Breast Cancer-Derived Microparticles Reduce Cancer Cell Adhesion, an Effect Augmented by Chemotherapy



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Extraction and Quantification of TMP
2.3. Modified Boyden Chamber Assay
2.4. Cell Spreading Assay
2.5. Cell Viability AlamarBlueTM Assay
2.6. Pillar Fabrication
2.7. CD44 Short Hairpin RNA
2.8. Western Blot
2.9. Tumor-Derived Microparticle Proteomics
2.10. Ex Vivo Pulmonary Metastasis Assay
2.11. Blood Samples from Cancer Patients
2.12. Immunostaining and Imaging
2.13. Statistical Analysis
3. Results
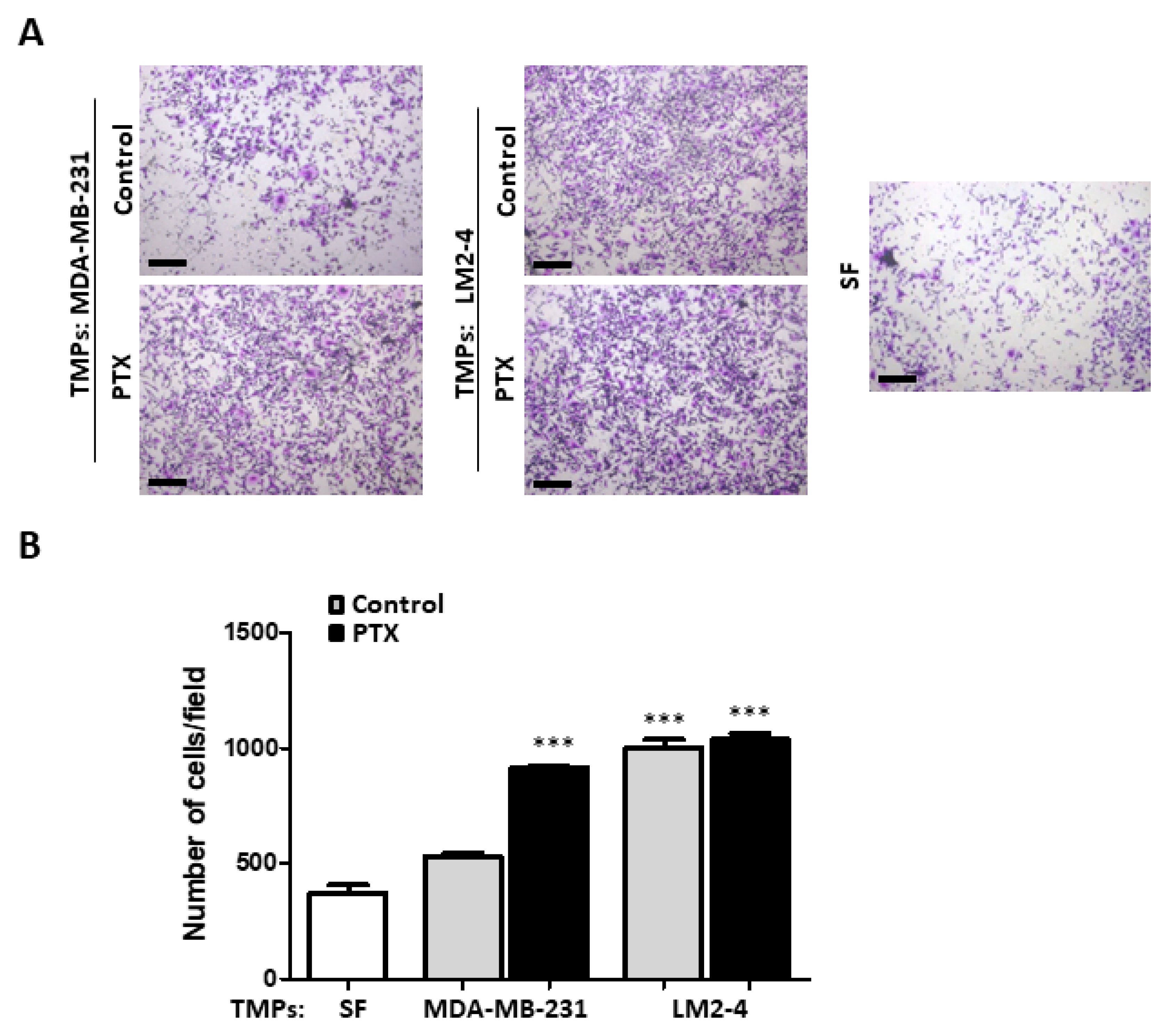
3.1. TMPs from Highly Metastatic or Chemotherapy-Treated Tumor Cells Induce Tumor Cell Invasion
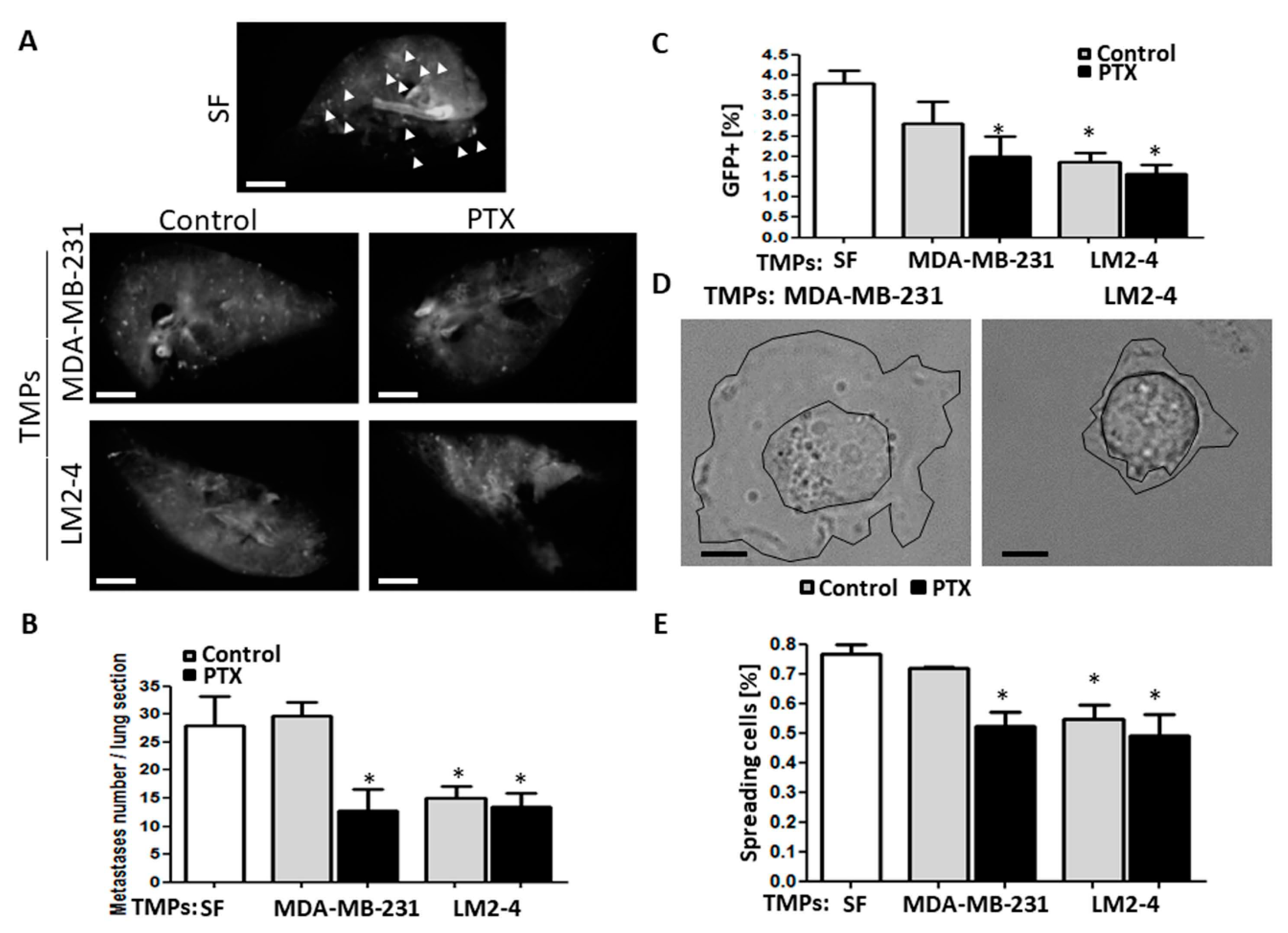
3.2. TMPs from Highly Metastatic or Chemotherapy-Treated Tumor Cells Inhibit Tumor Cell Seeding
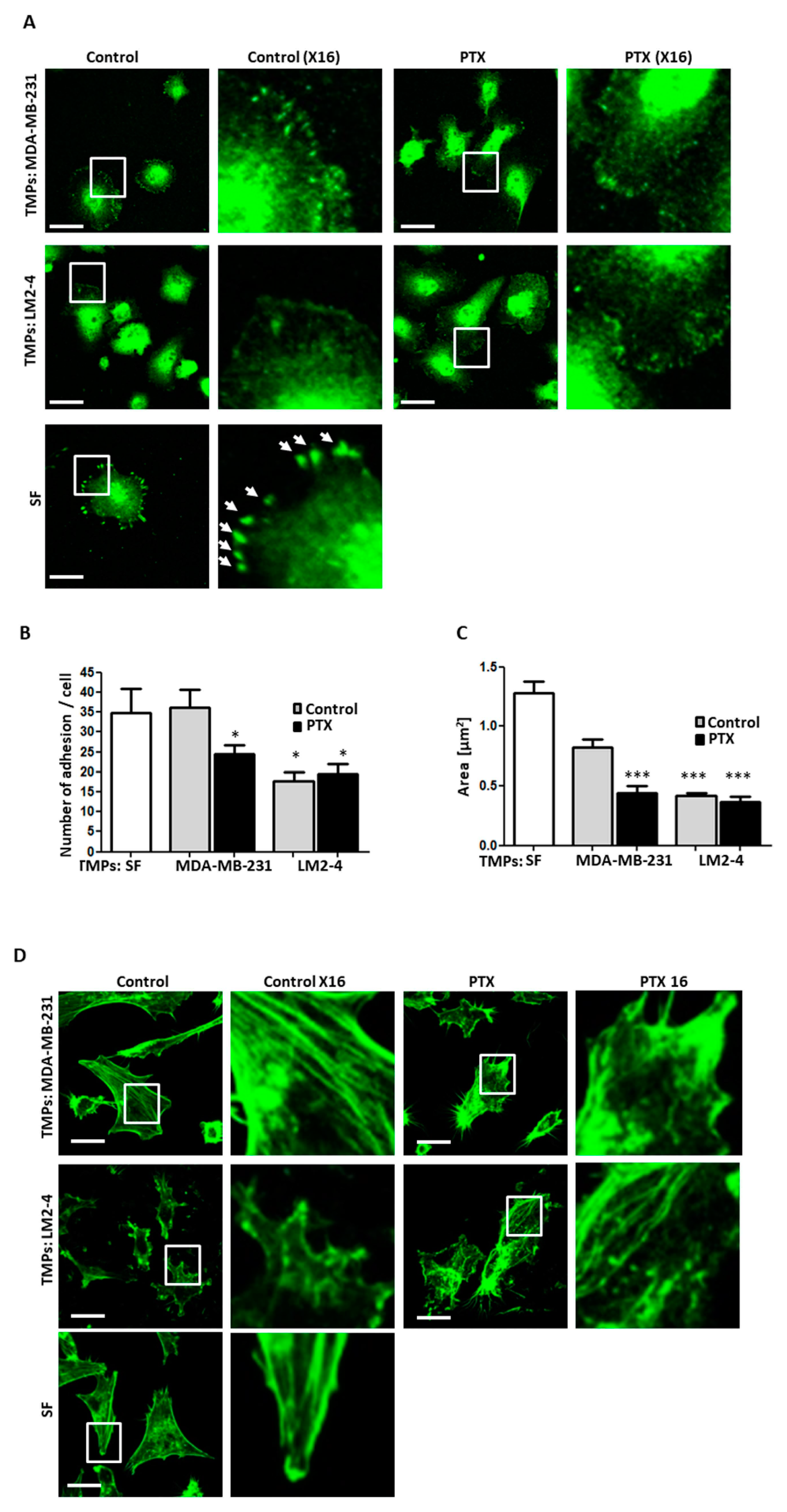
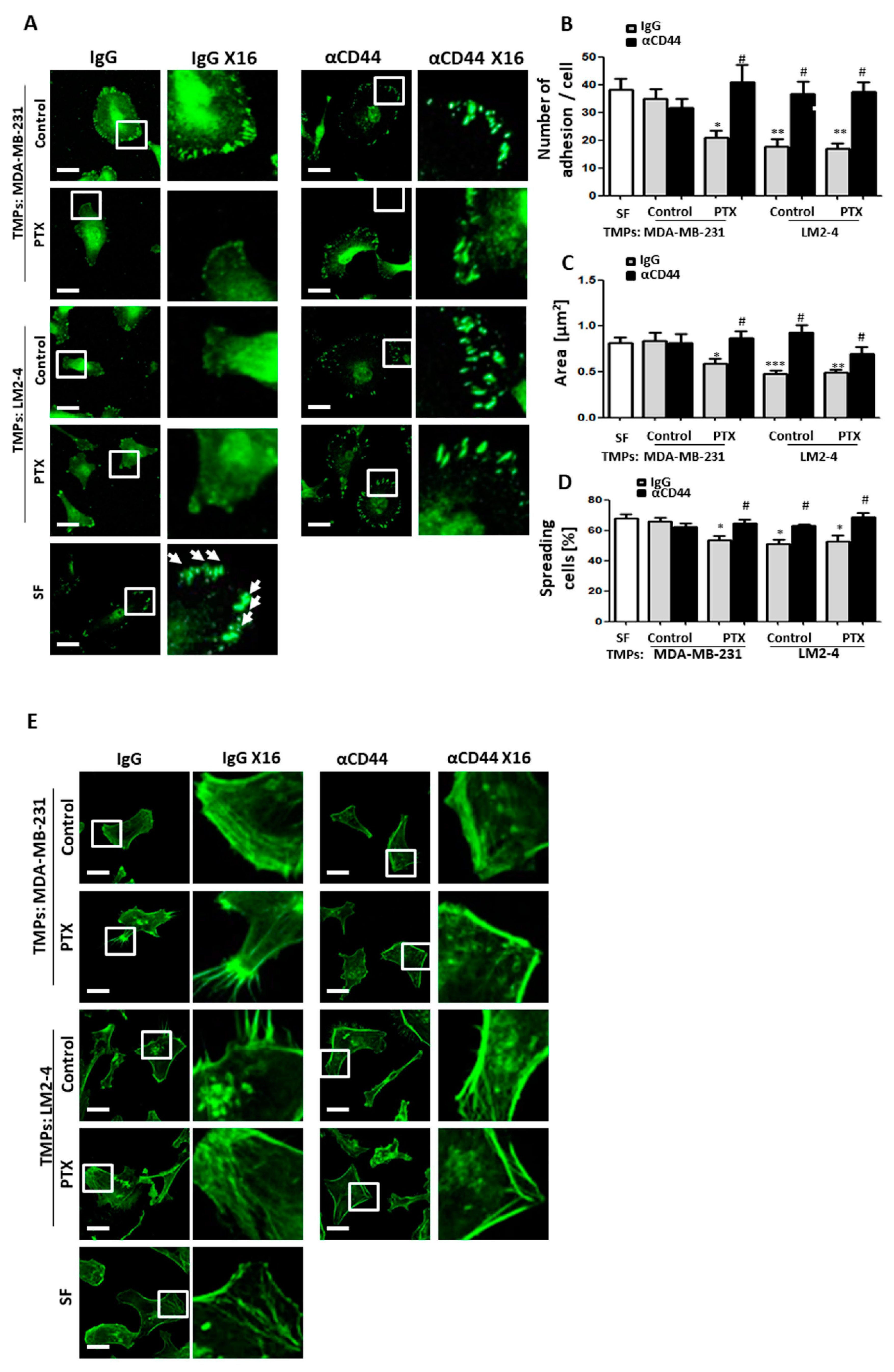
3.3. TMPs from Highly Metastatic or Chemotherapy-Treated Tumor Cells Inhibit Cellular Adhesion and Destroy Actin Filaments
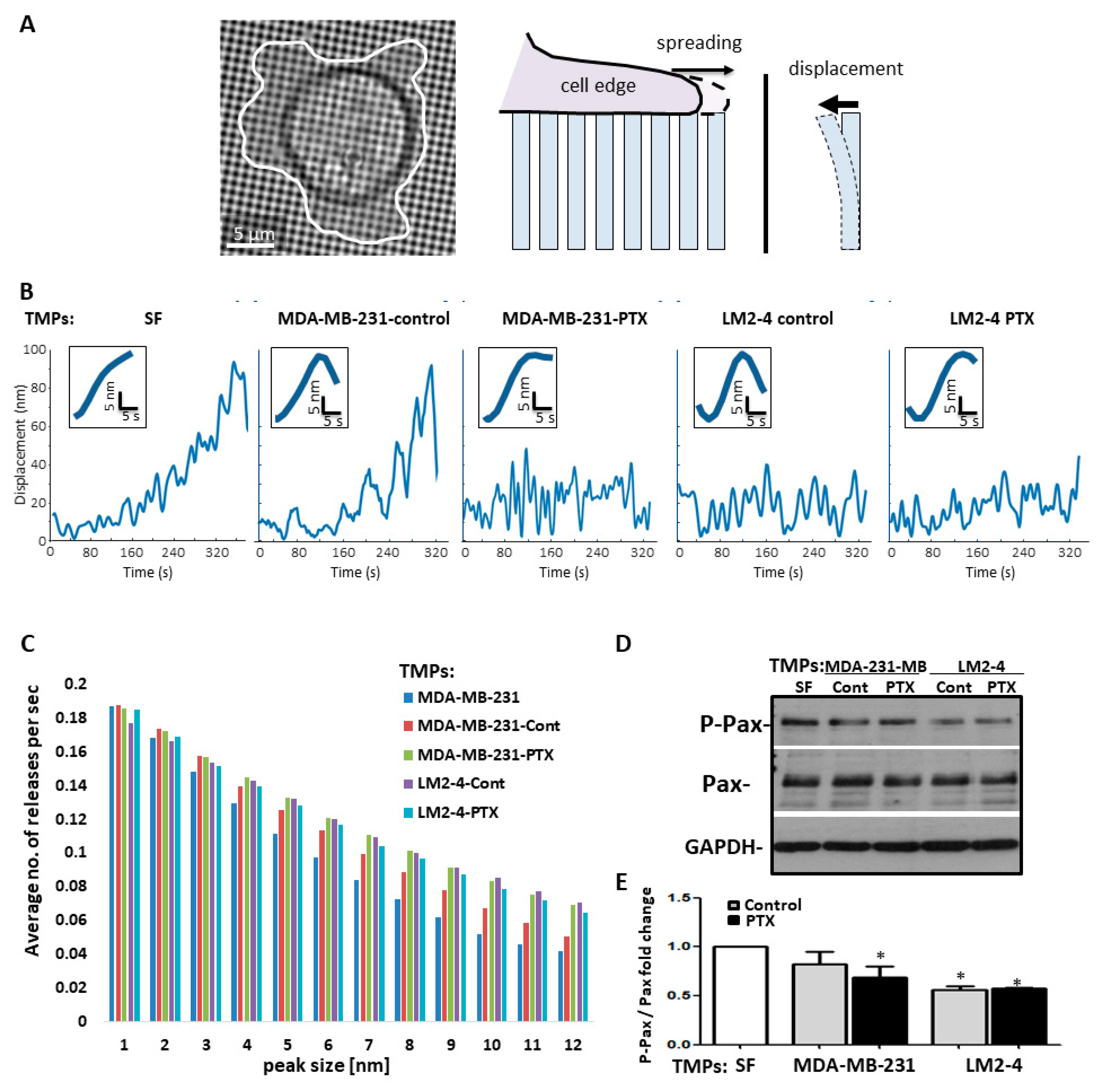
3.4. TMPs Increase the Pace of Metastatic Cell Biomechanical Forces
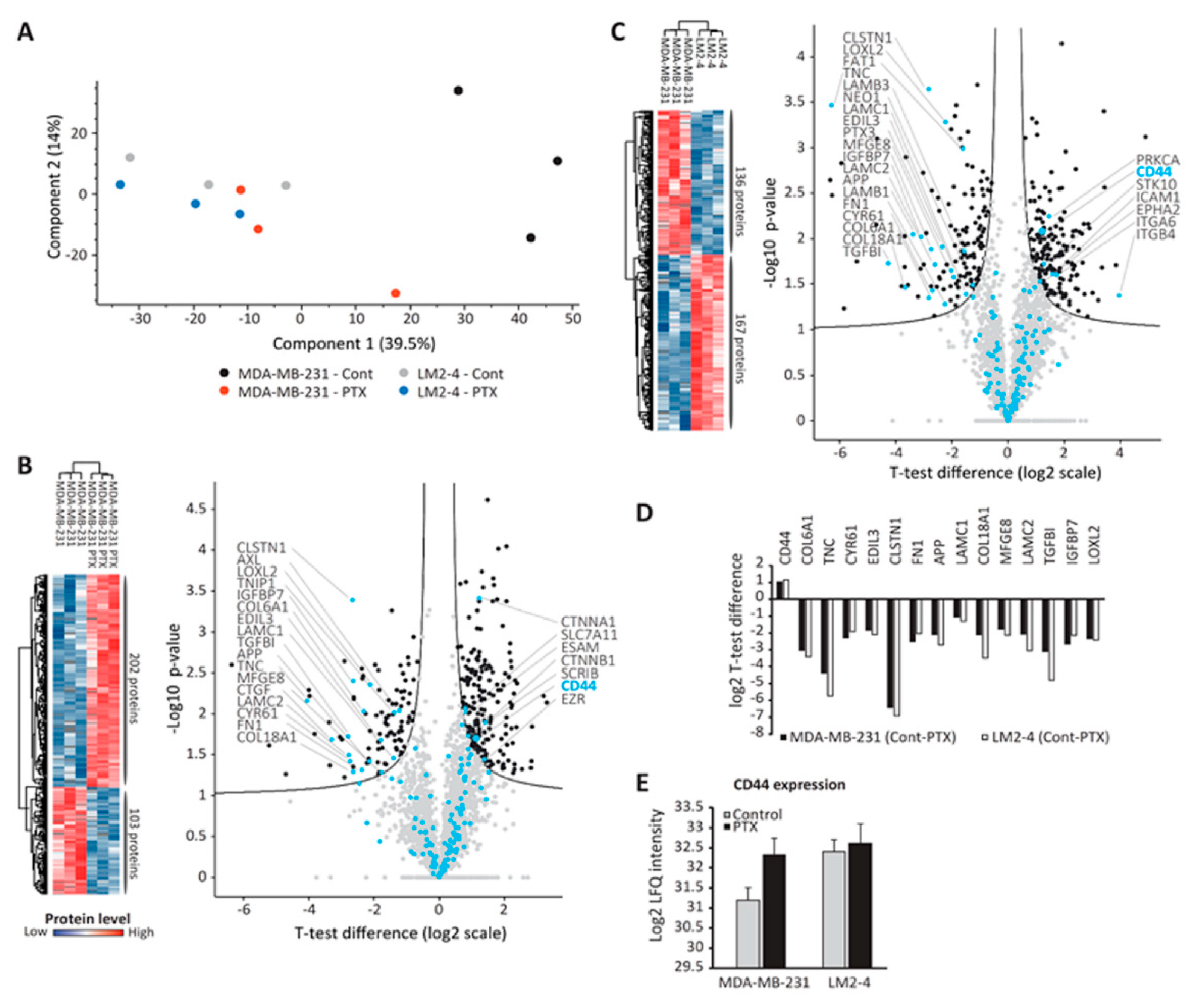
3.5. Proteomic Analysis of TMPs
3.6. CD44-Expressing TMPs Decrease Cell Adhesion and Support Tumor Cell Metastatic Properties
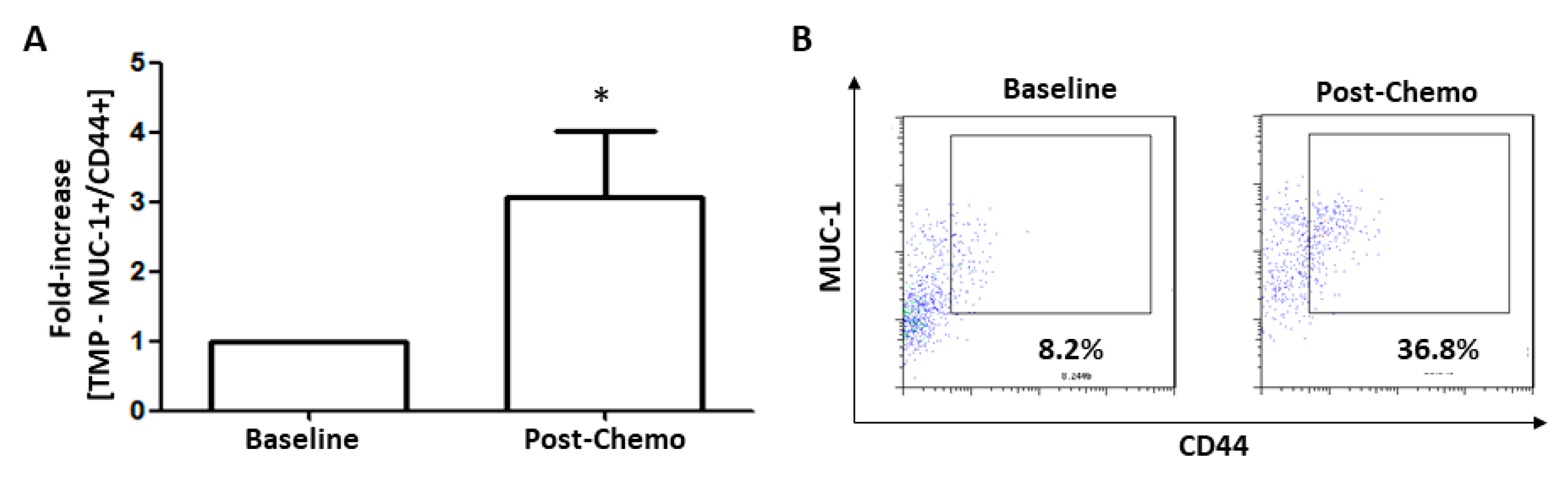
3.7. TMPs Expressing CD44 were Increased in Breast Cancer Patients Treated with Chemotherapy
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [PubMed]
- O’Shaughnessy, J. Extending survival with chemotherapy in metastatic breast cancer. Oncologist 2005, 10, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Sahai, E. Illuminating the metastatic process. Nat. Rev. Cancer 2007, 7, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Welch, D.R.; Hurst, D.R. Defining the Hallmarks of Metastasis. Cancer Res. 2019, 79, 3011–3027. [Google Scholar] [CrossRef] [PubMed]
- Van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Raposo, G.; Stahl, P.D. Extracellular vesicles: A new communication paradigm? Nat. Rev. Mol. Cell Biol. 2019, 20, 509–510. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.H.; D’Asti, E.; Magnus, N.; Al-Nedawi, K.; Meehan, B.; Rak, J. Microvesicles as mediators of intercellular communication in cancer--the emerging science of cellular ‘debris’. Semin Immunopathol 2011, 33, 455–467. [Google Scholar] [CrossRef]
- Hoshino, A.; Costa-Silva, B.; Shen, T.L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef]
- Peinado, H.; Aleckovic, M.; Lavotshkin, S.; Matei, I.; Costa-Silva, B.; Moreno-Bueno, G.; Hergueta-Redondo, M.; Williams, C.; Garcia-Santos, G.; Ghajar, C.; et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 2012, 18, 883–891. [Google Scholar] [CrossRef]
- Keklikoglou, I.; Cianciaruso, C.; Guc, E.; Squadrito, M.L.; Spring, L.M.; Tazzyman, S.; Lambein, L.; Poissonnier, A.; Ferraro, G.B.; Baer, C.; et al. Chemotherapy elicits pro-metastatic extracellular vesicles in breast cancer models. Nat. Cell Biol. 2019, 21, 190–202. [Google Scholar] [CrossRef]
- Al-Nedawi, K.; Meehan, B.; Micallef, J.; Lhotak, V.; May, L.; Guha, A.; Rak, J. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat. Cell Biol. 2008, 10, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, R.; Johnson, M.S.; Pokharel, D.; Krishnan, S.R.; Bebawy, M. Microparticles shed from multidrug resistant breast cancer cells provide a parallel survival pathway through immune evasion. BMC Cancer 2017, 17, 104. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, R.; Luk, F.; Dalla, P.V.; Grau, G.E.; Bebawy, M. Breast cancer-derived microparticles display tissue selectivity in the transfer of resistance proteins to cells. PLoS ONE 2013, 8, e61515. [Google Scholar] [CrossRef] [PubMed]
- Fremder, E.; Munster, M.; Aharon, A.; Miller, V.; Gingis-Velitski, S.; Voloshin, T.; Alishekevitz, D.; Bril, R.; Scherer, S.J.; Loven, D.; et al. Tumor-derived microparticles induce bone marrow-derived cell mobilization and tumor homing: A process regulated by osteopontin. Int. J. Cancer. J. Int. Du Cancer 2014, 135, 270–281. [Google Scholar] [CrossRef] [PubMed]
- Iczkowski, K.A. Cell adhesion molecule CD44: Its functional roles in prostate cancer. Am. J. Transl Res. 2010, 3, 1–7. [Google Scholar] [PubMed]
- Wang, S.J.; Wong, G.; de Heer, A.M.; Xia, W.; Bourguignon, L.Y. CD44 variant isoforms in head and neck squamous cell carcinoma progression. Laryngoscope 2009, 119, 1518–1530. [Google Scholar] [CrossRef] [PubMed]
- Heider, K.H.; Hofmann, M.; Hors, E.; van den Berg, F.; Ponta, H.; Herrlich, P.; Pals, S.T. A human homologue of the rat metastasis-associated variant of CD44 is expressed in colorectal carcinomas and adenomatous polyps. J. Cell Biol. 1993, 120, 227–233. [Google Scholar] [CrossRef]
- Senbanjo, L.T.; Chellaiah, M.A. CD44: A Multifunctional Cell Surface Adhesion Receptor Is a Regulator of Progression and Metastasis of Cancer Cells. Front. Cell Dev. Biol. 2017, 5, 18. [Google Scholar] [CrossRef]
- Wang, Z.; Zhao, K.; Hackert, T.; Zoller, M. CD44/CD44v6 a Reliable Companion in Cancer-Initiating Cell Maintenance and Tumor Progression. Front. Cell Dev. Biol. 2018, 6, 97. [Google Scholar] [CrossRef]
- Nakamura, K.; Sawada, K.; Kinose, Y.; Yoshimura, A.; Toda, A.; Nakatsuka, E.; Hashimoto, K.; Mabuchi, S.; Morishige, K.I.; Kurachi, H.; et al. Exosomes Promote Ovarian Cancer Cell Invasion through Transfer of CD44 to Peritoneal Mesothelial Cells. Mol. Cancer Res. 2017, 15, 78–92. [Google Scholar] [CrossRef]
- Issman, L.; Brenner, B.; Talmon, Y.; Aharon, A. Cryogenic transmission electron microscopy nanostructural study of shed microparticles. PLoS ONE 2013, 8, e83680. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.; Ji, T.; Chen, D.; Dong, W.; Zhang, H.; Yin, X.; Ma, J.; Liang, X.; Zhang, Y.; Shen, G.; et al. Tumor cell-derived microparticles polarize M2 tumor-associated macrophages for tumor progression. Oncoimmunology 2016, 5, e1118599. [Google Scholar] [CrossRef] [PubMed]
- Timaner, M.; Kotsofruk, R.; Raviv, Z.; Magidey, K.; Shechter, D.; Kan, T.; Nevelsky, A.; Daniel, S.; de Vries, E.G.E.; Zhang, T.; et al. Microparticles from tumors exposed to radiation promote immune evasion in part by PD-L1. Oncogene 2019. [Google Scholar] [CrossRef] [PubMed]
- Bachurski, D.; Schuldner, M.; Nguyen, P.H.; Malz, A.; Reiners, K.S.; Grenzi, P.C.; Babatz, F.; Schauss, A.C.; Hansen, H.P.; Hallek, M.; et al. Extracellular vesicle measurements with nanoparticle tracking analysis-An accuracy and repeatability comparison between NanoSight NS300 and ZetaView. J. Extracell Vesicles 2019, 8, 1596016. [Google Scholar] [CrossRef] [PubMed]
- Gingis-Velitski, S.; Loven, D.; Benayoun, L.; Munster, M.; Bril, R.; Voloshin, T.; Alishekevitz, D.; Bertolini, F.; Shaked, Y. Host response to short-term, single-agent chemotherapy induces matrix metalloproteinase-9 expression and accelerates metastasis in mice. Cancer Res. 2011, 71, 6986–6996. [Google Scholar] [CrossRef] [PubMed]
- Wolfenson, H.; Meacci, G.; Liu, S.; Stachowiak, M.R.; Iskratsch, T.; Ghassemi, S.; Roca-Cusachs, P.; O’Shaughnessy, B.; Hone, J.; Sheetz, M.P. Tropomyosin controls sarcomere-like contractions for rigidity sensing and suppressing growth on soft matrices. Nat. Cell Biol. 2016, 18, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Voloshin, T.; Gingis-Velitski, S.; Bril, R.; Benayoun, L.; Munster, M.; Milsom, C.; Man, S.; Kerbel, R.S.; Shaked, Y. G-CSF supplementation with chemotherapy can promote revascularization and subsequent tumor regrowth: Prevention by a CXCR4 antagonist. Blood 2011, 118, 3426–3435. [Google Scholar] [CrossRef] [PubMed]
- Ghassemi, S.; Meacci, G.; Liu, S.; Gondarenko, A.A.; Mathur, A.; Roca-Cusachs, P.; Sheetz, M.P.; Hone, J. Cells test substrate rigidity by local contractions on submicrometer pillars. Proc. Natl. Acad. Sci. USA 2012, 109, 5328–5333. [Google Scholar] [CrossRef]
- Schmitt, M.; Metzger, M.; Gradl, D.; Davidson, G.; Orian-Rousseau, V. CD44 functions in Wnt signaling by regulating LRP6 localization and activation. Cell Death Differ. 2015, 22, 677–689. [Google Scholar] [CrossRef]
- Harel, M.; Oren-Giladi, P.; Kaidar-Person, O.; Shaked, Y.; Geiger, T. Proteomics of microparticles with SILAC Quantification (PROMIS-Quan): A novel proteomic method for plasma biomarker quantification. Mol. Cell. Proteom. MCP 2015, 14, 1127–1136. [Google Scholar] [CrossRef]
- Rappsilber, J.; Mann, M.; Ishihama, Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2007, 2, 1896–1906. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Neuhauser, N.; Michalski, A.; Scheltema, R.A.; Olsen, J.V.; Mann, M. Andromeda: A peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 2011, 10, 1794–1805. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Cox, J. Perseus: A Bioinformatics Platform for Integrative Analysis of Proteomics Data in Cancer Research. Methods Mol. Biol. 2018, 1711, 133–148. [Google Scholar] [PubMed]
- Cox, J.; Hein, M.Y.; Luber, C.A.; Paron, I.; Nagaraj, N.; Mann, M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteom. MCP 2014, 13, 2513–2526. [Google Scholar] [CrossRef] [PubMed]
- Tusher, V.G.; Tibshirani, R.; Chu, G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. USA 2001, 98, 5116–5121. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, A.; Hong, S.H.; Osborne, T.; Khan, M.A.; Campbell, K.; Briggs, J.; Eleswarapu, A.; Buquo, L.; Ren, L.; Hewitt, S.M.; et al. Modeling metastasis biology and therapy in real time in the mouse lung. J. Clin. Investig. 2010, 120, 2979–2988. [Google Scholar] [CrossRef]
- Rachman-Tzemah, C.; Zaffryar-Eilot, S.; Grossman, M.; Ribero, D.; Timaner, M.; Maki, J.M.; Myllyharju, J.; Bertolini, F.; Hershkovitz, D.; Sagi, I.; et al. Blocking Surgically Induced Lysyl Oxidase Activity Reduces the Risk of Lung Metastases. Cell Rep. 2017, 19, 774–784. [Google Scholar] [CrossRef]
- Sultan, A. Five-minute preparation of platelet-poor plasma for routine coagulation testing. East. Mediterr Health J. 2010, 16, 233–236. [Google Scholar] [CrossRef]
- Hulspas, R.; O’Gorman, M.R.; Wood, B.L.; Gratama, J.W.; Sutherland, D.R. Considerations for the control of background fluorescence in clinical flow cytometry. Cytom. B Clin. Cytom. 2009, 76, 355–364. [Google Scholar] [CrossRef]
- Munoz, R.; Man, S.; Shaked, Y.; Lee, C.; Wong, J.; Francia, G.; Kerbel, R.S. Highly efficacious non-toxic treatment for advanced metastatic breast cancer using combination UFT-cyclophosphamide metronomic chemotherapy. Cancer Res. 2006, 66, 3386–3391. [Google Scholar] [CrossRef] [PubMed]
- Bachir, A.I.; Horwitz, A.R.; Nelson, W.J.; Bianchini, J.M. Actin-Based Adhesion Modules Mediate Cell Interactions with the Extracellular Matrix and Neighboring Cells. Cold Spring Harb. Perspect. Biol. 2017, 9, a023234. [Google Scholar] [CrossRef] [PubMed]
- Livne, A.; Geiger, B. The inner workings of stress fibers–from contractile machinery to focal adhesions and back. J. Cell Sci. 2016, 129, 1293–1304. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Tian, Y.; Yuan, X.; Wu, H.; Liu, Q.; Pestell, R.G.; Wu, K. The role of CD44 in epithelial-mesenchymal transition and cancer development. Onco Targets 2015, 8, 3783–3792. [Google Scholar]
- Orian-Rousseau, V. CD44, a therapeutic target for metastasising tumours. Eur. J. Cancer 2010, 46, 1271–1277. [Google Scholar] [CrossRef]
- Shaked, Y. Balancing efficacy of and host immune responses to cancer therapy: The yin and yang effects. Nat. Rev. Clin. Oncol. 2016, 13, 611–626. [Google Scholar] [CrossRef]
- Shaked, Y. The pro-tumorigenic host response to cancer therapies. Nat. Rev. Cancer 2019, 19, 667–685. [Google Scholar] [CrossRef]
- Janiszewska, M.; Primi, M.C.; Izard, T. Cell adhesion in cancer: Beyond the migration of single cells. J. Biol. Chem. 2020, 295, 2495–2505. [Google Scholar] [CrossRef]
- McFarlane, S.; McFarlane, C.; Montgomery, N.; Hill, A.; Waugh, D.J. CD44-mediated activation of alpha5beta1-integrin, cortactin and paxillin signaling underpins adhesion of basal-like breast cancer cells to endothelium and fibronectin-enriched matrices. Oncotarget 2015, 6, 36762–36773. [Google Scholar] [CrossRef]
- Ponta, H.; Sherman, L.; Herrlich, P.A. CD44: From adhesion molecules to signalling regulators. Nat. Rev. Mol. Cell Biol 2003, 4, 33–45. [Google Scholar] [CrossRef]
- Zwicker, J.I.; Liebman, H.A.; Neuberg, D.; Lacroix, R.; Bauer, K.A.; Furie, B.C.; Furie, B. Tumor-derived tissue factor-bearing microparticles are associated with venous thromboembolic events in malignancy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 6830–6840. [Google Scholar] [CrossRef] [PubMed]
- Karagiannis, G.S.; Pastoriza, J.M.; Wang, Y.; Harney, A.S.; Entenberg, D.; Pignatelli, J.; Sharma, V.P.; Xue, E.A.; Cheng, E.; D’Alfonso, T.M.; et al. Neoadjuvant chemotherapy induces breast cancer metastasis through a TMEM-mediated mechanism. Sci. Transl. Med. 2017, 9, eaan0026. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shechter, D.; Harel, M.; Mukherjee, A.; Sagredo, L.M.; Loven, D.; Prinz, E.; Avraham, S.; Orian-Rousseau, V.; Geiger, T.; Shaked, Y.; et al. Breast Cancer-Derived Microparticles Reduce Cancer Cell Adhesion, an Effect Augmented by Chemotherapy. Cells 2020, 9, 2269. https://doi.org/10.3390/cells9102269
Shechter D, Harel M, Mukherjee A, Sagredo LM, Loven D, Prinz E, Avraham S, Orian-Rousseau V, Geiger T, Shaked Y, et al. Breast Cancer-Derived Microparticles Reduce Cancer Cell Adhesion, an Effect Augmented by Chemotherapy. Cells. 2020; 9(10):2269. https://doi.org/10.3390/cells9102269
Chicago/Turabian StyleShechter, Dvir, Michal Harel, Abhishek Mukherjee, Leonel M. Sagredo, David Loven, Elad Prinz, Shimrit Avraham, Veronique Orian-Rousseau, Tamar Geiger, Yuval Shaked, and et al. 2020. "Breast Cancer-Derived Microparticles Reduce Cancer Cell Adhesion, an Effect Augmented by Chemotherapy" Cells 9, no. 10: 2269. https://doi.org/10.3390/cells9102269
APA StyleShechter, D., Harel, M., Mukherjee, A., Sagredo, L. M., Loven, D., Prinz, E., Avraham, S., Orian-Rousseau, V., Geiger, T., Shaked, Y., & Wolfenson, H. (2020). Breast Cancer-Derived Microparticles Reduce Cancer Cell Adhesion, an Effect Augmented by Chemotherapy. Cells, 9(10), 2269. https://doi.org/10.3390/cells9102269