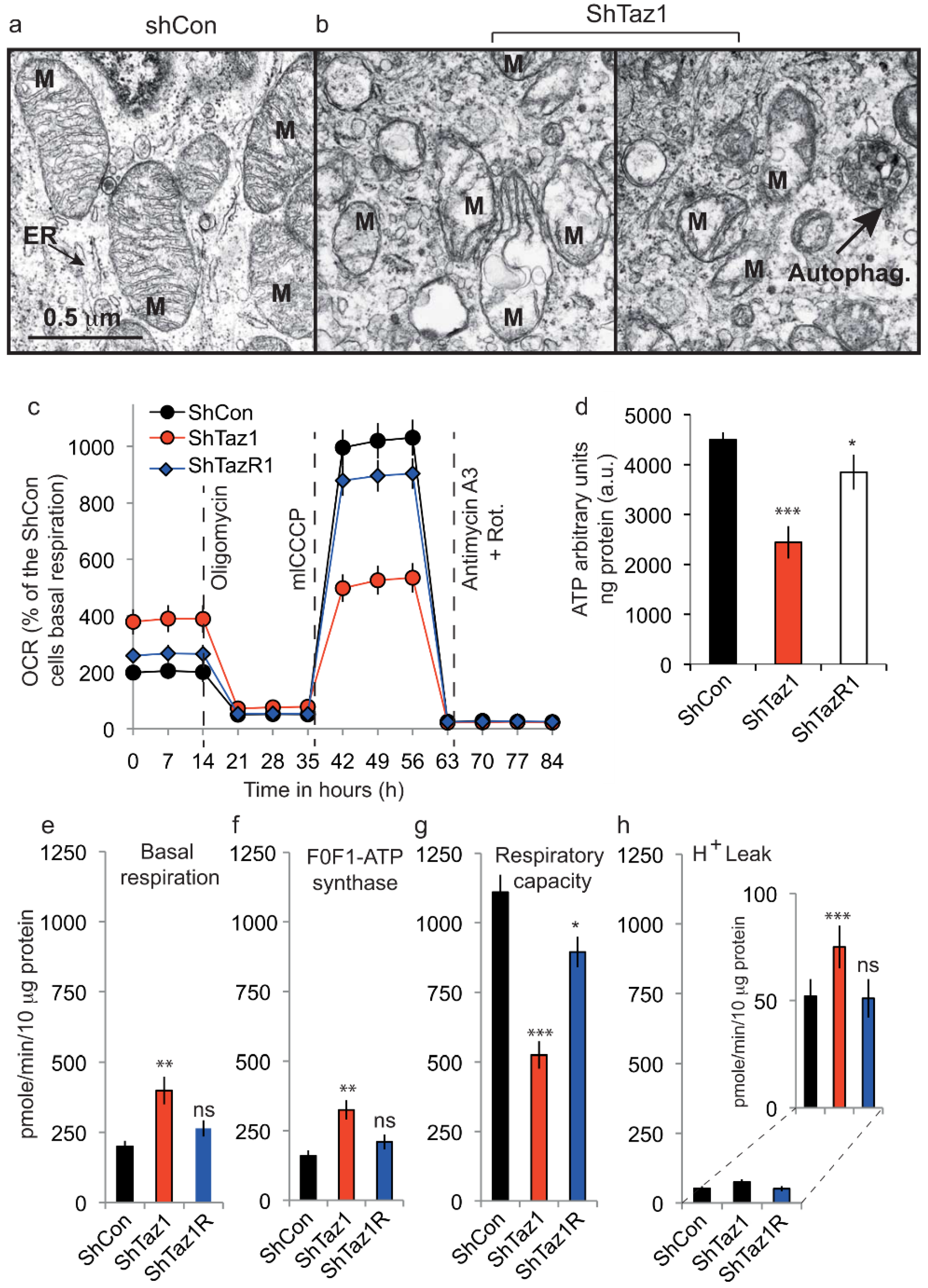
3.3. Cellular Bioenergetics
3.3.1. Electron Microscopy of the ShTaz1 Tafazzin Mutant
3.3.2. Cellular Light Scattering Properties and Confocal Images Revealed ShTaz1 Cell Hypertrophy
3.3.3. Seahorse Respiratory Measurements on intact Cells
3.4. Tafazzin Knockdown Activates AMPK and Increases Mitochondrial Density
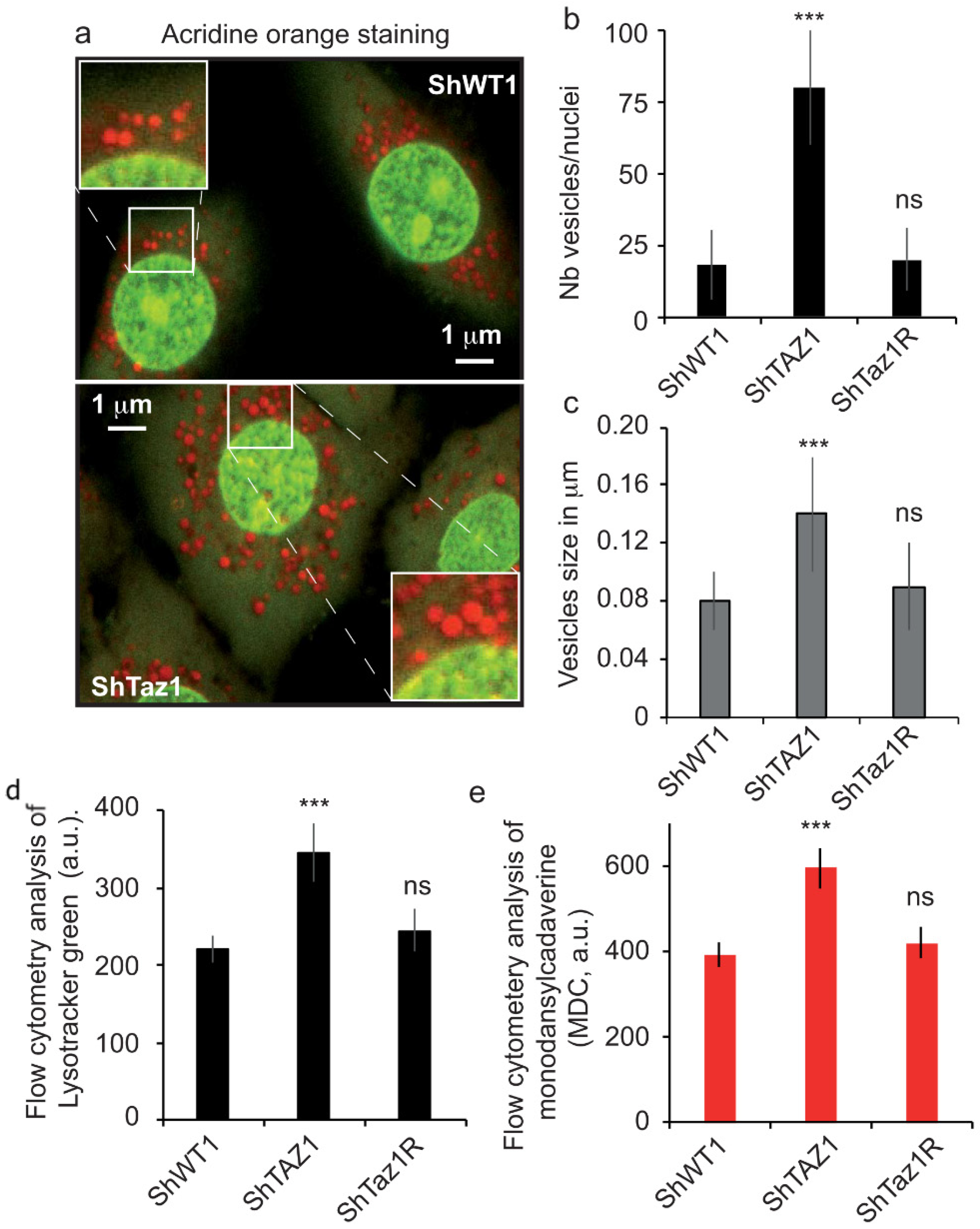
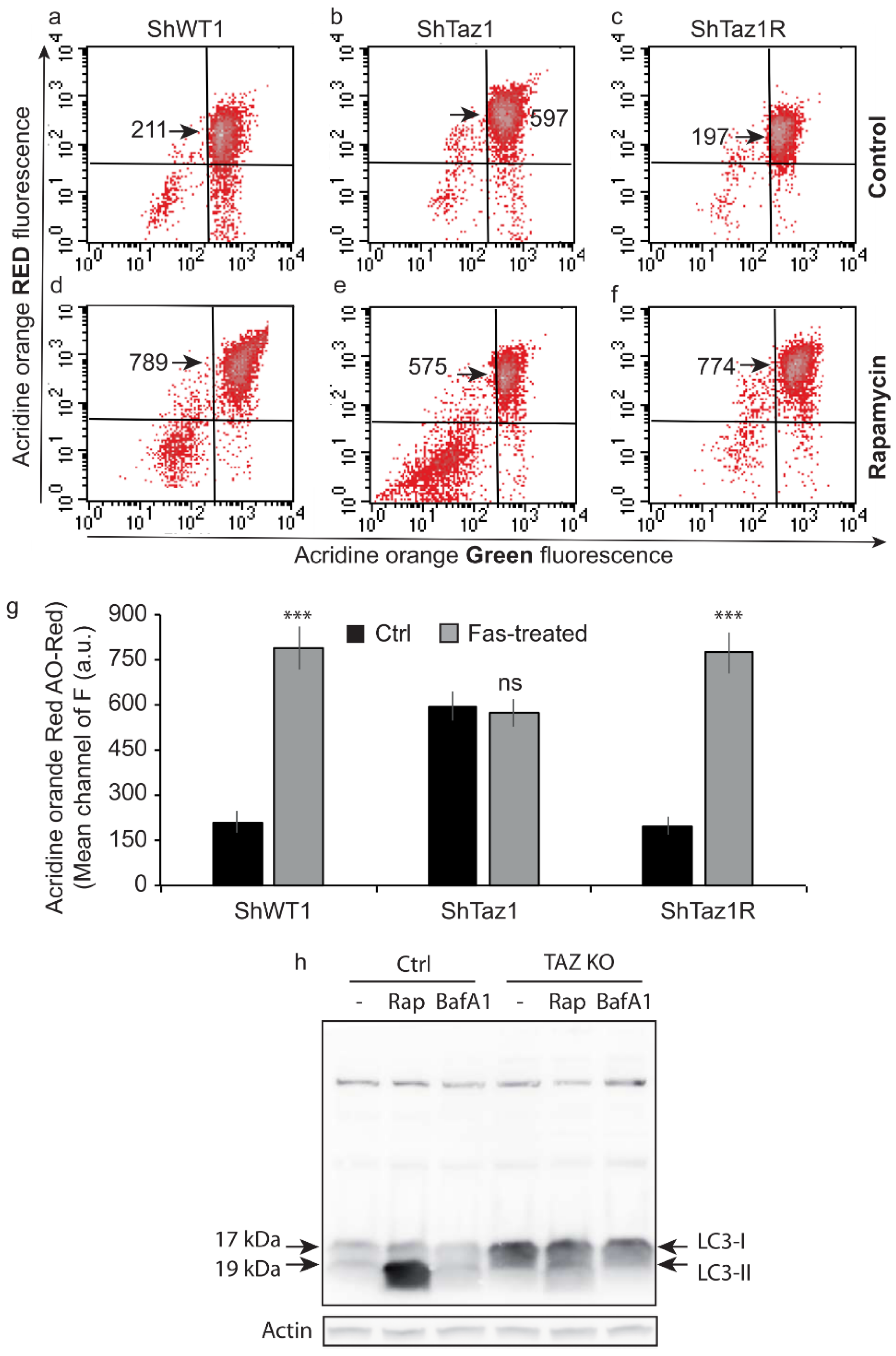
3.5. Acidic Compartments and Autophagy Inhibition
3.6. Abolition of Mitophagy in Tafazzin Cell Mutants
3.7. Changes of the Acidic Compartment in Normal and Rapamycin-Induced Autophagy
3.8. Assessment of Autophagic Processes with LC3-I and LC3-II
4. Discussion
4.1. Inhibition of the Extrinsic Apoptotic Pathway in Tafazzin-Deficient Cells
4.2. Alterations and Characteristics of Tafazzin-Knockdown HeLa Cell Proliferation
4.3. Bioenergetic Defects Lie in Mitochondrial Structure and Mitochondrial Homeostasis Destabilization
4.4. Mitophagy Inhibition Due to Defective Cardiolipin in the Tafazzin-Deficient HeLa Cell Mutant
4.5. Mitochondrial Dysfunctions are Compensated at the Cellular Level
4.6. Accumulation of Acidic Vesicles and Inhibition of Mitophagy
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Barth, P.; Scholte, H.; Berden, J.; Moorsel, J.V.D.K.-V.; Luyt-Houwen, I.; Veer-Korthof, E.V.; Van Der Harten, J.; Sobotka-Plojhar, M. An X-linked mitochondrial disease affecting cardiac muscle, skeletal muscle and neutrophil leucocytes. J. Neurol. Sci. 1983, 62, 327–355. [Google Scholar] [CrossRef]
- Clarke, S.L.; Bowron, A.; Gonzalez, I.L.; Groves, S.J.; Newbury-Ecob, R.; Clayton, N.; Martin, R.P.; Tsai-Goodman, B.; Garratt, V.; Ashworth, M.; et al. Barth syndrome. Orphanet J. Rare Dis. 2013, 8, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christodoulou, J.; McInnes, R.R.; Jay, V.; Wilson, G.; Becker, L.E.; Lehotay, D.C.; Platt, B.-A.; Bridge, P.J.; Robinson, B.H.; Clarke, J.T.R. Barth syndrome: Clinical observations and genetic linkage studies. Am. J. Med. Genet. 1994, 50, 255–264. [Google Scholar] [CrossRef]
- Johnston, J.; Kelley, R.I.; Feigenbaum, A.; Cox, G.F.; Iyer, G.S.; Funanage, V.L.; Proujansky, R. Mutation characterization and genotype-phenotype correlation in Barth syndrome. Am. J. Hum. Genet. 1997, 61, 1053–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronvelia, D.; Greenwood, J.; Platt, J.; Hakim, S.; Zaragoza, M.V. Intrafamilial variability for novel TAZ gene mutation: Barth syndrome with dilated cardiomyopathy and heart failure in an infant and left ventricular noncompaction in his great-uncle. Mol. Genet. Metab. 2012, 107, 428–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jefferies, J.L. Barth syndrome. Am. J. Med. Genet. C Semin. Med. Genet. 2013, 163C, 198–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bione, S.; D’Adamo, P.; Maestrini, E.; Gedeon, A.K.; Bolhuis, P.A.; Toniolo, D. A novel X-linked gene, G4.5. is responsible for Barth syndrome. Nat. Genet. 1996, 12, 385–389. [Google Scholar] [CrossRef]
- Kirwin, S.M.; Manolakos, A.; Barnett, S.S.; Gonzalez, I.L. Tafazzin splice variants and mutations in Barth syndrome. Mol. Genet. Metab. 2014, 111, 26–32. [Google Scholar] [CrossRef]
- Esaric, A.; Eandreau, K.; Earmand, A.-S.; Möller, I.M.; Petit, P.X. Barth Syndrome: From Mitochondrial Dysfunctions Associated with Aberrant Production of Reactive Oxygen Species to Pluripotent Stem Cell Studies. Front. Genet. 2015, 6, 359. [Google Scholar] [CrossRef] [Green Version]
- Greenberg, M.L.; Lopes, J.M. Genetic regulation of phospholipid biosynthesis in Saccharomyces cerevisiae. Microbiol. Rev. 1996, 60, 1–20. [Google Scholar] [CrossRef]
- Xu, Y.; Malhotra, A.; Ren, M.; Schlame, M. The Enzymatic Function of Tafazzin. J. Biol. Chem. 2006, 281, 39217–39224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, A.S.; Zhou, J.; Gohil, V.M.; Chen, S.; Greenberg, M.L. Cellular functions of cardiolipin in yeast. Biochim. Biophys. Acta BBA Bioenerg. 2009, 1793, 212–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrosillo, G.; Ruggiero, F.M.; Paradies, G. Role of reactive oxygen species and cardiolipin in the release of cytochrome c from mitochondria. FASEB J. 2003, 17, 2202–2208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ott, M.; Robertson, J.D.; Gogvadze, V.; Zhivotovsky, B.; Orrenius, S. Cytochrome c release from mitochondria proceeds by a two-step process. Proc. Natl. Acad. Sci. USA 2002, 99, 1259–1263. [Google Scholar] [CrossRef] [Green Version]
- Du, C.; Fang, M.; Li, Y.; Li, L.; Wang, X. Smac, a Mitochondrial Protein that Promotes Cytochrome c–Dependent Caspase Activation by Eliminating IAP Inhibition. Cell 2000, 102, 33–42. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Nijhawan, D.; Budihardjo, I.; Srinivasula, S.M.; Ahmad, M.; Alnemri, E.S.; Wang, X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 1997, 91, 479–489. [Google Scholar] [CrossRef] [Green Version]
- Chicco, A.J.; Sparagna, G.C. Role of cardiolipin alterations in mitochondrial dysfunction and disease. Am. J. Physiol. Cell Physiol. 2007, 292, C33–C44. [Google Scholar] [CrossRef] [Green Version]
- Jiang, F.; Ryan, M.T.; Schlame, M.; Zhao, M.; Gu, Z.; Klingenberg, M.; Pfanner, N.; Greenberg, M.L. Absence of Cardiolipin in thecrd1Null Mutant Results in Decreased Mitochondrial Membrane Potential and Reduced Mitochondrial Function. J. Biol. Chem. 2000, 275, 22387–22394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Böttinger, L.; Ellenrieder, L.; Becker, T. How lipids modulate mitochondrial protein import. J. Bioenerg. Biomembr. 2016, 48, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Bazán, S.; Mileykovskaya, E.; Mallampalli, V.K.P.S.; Heacock, P.; Sparagna, G.C.; Dowhan, W. Cardiolipin-dependent Reconstitution of Respiratory Supercomplexes from Purified Saccharomyces cerevisiae Complexes III and IV. J. Biol. Chem. 2013, 288, 401–411. [Google Scholar] [CrossRef] [Green Version]
- Ban, T.; Heymann, J.A.; Song, Z.; Hinshaw, J.E.; Chan, D.C. OPA1 disease alleles causing dominant optic atrophy have defects in cardiolipin-stimulated GTP hydrolysis and membrane tubulation. Hum. Mol. Genet. 2010, 19, 2113–2122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gebert, N.; Joshi, A.S.; Kutik, S.; Becker, T.; McKenzie, M.; Guan, X.L.; Mooga, V.P.; Stroud, D.A.; Kulkarni, G.; Wenk, M.R.; et al. Mitochondrial cardiolipin involved in outer-membrane protein biogenesis: Implications for Barth syndrome. Curr. Biol. 2009, 19, 2133–2139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, Z.; Valianpour, F.; Chen, S.; Vaz, F.M.; Hakkaart, G.A.; Wanders, R.J.A.; Greenberg, M.L. Aberrant cardiolipin metabolism in the yeast taz1 mutant: A model for Barth syndrome. Mol. Microbiol. 2004, 51, 149–158. [Google Scholar] [CrossRef]
- Moreadith, R.W.; Lehninger, A.L. The pathways of glutamate and glutamine oxidation by tumor cell mitochondria. Role of mitochondrial NAD(P)+-dependent malic enzyme. J. Biol. Chem. 1984, 259, 6215–6221. [Google Scholar] [PubMed]
- Gaspard, G.J.; McMaster, C.R. The Mitochondrial Quality Control Protein Yme1 Is Necessary to Prevent Defective Mitophagy in a Yeast Model of Barth Syndrome. J. Biol. Chem. 2015, 290, 9284–9298. [Google Scholar] [CrossRef] [Green Version]
- Xu, F.-P.; Chen, M.-S.; Wang, Y.-Z.; Yi, Q.; Lin, S.-B.; Chen, A.F.; Luo, J.-D. Leptin Induces Hypertrophy via Endothelin-1–Reactive Oxygen Species Pathway in Cultured Neonatal Rat Cardiomyocytes. Circulation 2004, 110, 1269–1275. [Google Scholar] [CrossRef] [Green Version]
- Khuchua, Z.; Yue, Z.; Batts, L.; Strauss, A.W. A Zebrafish Model of Human Barth Syndrome Reveals the Essential Role of Tafazzin in Cardiac Development and Function. Circ. Res. 2006, 99, 201–208. [Google Scholar] [CrossRef] [Green Version]
- Saini-Chohan, H.K.; Holmes, M.G.; Chicco, A.J.; Taylor, W.A.; Moore, R.L.; McCune, S.A.; Hickson-Bick, D.L.; Hatch, G.M.; Sparagna, G.C. Cardiolipin biosynthesis and remodeling enzymes are altered during development of heart failure. J. Lipid Res. 2009, 50, 1600–1608. [Google Scholar] [CrossRef] [Green Version]
- Chatzispyrou, I.A.; Guerrero-Castillo, S.; Held, N.M.; Ruiter, J.P.; Denis, S.W.; Ijlst, L.; Wanders, R.J.; Van Weeghel, M.; Ferdinandusse, S.; Vaz, F.M.; et al. Barth syndrome cells display widespread remodeling of mitochondrial complexes without affecting metabolic flux distribution. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2018, 1864, 3650–3658. [Google Scholar] [CrossRef]
- Pfeiffer, K.; Gohil, V.; Stuart, R.A.; Hunte, C.; Brandt, U.; Greenberg, M.L.; Schägger, H. Cardiolipin Stabilizes Respiratory Chain Supercomplexes. J. Biol. Chem. 2003, 278, 52873–52880. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Mileykovskaya, E.; Dowhan, W. Gluing the respiratory chain together. Cardiolipin is required for supercomplex formation in the inner mitochondrial membrane. J. Biol. Chem. 2002, 277, 43553–43556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeVay, R.M.; Dominguez-Ramirez, L.; Lackner, L.L.; Hoppins, S.; Stahlberg, H.; Nunnari, J. Coassembly of Mgm1 isoforms requires cardiolipin and mediates mitochondrial inner membrane fusion. J. Cell Biol. 2009, 186, 793–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, A.S.; Thompson, M.N.; Fei, N.; Hüttemann, M.; Greenberg, M.L. Cardiolipin and Mitochondrial Phosphatidylethanolamine Have Overlapping Functions in Mitochondrial Fusion inSaccharomyces cerevisiae. J. Biol. Chem. 2012, 287, 17589–17597. [Google Scholar] [CrossRef] [Green Version]
- Patil, V.A.; Fox, J.L.; Gohil, V.M.; Winge, D.R.; Greenberg, M.L. Loss of Cardiolipin Leads to Perturbation of Mitochondrial and Cellular Iron Homeostasis. J. Biol. Chem. 2013, 288, 1696–1705. [Google Scholar] [CrossRef] [Green Version]
- Ikon, N.; Su, B.; Hsu, F.-F.; Forteand, T.M.; Ryan, R.O. Exogenous cardiolipin localizes to mitochondria and prevents TAZ knockdown-induced apoptosis in myeloid progenitor cells. Biochem. Biophys. Res. Commun. 2015, 464, 580–585. [Google Scholar] [CrossRef] [Green Version]
- Li, X.-X.; Tsoi, B.; Li, Y.-F.; Kurihara, H.; He, R.-R. Cardiolipin and Its Different Properties in Mitophagy and Apoptosis. J. Histochem. Cytochem. 2015, 63, 301–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalvez, F.; D’Aurelio, M.; Boutant, M.; Moustapha, A.; Puech, J.-P.; Landes, T.; Arnauné-Pelloquin, L.; Vial, G.; Taleux, N.; Slomianny, C.; et al. Barth syndrome: Cellular compensation of mitochondrial dysfunction and apoptosis inhibition due to changes in cardiolipin remodeling linked to tafazzin (TAZ) gene mutation. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2013, 1832, 1194–1206. [Google Scholar] [CrossRef] [Green Version]
- Heit, B.; Yeung, T.; Grinstein, S. Changes in mitochondrial surface charge mediate recruitment of signaling molecules during apoptosis. Am. J. Physiol. Physiol. 2011, 300, C33–C41. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.-H.; Zhao, Y.; Ding, W.-X.; Na Shin, J.; He, X.; Seo, Y.-W.; Chen, J.; Rabinowich, H.; Amoscato, A.; Yin, X.-M. Bid-Cardiolipin Interaction at Mitochondrial Contact Site Contributes to Mitochondrial Cristae Reorganization and Cytochrome c Release. Mol. Biol. Cell 2004, 15, 3061–3072. [Google Scholar] [CrossRef] [Green Version]
- Manganelli, V.; Capozzi, A.; Recalchi, S.; Signore, M.; Mattei, V.; Garofalo, T.; Misasi, R.; Degli Esposti, M.; Sorice, M. Altered Traffic of Cardiolipin during Apoptosis: Exposure on the Cell Surface as a Trigger for “Antiphospholipid Antibodies”. J. Immunol. Res. 2015, 2015, 1–9. [Google Scholar] [CrossRef] [Green Version]
- McMillin, J.B.; Dowhan, W. Cardiolipin and apoptosis. Biochim. Biophys. Acta 2002, 1585, 97–107. [Google Scholar] [CrossRef]
- Chu, C.T.; Bayir, H.; Kagan, V.E. LC3 binds externalized cardiolipin on injured mitochondria to signal mitophagy in neurons: Implications for Parkinson disease. Autophagy 2014, 10, 376–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlame, M.; Rua, D.; Greenberg, M.L. The biosynthesis and functional role of cardiolipin. Prog. Lipid Res. 2000, 39, 257–288. [Google Scholar] [CrossRef]
- Koshkin, V.; Greenberg, M.L. Oxidative phosphorylation in cardiolipin-lacking yeast mitochondria. Biochem. J. 2000, 347, 687. [Google Scholar] [CrossRef]
- Kadenbach, B.; Mende, P.; Kolbe, H.; Stipani, I.; Palmieri, F. The mitochondrial phosphate carrier has an essential requirement for cardiolipin. FEBS Lett. 1982, 139, 109–112. [Google Scholar] [CrossRef] [Green Version]
- Robinson, N.C. Functional binding of cardiolipin to cytochrome c oxidase. J. Bioenerg. Biomembr. 1993, 25, 153–163. [Google Scholar] [CrossRef]
- Noel, H.; Pande, S.V. An essential requirement of cardiolipin for mitochondrial carnitine acylcarnitine translocase activity. Lipid requirement of carnitine acylcarnitine translocase. JBIC J. Biol. Inorg. Chem. 1986, 155, 99–102. [Google Scholar] [CrossRef]
- Vaz, F.; Houtkooper, R.H.; Valianpour, F.; Barth, P.G.; Wanders, R.J.A. Only One Splice Variant of the HumanTAZGene Encodes a Functional Protein with a Role in Cardiolipin Metabolism. J. Biol. Chem. 2003, 278, 43089–43094. [Google Scholar] [CrossRef] [Green Version]
- Brandner, K.; Mick, D.U.; Frazier, A.E.; Taylor, R.D.; Meisinger, C.; Rehling, P. Taz1, an Outer Mitochondrial Membrane Protein, Affects Stability and Assembly of Inner Membrane Protein Complexes: Implications for Barth Syndrome. Mol. Biol. Cell 2005, 16, 5202–5214. [Google Scholar] [CrossRef]
- Gonzalvez, F.; Schug, Z.T.; Houtkooper, R.H.; MacKenzie, E.D.; Brooks, D.G.; Wanders, R.J.; Petit, P.X.; Vaz, F.M.; Gottlieb, E. Cardiolipin provides an essential activating platform for caspase-8 on mitochondria. J. Cell Biol. 2008, 183, 681–696. [Google Scholar] [CrossRef] [Green Version]
- Gonzalvez, F.; Pariselli, F.; Jalmar, O.; Dupaigne, P.; Sureau, F.; Dellinger, M.; Hendrickson, E.A.; Bernard, S.; Petit, P.X. Mechanistic Issues of the Interaction of the Hairpin-Forming Domain of tBid with Mitochondrial Cardiolipin. PLoS ONE 2010, 5, e9342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valianpour, F.; Wanders, R.J.; Overmars, H.; Vreken, P.; Van Gennip, A.H.; Baas, F.; Plecko, B.; Santer, R.; Becker, K.; Barth, P.G. Cardiolipin deficiency in x-linked cardioskeletal myopathy and neutropenia (barth syndrome, mim 302060): A study in cultured skin fibroblasts. J. Pediatr. 2002, 141, 729–733. [Google Scholar] [CrossRef] [PubMed]
- Solly, K.; Wang, X.; Xu, X.; Strulovici, B.; Zheng, W. Application of real-time cell electronic sensing (RT-CES) technology to cell-based assays. Assay Drug Dev. Technol. 2004, 2, 363–372. [Google Scholar] [CrossRef]
- Sureau, F.; Chinsky, L.; Duquesne, M.; Laigle, A.; Turpin, P.; Amirand, C.; Ballini, J.; Vigny, P. Microspectrofluorimetric study of the kinetics of cellular uptake and metabolization of benzo(a)pyrene in human T 47 D mammary tumor cells: Evidence for cytochrome P1450 induction. Eur. Biophys. J. 1990, 18, 301–307. [Google Scholar] [CrossRef]
- De Oyanguren, F.J.S.; Rainey, N.E.; Moustapha, A.; Saric, A.; Sureau, F.; O’Connor, J.; Petit, P.X. Highlighting Curcumin-Induced Crosstalk between Autophagy and Apoptosis as Supported by Its Specific Subcellular Localization. Cells 2020, 9, 361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Traganos, F.; Darzynkiewicz, Z. Chapter 12 Lysosomal Proton Pump Activity: Supravital Cell Staining with Acridine Orange Differentiates Leukocyte Subpopulations. Methods Cell Biol. 1994, 41, 185–194. [Google Scholar] [CrossRef]
- Thomé, M.P.; Filippi-Chiela, E.C.; Villodre, E.S.; Migliavaca, C.B.; Onzi, G.R.; Felipe, K.B.; Lenz, G. Ratiometric analysis of Acridine Orange staining in the study of acidic organelles and autophagy. J. Cell Sci. 2016, 129, 4622–4632. [Google Scholar] [CrossRef] [Green Version]
- Gendron, M.C.; Schrantz, N.; Metivier, D.; Kroemer, G.; Maciorowska, Z.; Sureau, F.; Koester, S.; Petit, P.X. Oxidation of pyridine nucleotides during Fas- and ceramide-induced apoptosis in Jurkat cells: Correlation with changes in mitochondria, glutathione depletion, intracellular acidification and caspase 3 activation. Biochem. J. 2001, 353, 357–367. [Google Scholar]
- Niemann, A.; Takatsuki, A.; Elsässer, H.-P. The lysosomotropic agent monodansylcadaverine also acts as a solvent polarity probe. J. Histochem. Cytochem. 2000, 48, 251–258. [Google Scholar] [CrossRef] [Green Version]
- He, Q. Tafazzin knockdown causes hypertrophy of neonatal ventricular myocytes. Am. J. Physiol. Circ. Physiol. 2010, 299, H210–H216. [Google Scholar] [CrossRef] [Green Version]
- Harding, P.; Yang, X.-P.; Yang, J.; Shesely, E.; He, Q.; Lapointe, M.C. Gene expression profiling of dilated cardiomyopathy in older male EP4 knockout mice. Am. J. Physiol. Circ. Physiol. 2010, 298, H623–H632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winer, J.; Jung, C.K.; Shackel, I.; Williams, P.M. Development and validation of real-time quantitative reverse transcriptase-polymerase chain reaction for monitoring gene expression in cardiac myocytes in vitro. Anal. Biochem. 1999, 270, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Harding, P.; Carretero, O.A.; LaPointe, M.C. Effects of interleukin-1 beta and nitric oxide on cardiac myocytes. Hypertension 1995, 25, 421–430. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Harding, P.; Lapointe, M.C. PKA, Rap1, ERK1/2, and p90RSK mediate PGE2 and EP4 signaling in neonatal ventricular myocytes. Am. J. Physiol. Circ. Physiol. 2009, 298, H136–H143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lapointe, M.C.; Wu, G.; Garami, M.; Yang, X.-P.; Gardner, D.G. Tissue-Specific Expression of the Human Brain Natriuretic Peptide Gene in Cardiac Myocytes. Hypertension 1996, 27, 715–722. [Google Scholar] [CrossRef] [PubMed]
- Kho, D.; Macdonald, C.; Johnson, R.; Unsworth, C.P.; O’Carroll, S.J.; Du Mez, E.; Angel, C.E.; Graham, E.S. Application of xCELLigence RTCA Biosensor Technology for Revealing the Profile and Window of Drug Responsiveness in Real Time. Biosensors 2015, 5, 199–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darzynkiewicz, Z. Cytometry of the cell cycle: In search for perfect methodology for DNA content analysis in tissue specimens. Cell Cycle 2010, 9, 3395–3396. [Google Scholar] [CrossRef] [PubMed]
- Biederbick, A.; Kern, H.F.; Elsässer, H.P. Monodansylcadaverine (MDC) is a specific in vivo marker for autophagic vacuoles. Eur. J. Cell Biol. 1995, 66, 3–14. [Google Scholar]
- Shimizu, S. Biological Roles of Alternative Autophagy. Mol. Cells 2018, 41, 50–54. [Google Scholar]
- Chu, C.T. A pivotal role for PINK1 and autophagy in mitochondrial quality control: Implications for Parkinson disease. Hum. Mol. Genet. 2010, 19, R28–R37. [Google Scholar] [CrossRef] [Green Version]
- Chu, C.T.; Ji, J.; Dagda, R.K.; Jiang, J.F.; Tyurina, Y.Y.; Kapralov, A.A.; Tyurin, V.A.; Yanamala, N.; Shrivastava, I.H.; Mohammadyani, D.; et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat. Cell Biol. 2013, 15, 1197–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; McCain, M.L.; Yang, L.; He, A.; Pasqualini, F.S.; Agarwal, A.; Yuan, H.; Jiang, D.; Zhang, D.; Zangi, L.; et al. Modeling the mitochondrial cardiomyopathy of Barth syndrome with induced pluripotent stem cell and heart-on-chip technologies. Nat. Med. 2014, 20, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Vreken, P.; Valianpour, F.; Nijtmans, L.G.; Grivell, L.A.; Plecko, B.; Wanders, R.J.; Barth, P.G. Defective Remodeling of Cardiolipin and Phosphatidylglycerol in Barth Syndrome. Biochem. Biophys. Res. Commun. 2000, 279, 378–382. [Google Scholar] [CrossRef] [PubMed]
- Le, C.H.; Benage, L.G.; Specht, K.S.; Li Puma, L.C.; Mulligan, C.M.; Heuberger, A.L.; Prenni, J.E.; Claypool, S.M.; Chatfield, K.C.; Sparagna, G.; et al. Taffazin deficiency impair CoA-dependent oxidative metabolism in cardiac mitochondria. J. Biol. Chem. 2020, 295, 12485–12497. [Google Scholar] [CrossRef] [PubMed]
- Vayssiere, J.L.; Petit, P.X.; Risler, Y.; Mignotte, B. Commitment to apoptosis is associated with changes in mitochondrial biogenesis and activity in cell lines conditionally immortalized with simian virus 40. Proc. Natl. Acad. Sci. USA 1994, 91, 11752–11756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petit, P.X.; Lecoeur, H.; Zorn, E.; Dauguet, C.; Mignotte, B.; Gougeon, M.L. Alterations in mitochondrial structure and function are early events of dexamethasone-induced thymocyte apoptosis. J. Cell Biol. 1995, 130, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Zamzami, N.; Marchetti, P.; Castedo, M.; Zanin, C.; Vayssière, J.L.; Petit, P.X.; Kroemer, G. Reduction in mitochondrial potential constitutes an early irreversible step of programmed lymphocyte death in vivo. J. Exp. Med. 1995, 181, 1661–1672. [Google Scholar] [CrossRef] [Green Version]
- Zamzami, N.; Marchetti, P.; Castedo, M.; Decaudin, D.; Macho, A.; Hirsch, T.; Susin, S.A.; Petit, P.X.; Mignotte, B.; Kroemer, G. Sequential reduction of mitochondrial transmembrane potential and generation of reactive oxygen species in early programmed cell death. J. Exp. Med. 1995, 182, 367–377. [Google Scholar] [CrossRef]
- Gonzalvez, F.; Pariselli, F.; Dupaigne, P.; Budihardjo, I.; Lutter, M.; Antonsson, B.; Diolez, P.; Manon, S.; Martinou, J.-C.; Goubern, M.; et al. tBid interaction with cardiolipin primarily orchestrates mitochondrial dysfunctions and subsequently activates Bax and Bak. Cell Death Differ. 2005, 12, 614–626. [Google Scholar] [CrossRef] [Green Version]
- Gonzalvez, F.; Bessoule, J.-J.; Rocchiccioli, F.; Manon, S.; Petit, P.X. Role of cardiolipin on tBid and tBid/Bax synergistic effects on yeast mitochondria. Cell Death Differ. 2005, 12, 659–667. [Google Scholar] [CrossRef] [Green Version]
- Richter, E.A.; Ruderman, N.B. AMPK and the biochemistry of exercise: Implications for human health and disease. Biochem. J. 2009, 418, 261–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardie, D.G.; Hardie, D.G. AMPK: A key regulator of energy balance in the single cell and the whole organism. Int. J. Obes. 2008, 32, S7–S12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jäger, S.; Handschin, C.; St.-Pierre, J.; Spiegelman, B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1. Proc. Natl. Acad. Sci. USA 2007, 104, 12017–12022. [Google Scholar] [CrossRef] [Green Version]
- Watt, V.M.; Yip, C.C. HeLa cells contain the atrial natriuretic peptide receptor with guanylate cyclase activity. Biochem. Biophys. Res. Commun. 1989, 164, 671–677. [Google Scholar] [CrossRef]
- Guo, J.Y.; Karsli-Uzunbas, G.; Mathew, R.; Aisner, S.C.; Kamphorst, J.J.; Strohecker, A.M.; Chen, G.; Price, S.; Lu, W.; Teng, X.; et al. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev. 2013, 27, 1447–1461. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Liu, H.-H.; Cao, Y.-T.; Zhang, L.-L.; Huang, F.; Yi, C. The Role of Mitochondrial Dynamics and Mitophagy in Carcinogenesis, Metastasis and Therapy. Front. Cell Dev. Biol. 2020, 8, 413. [Google Scholar] [CrossRef]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial dynamics: Overview of molecular mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar] [CrossRef] [Green Version]
- Wai, T.; Langer, T. Mitochondrial Dynamics and Metabolic Regulation. Trends Endocrinol. Metab. 2016, 27, 105–117. [Google Scholar] [CrossRef]
- Mishra, P.; Chan, D.C. Metabolic regulation of mitochondrial dynamics. J. Cell Biol. 2016, 212, 379–387. [Google Scholar] [CrossRef] [Green Version]
- Chan, D.C. Mitochondrial Dynamics and Its Involvement in Disease. Annu. Rev. Pathol. Mech. Dis. 2020, 15, 235–259. [Google Scholar] [CrossRef] [Green Version]
- Pernas, L.; Scorrano, L. Mito-Morphosis: Mitochondrial Fusion, Fission, and Cristae Remodeling as Key Mediators of Cellular Function. Annu. Rev. Physiol. 2016, 78, 505–531. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C. Fusion and Fission: Interlinked Processes Critical for Mitochondrial Health. Annu. Rev. Genet. 2012, 46, 265–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, D.C. Mitochondrial Fusion and Fission in Mammals. Annu. Rev. Cell Dev. Biol. 2006, 22, 79–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.-F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 Is Selectively Stabilized on Impaired Mitochondria to Activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jalmar, O.; François-Moutal, L.; García-Sáez, A.-J.; Perry, M.; Granjon, T.; Gonzalvez, F.; Gottlieb, E.; Ayala-Sanmartin, J.; Klösgen, B.; Schwille, P.; et al. Caspase-8 Binding to Cardiolipin in Giant Unilamellar Vesicles Provides a Functional Docking Platform for Bid. PLoS ONE 2013, 8, e55250. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Type | Side Scatter (90° Light Scatter) | Forward Low-Angle Light Scatter (10–21° Angle) |
|---|---|---|
| ShWT1 | 257 ± 35 | 476 ± 18 |
| ShTaz1 | 207 ± 29 | 442 ± 12 |
| ShTaz1R | 230 ± 31 | 458 ± 15 |
| Activities. nmol/min/mg | ATP Synthesis Activity | Citrate Synthase Activity | ATP/CS |
|---|---|---|---|
| ShWT1 | 25.8 ± 1.5 | 182.3 ± 24.6 | 0.141 |
| ShTaz1 | 17.4 ± 1.2 * | 287.5 ± 9.0 * | 0.060 * |
| ShTaz1R | 23.9 ± 1.4 | 201.6 ± 19.3 | 0.118 |
| Coupled Respiration | Uncoupled Respiration | CS/Coupled | |
| ShWT1 | 7.7 ± 0.6 | 19.7 ± 0.8 | 0.042 ± 0.004 |
| ShTaz1 | 6.9 ± 0.9 | 19.6 ± 1.8 | 0.024 ± 0.006 * |
| ShTaz1R | 7.5 ± 0.6 | 19.8 ± 1.3 | 0.037 ± 0.007 |
| ATP (pmole/mg protein) | Cell Culture on Galactose | Cell Culture on Glucose |
|---|---|---|
| ShWT1 | 61 ± 4 | 58 ± 8 |
| ShTaz1 | 21 ± 5 | 48 ± 5 |
| ShTazR1 | 55 ± 7 | 56 ± 6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petit, P.X.; Ardilla-Osorio, H.; Penalvia, L.; Rainey, N.E. Tafazzin Mutation Affecting Cardiolipin Leads to Increased Mitochondrial Superoxide Anions and Mitophagy Inhibition in Barth Syndrome. Cells 2020, 9, 2333. https://doi.org/10.3390/cells9102333
Petit PX, Ardilla-Osorio H, Penalvia L, Rainey NE. Tafazzin Mutation Affecting Cardiolipin Leads to Increased Mitochondrial Superoxide Anions and Mitophagy Inhibition in Barth Syndrome. Cells. 2020; 9(10):2333. https://doi.org/10.3390/cells9102333
Chicago/Turabian StylePetit, Patrice X., Hector Ardilla-Osorio, Lucile Penalvia, and Nathan E. Rainey. 2020. "Tafazzin Mutation Affecting Cardiolipin Leads to Increased Mitochondrial Superoxide Anions and Mitophagy Inhibition in Barth Syndrome" Cells 9, no. 10: 2333. https://doi.org/10.3390/cells9102333
APA StylePetit, P. X., Ardilla-Osorio, H., Penalvia, L., & Rainey, N. E. (2020). Tafazzin Mutation Affecting Cardiolipin Leads to Increased Mitochondrial Superoxide Anions and Mitophagy Inhibition in Barth Syndrome. Cells, 9(10), 2333. https://doi.org/10.3390/cells9102333