Cell Death in the Tumor Microenvironment: Implications for Cancer Immunotherapy

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
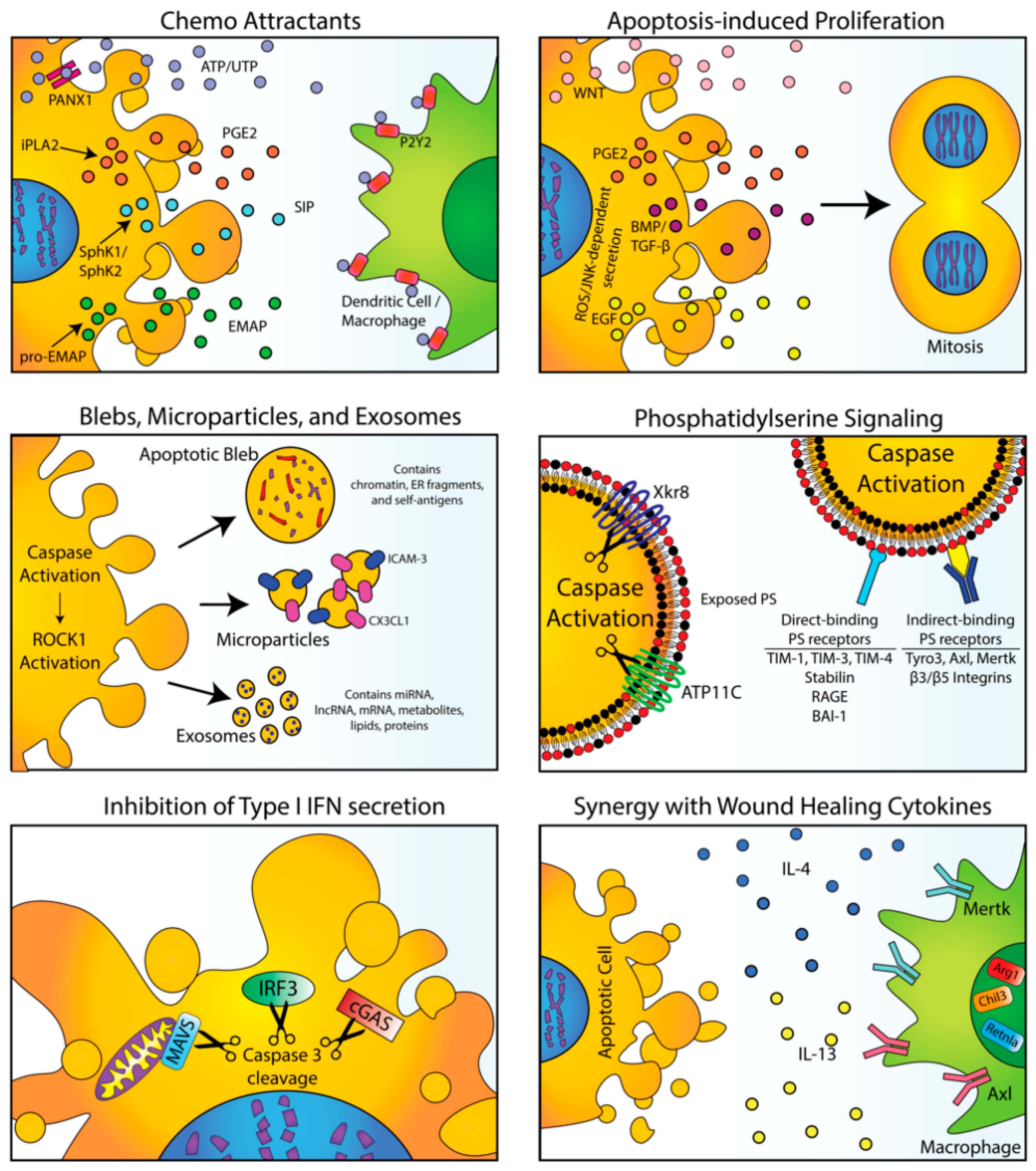
1.1. Apoptosis and Efferocytosis Provide Homeostatic and Tolerogenic Signals Under Physiological Conditions
1.2. Physiological Signals from Apoptotic and Efferocytic Cells Promote Compensatory Proliferation and Modify the Local Immune Microenvironment
2. Physiological Signals from Efferocytosis in Normal Tissues
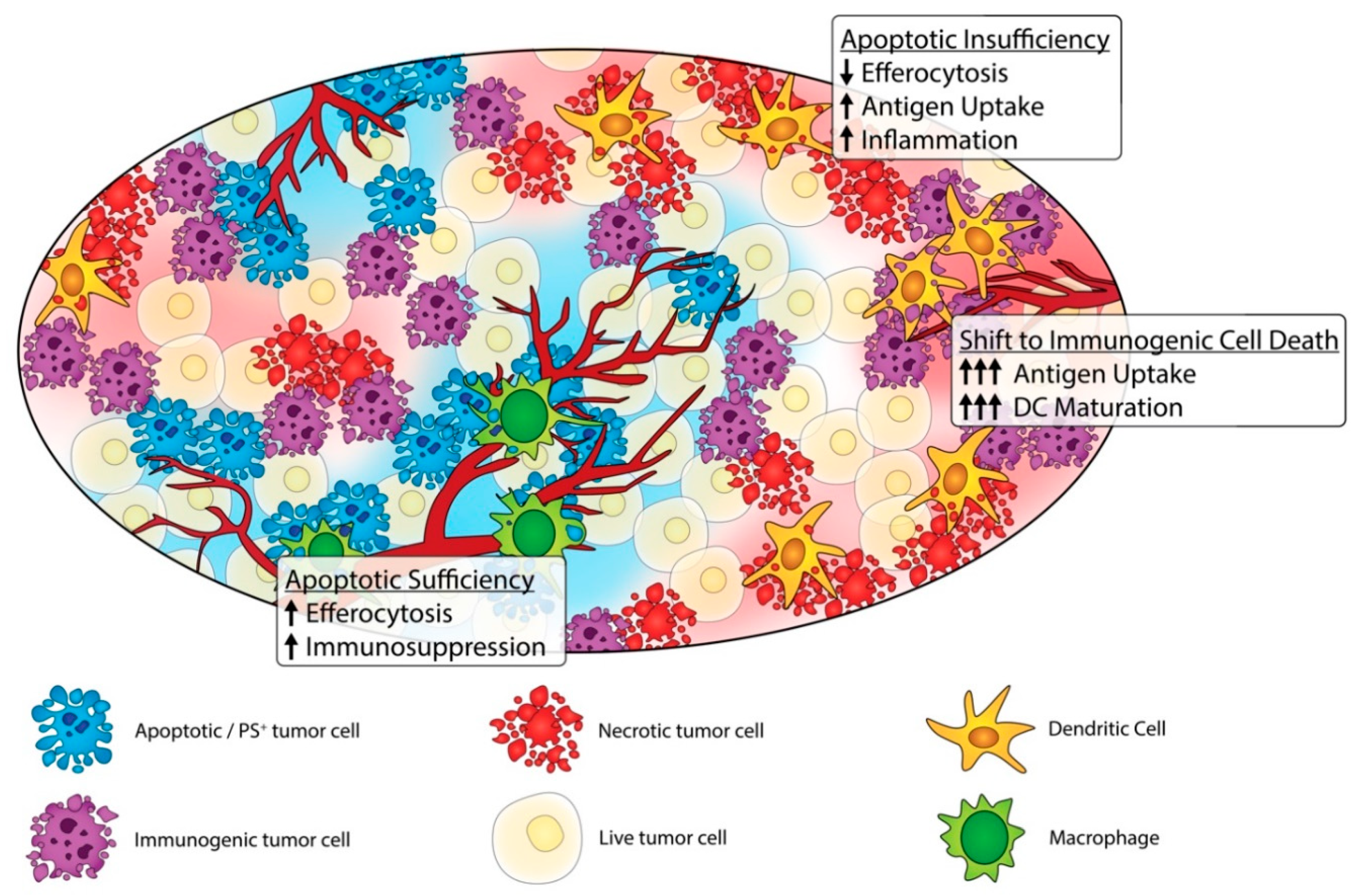
3. The Dark Side of Apoptosis and Efferocytosis in Cancer Progression
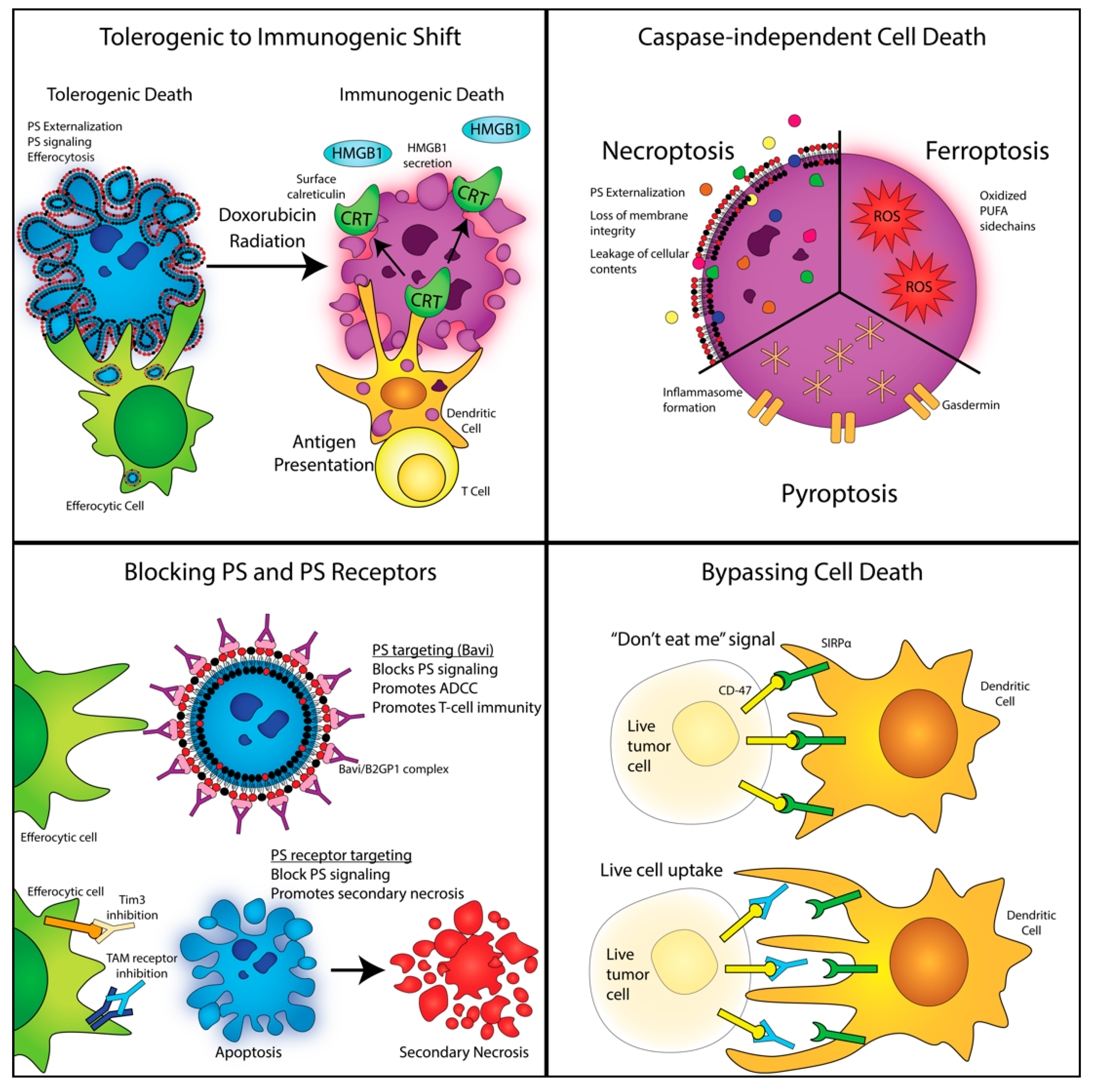
4. Shifting Classical Apoptosis towards Immunogenic Cell Death and Non-Caspase-Mediated Forms of Cell Death
5. Non-Caspase-Mediated Alternative Forms of Cell Death
6. Modulation of PS Signaling and Efferocytosis as an Anti-Tumor Strategy
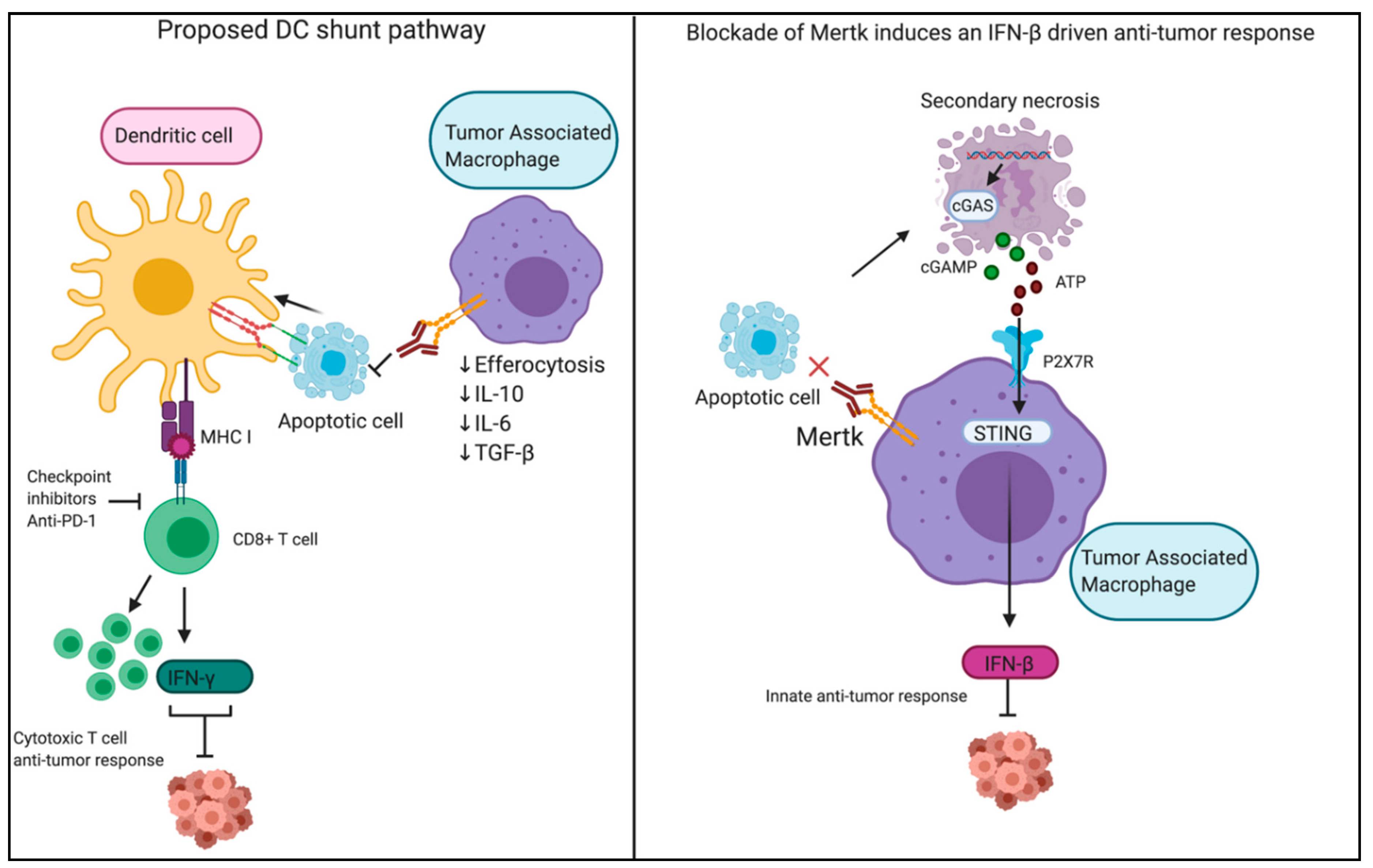
7. Targeting PS Receptors in Immuno-Oncology
8. PS Exposure Occurs on Viable Activated Cells and During Non-Apoptotic Forms of Cell Death
9. Conclusions
Funding
Conflicts of Interest
References
- Bianconi, E.; Piovesan, A.; Facchin, F.; Beraudi, A.; Casadei, R.; Frabetti, F.; Vitale, L.; Pelleri, M.C.; Tassani, S.; Piva, F.; et al. An estimation of the number of cells in the human body. Ann. Hum. Biol. 2013, 40, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Arandjelovic, S.; Ravichandran, K.S. Phagocytosis of apoptotic cells in homeostasis. Nat. Immunol. 2015, 16, 907–917. [Google Scholar] [CrossRef] [PubMed]
- Rothlin, C.V.; Ghosh, S.; Zuniga, E.I.; Oldstone, M.B.; Lemke, G. TAM receptors are pleiotropic inhibitors of the innate immune response. Cell 2007, 131, 1124–1136. [Google Scholar] [CrossRef] [PubMed]
- Medina, C.B.; Mehrotra, P.; Arandjelovic, S.; Perry, J.S.A.; Guo, Y.; Morioka, S.; Barron, B.; Walk, S.F.; Ghesquiere, B.; Krupnick, A.S.; et al. Metabolites released from apoptotic cells act as tissue messengers. Nature 2020, 580, 130–135. [Google Scholar] [CrossRef]
- Morioka, S.; Maueroder, C.; Ravichandran, K.S. Living on the Edge: Efferocytosis at the Interface of Homeostasis and Pathology. Immunity 2019, 50, 1149–1162. [Google Scholar] [CrossRef]
- Henson, P.M. Cell Removal: Efferocytosis. Annu. Rev. Cell Dev. Biol. 2017, 33, 127–144. [Google Scholar] [CrossRef]
- Kumar, S.; Calianese, D.; Birge, R.B. Efferocytosis of dying cells differentially modulate immunological outcomes in tumor microenvironment. Immunol. Rev. 2017, 280, 149–164. [Google Scholar] [CrossRef]
- Kimani, S.G.; Geng, K.; Kasikara, C.; Kumar, S.; Sriram, G.; Wu, Y.; Birge, R.B. Contribution of Defective PS Recognition and Efferocytosis to Chronic Inflammation and Autoimmunity. Front. Immunol. 2014, 5, 566. [Google Scholar] [CrossRef]
- Kumar, S.; Birge, R.B. Efferocytosis. Curr. Biol. 2016, 26, R558–R559. [Google Scholar] [CrossRef]
- Kawano, M.; Nagata, S. Efferocytosis and autoimmune disease. Int. Immunol. 2018, 30, 551–558. [Google Scholar] [CrossRef]
- Kourtzelis, I.; Hajishengallis, G.; Chavakis, T. Phagocytosis of Apoptotic Cells in Resolution of Inflammation. Front. Immunol. 2020, 11, 553. [Google Scholar] [CrossRef] [PubMed]
- Szondy, Z.; Garabuczi, E.; Joos, G.; Tsay, G.J.; Sarang, Z. Impaired clearance of apoptotic cells in chronic inflammatory diseases: Therapeutic implications. Front. Immunol. 2014, 5, 354. [Google Scholar] [CrossRef]
- Levine, J.S.; Ucker, D.S. Voices from the dead: The complex vocabulary and intricate grammar of dead cells. Adv. Protein Chem. Struct. Biol. 2019, 116, 1–90. [Google Scholar] [CrossRef] [PubMed]
- Mollereau, B.; Perez-Garijo, A.; Bergmann, A.; Miura, M.; Gerlitz, O.; Ryoo, H.D.; Steller, H.; Morata, G. Compensatory proliferation and apoptosis-induced proliferation: A need for clarification. Cell Death Differ. 2013, 20, 181. [Google Scholar] [CrossRef] [PubMed]
- Ryoo, H.D.; Gorenc, T.; Steller, H. Apoptotic cells can induce compensatory cell proliferation through the JNK and the Wingless signaling pathways. Dev. Cell 2004, 7, 491–501. [Google Scholar] [CrossRef]
- Bosurgi, L.; Cao, Y.G.; Cabeza-Cabrerizo, M.; Tucci, A.; Hughes, L.D.; Kong, Y.; Weinstein, J.S.; Licona-Limon, P.; Schmid, E.T.; Pelorosso, F.; et al. Macrophage function in tissue repair and remodeling requires IL-4 or IL-13 with apoptotic cells. Science 2017, 356, 1072–1076. [Google Scholar] [CrossRef]
- Bosurgi, L.; Hughes, L.D.; Rothlin, C.V.; Ghosh, S. Death begets a new beginning. Immunol. Rev. 2017, 280, 8–25. [Google Scholar] [CrossRef]
- Savill, J.; Fadok, V. Corpse clearance defines the meaning of cell death. Nature 2000, 407, 784–788. [Google Scholar] [CrossRef]
- Birge, R.B.; Ucker, D.S. Innate apoptotic immunity: The calming touch of death. Cell Death Differ. 2008, 15, 1096–1102. [Google Scholar] [CrossRef]
- Doran, A.C.; Yurdagul, A., Jr.; Tabas, I. Efferocytosis in health and disease. Nat. Rev. Immunol. 2020, 20, 254–267. [Google Scholar] [CrossRef]
- Ucker, D.S.; Levine, J.S. Exploitation of Apoptotic Regulation in Cancer. Front. Immunol. 2018, 9, 241. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Li, F.; Liu, X.; Li, W.; Shi, W.; Liu, F.F.; O’Sullivan, B.; He, Z.; Peng, Y.; Tan, A.C.; et al. Caspase 3-mediated stimulation of tumor cell repopulation during cancer radiotherapy. Nat. Med. 2011, 17, 860–866. [Google Scholar] [CrossRef] [PubMed]
- Lauber, K.; Ernst, A.; Orth, M.; Herrmann, M.; Belka, C. Dying cell clearance and its impact on the outcome of tumor radiotherapy. Front. Oncol. 2012, 2, 116. [Google Scholar] [CrossRef]
- Poon, I.K.; Lucas, C.D.; Rossi, A.G.; Ravichandran, K.S. Apoptotic cell clearance: Basic biology and therapeutic potential. Nat. Rev. Immunol. 2014, 14, 166–180. [Google Scholar] [CrossRef] [PubMed]
- Lauber, K.; Bohn, E.; Krober, S.M.; Xiao, Y.J.; Blumenthal, S.G.; Lindemann, R.K.; Marini, P.; Wiedig, C.; Zobywalski, A.; Baksh, S.; et al. Apoptotic cells induce migration of phagocytes via caspase-3-mediated release of a lipid attraction signal. Cell 2003, 113, 717–730. [Google Scholar] [CrossRef]
- Peter, C.; Waibel, M.; Keppeler, H.; Lehmann, R.; Xu, G.; Halama, A.; Adamski, J.; Schulze-Osthoff, K.; Wesselborg, S.; Lauber, K. Release of lysophospholipid ‘find-me’ signals during apoptosis requires the ATP-binding cassette transporter A1. Autoimmunity 2012, 45, 568–573. [Google Scholar] [CrossRef]
- Elliott, M.R.; Chekeni, F.B.; Trampont, P.C.; Lazarowski, E.R.; Kadl, A.; Walk, S.F.; Park, D.; Woodson, R.I.; Ostankovich, M.; Sharma, P.; et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 2009, 461, 282–286. [Google Scholar] [CrossRef]
- Medina, C.B.; Ravichandran, K.S. Do not let death do us part: ‘find-me’ signals in communication between dying cells and the phagocytes. Cell Death Differ. 2016, 23, 979–989. [Google Scholar] [CrossRef]
- Mao, P.; Smith, L.; Xie, W.; Wang, M. Dying endothelial cells stimulate proliferation of malignant glioma cells via a caspase 3-mediated pathway. Oncol. Lett. 2013, 5, 1615–1620. [Google Scholar] [CrossRef]
- Gude, D.R.; Alvarez, S.E.; Paugh, S.W.; Mitra, P.; Yu, J.; Griffiths, R.; Barbour, S.E.; Milstien, S.; Spiegel, S. Apoptosis induces expression of sphingosine kinase 1 to release sphingosine-1-phosphate as a “come-and-get-me” signal. FASEB J. 2008, 22, 2629–2638. [Google Scholar] [CrossRef]
- Knies, U.E.; Behrensdorf, H.A.; Mitchell, C.A.; Deutsch, U.; Risau, W.; Drexler, H.C.; Clauss, M. Regulation of endothelial monocyte-activating polypeptide II release by apoptosis. Proc. Natl. Acad. Sci. USA 1998, 95, 12322–12327. [Google Scholar] [CrossRef] [PubMed]
- Krysko, D.V.; Garg, A.D.; Kaczmarek, A.; Krysko, O.; Agostinis, P.; Vandenabeele, P. Immunogenic cell death and DAMPs in cancer therapy. Nat. Rev. Cancer 2012, 12, 860–875. [Google Scholar] [CrossRef] [PubMed]
- Diwanji, N.; Bergmann, A. Two Sides of the Same Coin-Compensatory Proliferation in Regeneration and Cancer. Adv. Exp. Med. Biol. 2019, 1167, 65–85. [Google Scholar] [CrossRef]
- Fan, Y.; Bergmann, A. Apoptosis-induced compensatory proliferation. The Cell is dead. Long live the Cell! Trends Cell Biol. 2008, 18, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.; Witek, R.P.; Syn, W.K.; Choi, S.S.; Omenetti, A.; Premont, R.; Guy, C.D.; Diehl, A.M. Signals from dying hepatocytes trigger growth of liver progenitors. Gut 2010, 59, 655–665. [Google Scholar] [CrossRef]
- Li, F.; Huang, Q.; Chen, J.; Peng, Y.; Roop, D.R.; Bedford, J.S.; Li, C.Y. Apoptotic cells activate the “phoenix rising” pathway to promote wound healing and tissue regeneration. Sci. Signal. 2010, 3, ra13. [Google Scholar] [CrossRef]
- Li, H.; Yang, B.; Huang, J.; Lin, Y.; Xiang, T.; Wan, J.; Li, H.; Chouaib, S.; Ren, G. Cyclooxygenase-2 in tumor-associated macrophages promotes breast cancer cell survival by triggering a positive-feedback loop between macrophages and cancer cells. Oncotarget 2015, 6, 29637–29650. [Google Scholar] [CrossRef]
- Fogarty, C.E.; Bergmann, A. Killers creating new life: Caspases drive apoptosis-induced proliferation in tissue repair and disease. Cell Death Differ. 2017, 24, 1390–1400. [Google Scholar] [CrossRef]
- Niethammer, P.; Grabher, C.; Look, A.T.; Mitchison, T.J. A tissue-scale gradient of hydrogen peroxide mediates rapid wound detection in zebrafish. Nature 2009, 459, 996–999. [Google Scholar] [CrossRef]
- Razzell, W.; Evans, I.R.; Martin, P.; Wood, W. Calcium flashes orchestrate the wound inflammatory response through DUOX activation and hydrogen peroxide release. Curr. Biol. 2013, 23, 424–429. [Google Scholar] [CrossRef]
- Fogarty, C.E.; Diwanji, N.; Lindblad, J.L.; Tare, M.; Amcheslavsky, A.; Makhijani, K.; Bruckner, K.; Fan, Y.; Bergmann, A. Extracellular Reactive Oxygen Species Drive Apoptosis-Induced Proliferation via Drosophila Macrophages. Curr. Biol. 2016, 26, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Lynch, C.; Panagopoulou, M.; Gregory, C.D. Extracellular Vesicles Arising from Apoptotic Cells in Tumors: Roles in Cancer Pathogenesis and Potential Clinical Applications. Front. Immunol. 2017, 8, 1174. [Google Scholar] [CrossRef] [PubMed]
- Truman, L.A.; Ford, C.A.; Pasikowska, M.; Pound, J.D.; Wilkinson, S.J.; Dumitriu, I.E.; Melville, L.; Melrose, L.A.; Ogden, C.A.; Nibbs, R.; et al. CX3CL1/fractalkine is released from apoptotic lymphocytes to stimulate macrophage chemotaxis. Blood 2008, 112, 5026–5036. [Google Scholar] [CrossRef]
- Torr, E.E.; Gardner, D.H.; Thomas, L.; Goodall, D.M.; Bielemeier, A.; Willetts, R.; Griffiths, H.R.; Marshall, L.J.; Devitt, A. Apoptotic cell-derived ICAM-3 promotes both macrophage chemoattraction to and tethering of apoptotic cells. Cell Death Differ. 2012, 19, 671–679. [Google Scholar] [CrossRef]
- Coleman, M.L.; Sahai, E.A.; Yeo, M.; Bosch, M.; Dewar, A.; Olson, M.F. Membrane blebbing during apoptosis results from caspase-mediated activation of ROCK I. Nat. Cell Biol. 2001, 3, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Tixeira, R.; Phan, T.K.; Caruso, S.; Shi, B.; Atkin-Smith, G.K.; Nedeva, C.; Chow, J.D.Y.; Puthalakath, H.; Hulett, M.D.; Herold, M.J.; et al. ROCK1 but not LIMK1 or PAK2 is a key regulator of apoptotic membrane blebbing and cell disassembly. Cell Death Differ. 2020, 27, 102–116. [Google Scholar] [CrossRef] [PubMed]
- Segundo, C.; Medina, F.; Rodriguez, C.; Martinez-Palencia, R.; Leyva-Cobian, F.; Brieva, J.A. Surface molecule loss and bleb formation by human germinal center B cells undergoing apoptosis: Role of apoptotic blebs in monocyte chemotaxis. Blood 1999, 94, 1012–1020. [Google Scholar] [CrossRef]
- Lane, J.D.; Allan, V.J.; Woodman, P.G. Active relocation of chromatin and endoplasmic reticulum into blebs in late apoptotic cells. J. Cell Sci. 2005, 118, 4059–4071. [Google Scholar] [CrossRef]
- Ruben, J.M.; van den Ancker, W.; Bontkes, H.J.; Westers, T.M.; Hooijberg, E.; Ossenkoppele, G.J.; de Gruijl, T.D.; van de Loosdrecht, A.A. Apoptotic blebs from leukemic cells as a preferred source of tumor-associated antigen for dendritic cell-based vaccines. Cancer Immunol. Immunother. 2014, 63, 335–345. [Google Scholar] [CrossRef]
- Gregory, C.D.; Paterson, M. An apoptosis-driven ‘onco-regenerative niche’: Roles of tumour-associated macrophages and extracellular vesicles. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 373. [Google Scholar] [CrossRef]
- Nagata, S. Apoptosis and Clearance of Apoptotic Cells. Annu. Rev. Immunol. 2018, 36, 489–517. [Google Scholar] [CrossRef] [PubMed]
- Birge, R.B.; Boeltz, S.; Kumar, S.; Carlson, J.; Wanderley, J.; Calianese, D.; Barcinski, M.; Brekken, R.A.; Huang, X.; Hutchins, J.T.; et al. Phosphatidylserine is a global immunosuppressive signal in efferocytosis, infectious disease, and cancer. Cell Death Differ. 2016, 23, 962–978. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, J.; Denning, D.P.; Imanishi, E.; Horvitz, H.R.; Nagata, S. Xk-related protein 8 and CED-8 promote phosphatidylserine exposure in apoptotic cells. Science 2013, 341, 403–406. [Google Scholar] [CrossRef]
- Segawa, K.; Kurata, S.; Nagata, S. Human Type IV P-type ATPases That Work as Plasma Membrane Phospholipid Flippases and Their Regulation by Caspase and Calcium. J. Biol. Chem. 2016, 291, 762–772. [Google Scholar] [CrossRef]
- Segawa, K.; Kurata, S.; Yanagihashi, Y.; Brummelkamp, T.R.; Matsuda, F.; Nagata, S. Caspase-mediated cleavage of phospholipid flippase for apoptotic phosphatidylserine exposure. Science 2014, 344, 1164–1168. [Google Scholar] [CrossRef] [PubMed]
- Ravichandran, K.S. Find-me and eat-me signals in apoptotic cell clearance: Progress and conundrums. J. Exp. Med. 2010, 207, 1807–1817. [Google Scholar] [CrossRef]
- Nishi, C.; Toda, S.; Segawa, K.; Nagata, S. Tim4- and MerTK-mediated engulfment of apoptotic cells by mouse resident peritoneal macrophages. Mol. Cell Biol. 2014, 34, 1512–1520. [Google Scholar] [CrossRef]
- Nishi, C.; Yanagihashi, Y.; Segawa, K.; Nagata, S. MERTK tyrosine kinase receptor together with TIM4 phosphatidylserine receptor mediates distinct signal transduction pathways for efferocytosis and cell proliferation. J. Biol. Chem. 2019, 294, 7221–7230. [Google Scholar] [CrossRef]
- Miyanishi, M.; Segawa, K.; Nagata, S. Synergistic effect of Tim4 and MFG-E8 null mutations on the development of autoimmunity. Int. Immunol. 2012, 24, 551–559. [Google Scholar] [CrossRef]
- Toda, S.; Hanayama, R.; Nagata, S. Two-step engulfment of apoptotic cells. Mol. Cell Biol. 2012, 32, 118–125. [Google Scholar] [CrossRef]
- Wu, Y.; Singh, S.; Georgescu, M.M.; Birge, R.B. A role for Mer tyrosine kinase in alphavbeta5 integrin-mediated phagocytosis of apoptotic cells. J. Cell Sci. 2005, 118, 539–553. [Google Scholar] [CrossRef]
- Hu, B.; Jennings, J.H.; Sonstein, J.; Floros, J.; Todt, J.C.; Polak, T.; Curtis, J.L. Resident murine alveolar and peritoneal macrophages differ in adhesion of apoptotic thymocytes. Am. J. Respir. Cell Mol. Biol. 2004, 30, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Punturieri, A.; Todt, J.; Sonstein, J.; Polak, T.; Curtis, J.L. Recognition and phagocytosis of apoptotic T cells by resident murine tissue macrophages require multiple signal transduction events. J. Leukoc. Biol. 2002, 71, 881–889. [Google Scholar] [PubMed]
- Kasikara, C.; Davra, V.; Calianese, D.; Geng, K.; Spires, T.E.; Quigley, M.; Wichroski, M.; Sriram, G.; Suarez-Lopez, L.; Yaffe, M.B.; et al. Pan-TAM Tyrosine Kinase Inhibitor BMS-777607 Enhances Anti-PD-1 mAb Efficacy in a Murine Model of Triple-Negative Breast Cancer. Cancer Res. 2019, 79, 2669–2683. [Google Scholar] [CrossRef] [PubMed]
- Zagorska, A.; Traves, P.G.; Lew, E.D.; Dransfield, I.; Lemke, G. Diversification of TAM receptor tyrosine kinase function. Nat. Immunol. 2014, 15, 920–928. [Google Scholar] [CrossRef]
- Cai, B.; Thorp, E.B.; Doran, A.C.; Sansbury, B.E.; Daemen, M.J.; Dorweiler, B.; Spite, M.; Fredman, G.; Tabas, I. MerTK receptor cleavage promotes plaque necrosis and defective resolution in atherosclerosis. J. Clin. Investig. 2017, 127, 564–568. [Google Scholar] [CrossRef]
- Thorp, E.; Vaisar, T.; Subramanian, M.; Mautner, L.; Blobel, C.; Tabas, I. Shedding of the Mer tyrosine kinase receptor is mediated by ADAM17 protein through a pathway involving reactive oxygen species, protein kinase Cdelta, and p38 mitogen-activated protein kinase (MAPK). J. Biol. Chem. 2011, 286, 33335–33344. [Google Scholar] [CrossRef]
- Cvetanovic, M.; Mitchell, J.E.; Patel, V.; Avner, B.S.; Su, Y.; van der Saag, P.T.; Witte, P.L.; Fiore, S.; Levine, J.S.; Ucker, D.S. Specific recognition of apoptotic cells reveals a ubiquitous and unconventional innate immunity. J. Biol. Chem. 2006, 281, 20055–20067. [Google Scholar] [CrossRef]
- Cvetanovic, M.; Ucker, D.S. Innate immune discrimination of apoptotic cells: Repression of proinflammatory macrophage transcription is coupled directly to specific recognition. J. Immunol. 2004, 172, 880–889. [Google Scholar] [CrossRef]
- Tibrewal, N.; Wu, Y.; D’Mello, V.; Akakura, R.; George, T.C.; Varnum, B.; Birge, R.B. Autophosphorylation docking site Tyr-867 in Mer receptor tyrosine kinase allows for dissociation of multiple signaling pathways for phagocytosis of apoptotic cells and down-modulation of lipopolysaccharide-inducible NF-kappaB transcriptional activation. J. Biol. Chem. 2008, 283, 3618–3627. [Google Scholar] [CrossRef]
- Park, D.; Han, C.Z.; Elliott, M.R.; Kinchen, J.M.; Trampont, P.C.; Das, S.; Collins, S.; Lysiak, J.J.; Hoehn, K.L.; Ravichandran, K.S. Continued clearance of apoptotic cells critically depends on the phagocyte Ucp2 protein. Nature 2011, 477, 220–224. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Subramanian, M.; Yurdagul, A., Jr.; Barbosa-Lorenzi, V.C.; Cai, B.; de Juan-Sanz, J.; Ryan, T.A.; Nomura, M.; Maxfield, F.R.; Tabas, I. Mitochondrial Fission Promotes the Continued Clearance of Apoptotic Cells by Macrophages. Cell 2017, 171, 331–345.e322. [Google Scholar] [CrossRef]
- Boada-Romero, E.; Martinez, J.; Heckmann, B.L.; Green, D.R. The clearance of dead cells by efferocytosis. Nat. Rev. Mol. Cell Biol. 2020, 21, 398–414. [Google Scholar] [CrossRef] [PubMed]
- Cunha, L.D.; Yang, M.; Carter, R.; Guy, C.; Harris, L.; Crawford, J.C.; Quarato, G.; Boada-Romero, E.; Kalkavan, H.; Johnson, M.D.L.; et al. LC3-Associated Phagocytosis in Myeloid Cells Promotes Tumor Immune Tolerance. Cell 2018, 175, 429–441. [Google Scholar] [CrossRef] [PubMed]
- Heatley, M.K. A high apoptotic index occurs in subtypes of endometrial adenocarcinoma associated with a poor prognosis. Pathology 1997, 29, 272–275. [Google Scholar] [CrossRef] [PubMed]
- Leoncini, L.; Del Vecchio, M.T.; Megha, T.; Barbini, P.; Galieni, P.; Pileri, S.; Sabattini, E.; Gherlinzoni, F.; Tosi, P.; Kraft, R.; et al. Correlations between apoptotic and proliferative indices in malignant non-Hodgkin’s lymphomas. Am. J. Pathol. 1993, 142, 755–763. [Google Scholar] [PubMed]
- Naresh, K.N.; Lakshminarayanan, K.; Pai, S.A.; Borges, A.M. Apoptosis index is a predictor of metastatic phenotype in patients with early stage squamous carcinoma of the tongue: A hypothesis to support this paradoxical association. Cancer 2001, 91, 578–584. [Google Scholar] [CrossRef]
- Gregory, C.D.; Pound, J.D. Microenvironmental influences of apoptosis in vivo and in vitro. Apoptosis 2010, 15, 1029–1049. [Google Scholar] [CrossRef]
- Vaught, D.B.; Stanford, J.C.; Cook, R.S. Efferocytosis creates a tumor microenvironment supportive of tumor survival and metastasis. Cancer Cell Microenviron. 2015, 2. [Google Scholar] [CrossRef]
- Wong, W.Y.; Allie, S.; Limesand, K.H. PKCzeta and JNK signaling regulate radiation-induced compensatory proliferation in parotid salivary glands. PLoS ONE 2019, 14, e0219572. [Google Scholar] [CrossRef]
- Ford, C.A.; Petrova, S.; Pound, J.D.; Voss, J.J.; Melville, L.; Paterson, M.; Farnworth, S.L.; Gallimore, A.M.; Cuff, S.; Wheadon, H.; et al. Oncogenic properties of apoptotic tumor cells in aggressive B cell lymphoma. Curr. Biol. 2015, 25, 577–588. [Google Scholar] [CrossRef]
- Lauber, K.; Herrmann, M. Tumor biology: With a little help from my dying friends. Curr. Biol. 2015, 25, R198–R201. [Google Scholar] [CrossRef]
- Lemke, G. Phosphatidylserine Is the Signal for TAM Receptors and Their Ligands. Trends Biochem. Sci. 2017, 42, 738–748. [Google Scholar] [CrossRef]
- Casares, N.; Pequignot, M.O.; Tesniere, A.; Ghiringhelli, F.; Roux, S.; Chaput, N.; Schmitt, E.; Hamai, A.; Hervas-Stubbs, S.; Obeid, M.; et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J. Exp. Med. 2005, 202, 1691–1701. [Google Scholar] [CrossRef] [PubMed]
- Chaput, N.; De Botton, S.; Obeid, M.; Apetoh, L.; Ghiringhelli, F.; Panaretakis, T.; Flament, C.; Zitvogel, L.; Kroemer, G. Molecular determinants of immunogenic cell death: Surface exposure of calreticulin makes the difference. J. Mol. Med. 2007, 85, 1069–1076. [Google Scholar] [CrossRef]
- Galluzzi, L.; Petroni, G.; Kroemer, G. Immunogenicity of cell death driven by immune effectors. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Kasikova, L.; Hensler, M.; Truxova, I.; Skapa, P.; Laco, J.; Belicova, L.; Praznovec, I.; Vosahlikova, S.; Halaska, M.J.; Brtnicky, T.; et al. Calreticulin exposure correlates with robust adaptive antitumor immunity and favorable prognosis in ovarian carcinoma patients. J. Immunother. Cancer 2019, 7, 312. [Google Scholar] [CrossRef]
- Obeid, M.; Tesniere, A.; Panaretakis, T.; Tufi, R.; Joza, N.; van Endert, P.; Ghiringhelli, F.; Apetoh, L.; Chaput, N.; Flament, C.; et al. Ecto-calreticulin in immunogenic chemotherapy. Immunol. Rev. 2007, 220, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Kepp, O.; Menger, L.; Vacchelli, E.; Locher, C.; Adjemian, S.; Yamazaki, T.; Martins, I.; Sukkurwala, A.Q.; Michaud, M.; Senovilla, L.; et al. Crosstalk between ER stress and immunogenic cell death. Cytokine Growth Factor Rev. 2013, 24, 311–318. [Google Scholar] [CrossRef]
- Garg, A.D.; Krysko, D.V.; Verfaillie, T.; Kaczmarek, A.; Ferreira, G.B.; Marysael, T.; Rubio, N.; Firczuk, M.; Mathieu, C.; Roebroek, A.J.; et al. A novel pathway combining calreticulin exposure and ATP secretion in immunogenic cancer cell death. EMBO J. 2012, 31, 1062–1079. [Google Scholar] [CrossRef]
- Panaretakis, T.; Joza, N.; Modjtahedi, N.; Tesniere, A.; Vitale, I.; Durchschlag, M.; Fimia, G.M.; Kepp, O.; Piacentini, M.; Froehlich, K.U.; et al. The co-translocation of ERp57 and calreticulin determines the immunogenicity of cell death. Cell Death Differ. 2008, 15, 1499–1509. [Google Scholar] [CrossRef] [PubMed]
- Panaretakis, T.; Kepp, O.; Brockmeier, U.; Tesniere, A.; Bjorklund, A.C.; Chapman, D.C.; Durchschlag, M.; Joza, N.; Pierron, G.; van Endert, P.; et al. Mechanisms of pre-apoptotic calreticulin exposure in immunogenic cell death. EMBO J. 2009, 28, 578–590. [Google Scholar] [CrossRef] [PubMed]
- Dudek-Peric, A.M.; Ferreira, G.B.; Muchowicz, A.; Wouters, J.; Prada, N.; Martin, S.; Kiviluoto, S.; Winiarska, M.; Boon, L.; Mathieu, C.; et al. Antitumor immunity triggered by melphalan is potentiated by melanoma cell surface-associated calreticulin. Cancer Res. 2015, 75, 1603–1614. [Google Scholar] [CrossRef]
- Garg, A.D.; Agostinis, P. Cell death and immunity in cancer: From danger signals to mimicry of pathogen defense responses. Immunol. Rev. 2017, 280, 126–148. [Google Scholar] [CrossRef] [PubMed]
- Vanmeerbeek, I.; Sprooten, J.; De Ruysscher, D.; Tejpar, S.; Vandenberghe, P.; Fucikova, J.; Spisek, R.; Zitvogel, L.; Kroemer, G.; Galluzzi, L.; et al. Trial watch: Chemotherapy-induced immunogenic cell death in immuno-oncology. Oncoimmunology 2020, 9, 1703449. [Google Scholar] [CrossRef] [PubMed]
- Apetoh, L.; Ghiringhelli, F.; Tesniere, A.; Obeid, M.; Ortiz, C.; Criollo, A.; Mignot, G.; Maiuri, M.C.; Ullrich, E.; Saulnier, P.; et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat. Med. 2007, 13, 1050–1059. [Google Scholar] [CrossRef]
- Yamazaki, T.; Hannani, D.; Poirier-Colame, V.; Ladoire, S.; Locher, C.; Sistigu, A.; Prada, N.; Adjemian, S.; Catani, J.P.; Freudenberg, M.; et al. Defective immunogenic cell death of HMGB1-deficient tumors: Compensatory therapy with TLR4 agonists. Cell Death Differ. 2014, 21, 69–78. [Google Scholar] [CrossRef]
- Garg, A.D.; Krysko, D.V.; Vandenabeele, P.; Agostinis, P. Extracellular ATP and P(2)X(7) receptor exert context-specific immunogenic effects after immunogenic cancer cell death. Cell Death Dis. 2016, 7, e2097. [Google Scholar] [CrossRef]
- Sistigu, A.; Yamazaki, T.; Vacchelli, E.; Chaba, K.; Enot, D.P.; Adam, J.; Vitale, I.; Goubar, A.; Baracco, E.E.; Remedios, C.; et al. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat. Med. 2014, 20, 1301–1309. [Google Scholar] [CrossRef]
- Vanpouille-Box, C.; Hoffmann, J.A.; Galluzzi, L. Pharmacological modulation of nucleic acid sensors-therapeutic potential and persisting obstacles. Nat. Rev. Drug Discov. 2019, 18, 845–867. [Google Scholar] [CrossRef]
- Arur, S.; Uche, U.E.; Rezaul, K.; Fong, M.; Scranton, V.; Cowan, A.E.; Mohler, W.; Han, D.K. Annexin I is an endogenous ligand that mediates apoptotic cell engulfment. Dev. Cell 2003, 4, 587–598. [Google Scholar] [CrossRef]
- Scannell, M.; Flanagan, M.B.; deStefani, A.; Wynne, K.J.; Cagney, G.; Godson, C.; Maderna, P. Annexin-1 and peptide derivatives are released by apoptotic cells and stimulate phagocytosis of apoptotic neutrophils by macrophages. J. Immunol. 2007, 178, 4595–4605. [Google Scholar] [CrossRef]
- Vacchelli, E.; Ma, Y.; Baracco, E.E.; Sistigu, A.; Enot, D.P.; Pietrocola, F.; Yang, H.; Adjemian, S.; Chaba, K.; Semeraro, M.; et al. Chemotherapy-induced antitumor immunity requires formyl peptide receptor 1. Science 2015, 350, 972–978. [Google Scholar] [CrossRef] [PubMed]
- Krysko, O.; Aaes, T.L.; Kagan, V.E.; D’Herde, K.; Bachert, C.; Leybaert, L.; Vandenabeele, P.; Krysko, D.V. Necroptotic cell death in anti-cancer therapy. Immunol. Rev. 2017, 280, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Sprooten, J.; De Wijngaert, P.; Vanmeerbeerk, I.; Martin, S.; Vangheluwe, P.; Schlenner, S.; Krysko, D.V.; Parys, J.B.; Bultynck, G.; Vandenabeele, P.; et al. Necroptosis in Immuno-Oncology and Cancer Immunotherapy. Cells 2020, 9, 1823. [Google Scholar] [CrossRef]
- Messmer, M.N.; Snyder, A.G.; Oberst, A. Comparing the effects of different cell death programs in tumor progression and immunotherapy. Cell Death Differ. 2019, 26, 115–129. [Google Scholar] [CrossRef]
- He, G.W.; Gunther, C.; Thonn, V.; Yu, Y.Q.; Martini, E.; Buchen, B.; Neurath, M.F.; Sturzl, M.; Becker, C. Regression of apoptosis-resistant colorectal tumors by induction of necroptosis in mice. J. Exp. Med. 2017, 214, 1655–1662. [Google Scholar] [CrossRef]
- Oberst, A.; Dillon, C.P.; Weinlich, R.; McCormick, L.L.; Fitzgerald, P.; Pop, C.; Hakem, R.; Salvesen, G.S.; Green, D.R. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 2011, 471, 363–367. [Google Scholar] [CrossRef]
- Ning, X.; Wang, Y.; Jing, M.; Sha, M.; Lv, M.; Gao, P.; Zhang, R.; Huang, X.; Feng, J.M.; Jiang, Z. Apoptotic Caspases Suppress Type I Interferon Production via the Cleavage of cGAS, MAVS, and IRF3. Mol. Cell 2019, 74, 19–31. [Google Scholar] [CrossRef]
- Koppe, C.; Reisinger, F.; Wehr, K.; Vucur, M.; Trautwein, C.; Tacke, F.; Heikenwalder, M.; Luedde, T. An NF-kappaB- and IKK-Independent Function of NEMO Prevents Hepatocarcinogenesis by Suppressing Compensatory Liver Regeneration. Cancers 2019, 11, 999. [Google Scholar] [CrossRef]
- Vucur, M.; Reisinger, F.; Gautheron, J.; Janssen, J.; Roderburg, C.; Cardenas, D.V.; Kreggenwinkel, K.; Koppe, C.; Hammerich, L.; Hakem, R.; et al. RIP3 inhibits inflammatory hepatocarcinogenesis but promotes cholestasis by controlling caspase-8- and JNK-dependent compensatory cell proliferation. Cell Rep. 2013, 4, 776–790. [Google Scholar] [CrossRef]
- Yatim, N.; Jusforgues-Saklani, H.; Orozco, S.; Schulz, O.; Barreira da Silva, R.; Reis e Sousa, C.; Green, D.R.; Oberst, A.; Albert, M.L. RIPK1 and NF-kappaB signaling in dying cells determines cross-priming of CD8(+) T cells. Science 2015, 350, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Aaes, T.L.; Kaczmarek, A.; Delvaeye, T.; De Craene, B.; De Koker, S.; Heyndrickx, L.; Delrue, I.; Taminau, J.; Wiernicki, B.; De Groote, P.; et al. Vaccination with Necroptotic Cancer Cells Induces Efficient Anti-tumor Immunity. Cell Rep. 2016, 15, 274–287. [Google Scholar] [CrossRef]
- Snyder, A.G.; Hubbard, N.W.; Messmer, M.N.; Kofman, S.B.; Hagan, C.E.; Orozco, S.L.; Chiang, K.; Daniels, B.P.; Baker, D.; Oberst, A. Intratumoral activation of the necroptotic pathway components RIPK1 and RIPK3 potentiates antitumor immunity. Sci. Immunol. 2019, 4. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Frank, D.; Vince, J.E. Pyroptosis versus necroptosis: Similarities, differences, and crosstalk. Cell Death Differ. 2019, 26, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Tang, R.; Xu, J.; Zhang, B.; Liu, J.; Liang, C.; Hua, J.; Meng, Q.; Yu, X.; Shi, S. Ferroptosis, necroptosis, and pyroptosis in anticancer immunity. J. Hematol. Oncol. 2020, 13, 110. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Liu, X.L.; Zhao, R. Induction of Pyroptosis and Its Implications in Cancer Management. Front. Oncol. 2019, 9, 971. [Google Scholar] [CrossRef]
- Wang, Y.; Gao, W.; Shi, X.; Ding, J.; Liu, W.; He, H.; Wang, K.; Shao, F. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 2017, 547, 99–103. [Google Scholar] [CrossRef]
- Lang, X.; Green, M.D.; Wang, W.; Yu, J.; Choi, J.E.; Jiang, L.; Liao, P.; Zhou, J.; Zhang, Q.; Dow, A.; et al. Radiotherapy and Immunotherapy Promote Tumoral Lipid Oxidation and Ferroptosis via Synergistic Repression of SLC7A11. Cancer Discov. 2019, 9, 1673–1685. [Google Scholar] [CrossRef]
- Wang, W.; Green, M.; Choi, J.E.; Gijon, M.; Kennedy, P.D.; Johnson, J.K.; Liao, P.; Lang, X.; Kryczek, I.; Sell, A.; et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 2019, 569, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Silva, L.; Quevedo, L.; Varela, I. Tumor Functional Heterogeneity Unraveled by scRNA-seq Technologies. Trends Cancer 2020, 6, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [PubMed]
- Kubota, Y. Tumor angiogenesis and anti-angiogenic therapy. Keio J. Med. 2012, 61, 47–56. [Google Scholar] [CrossRef]
- Saharinen, P.; Eklund, L.; Alitalo, K. Therapeutic targeting of the angiopoietin-TIE pathway. Nat. Rev. Drug Discov. 2017, 16, 635–661. [Google Scholar] [CrossRef]
- Yamamoto, K.; Venida, A.; Yano, J.; Biancur, D.E.; Kakiuchi, M.; Gupta, S.; Sohn, A.S.W.; Mukhopadhyay, S.; Lin, E.Y.; Parker, S.J.; et al. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature 2020, 581, 100–105. [Google Scholar] [CrossRef]
- Blume, K.E.; Soeroes, S.; Waibel, M.; Keppeler, H.; Wesselborg, S.; Herrmann, M.; Schulze-Osthoff, K.; Lauber, K. Cell surface externalization of annexin A1 as a failsafe mechanism preventing inflammatory responses during secondary necrosis. J. Immunol. 2009, 183, 8138–8147. [Google Scholar] [CrossRef]
- Bondanza, A.; Zimmermann, V.S.; Rovere-Querini, P.; Turnay, J.; Dumitriu, I.E.; Stach, C.M.; Voll, R.E.; Gaipl, U.S.; Bertling, W.; Poschl, E.; et al. Inhibition of phosphatidylserine recognition heightens the immunogenicity of irradiated lymphoma cells in vivo. J. Exp. Med. 2004, 200, 1157–1165. [Google Scholar] [CrossRef]
- Stach, C.M.; Turnay, X.; Voll, R.E.; Kern, P.M.; Kolowos, W.; Beyer, T.D.; Kalden, J.R.; Herrmann, M. Treatment with annexin V increases immunogenicity of apoptotic human T-cells in Balb/c mice. Cell Death Differ. 2000, 7, 911–915. [Google Scholar] [CrossRef]
- Belzile, O.; Huang, X.; Gong, J.; Carlson, J.; Schroit, A.J.; Brekken, R.A.; Freimark, B.D. Antibody targeting of phosphatidylserine for the detection and immunotherapy of cancer. Immunotargets Ther. 2018, 7, 1–14. [Google Scholar] [CrossRef]
- Dayoub, A.S.; Brekken, R.A. TIMs, TAMs, and PS- antibody targeting: Implications for cancer immunotherapy. Cell Commun. Signal. 2020, 18, 29. [Google Scholar] [CrossRef]
- Luster, T.A.; He, J.; Huang, X.; Maiti, S.N.; Schroit, A.J.; de Groot, P.G.; Thorpe, P.E. Plasma protein beta-2-glycoprotein 1 mediates interaction between the anti-tumor monoclonal antibody 3G4 and anionic phospholipids on endothelial cells. J. Biol. Chem. 2006, 281, 29863–29871. [Google Scholar] [CrossRef]
- Gerber, D.E.; Horn, L.; Boyer, M.; Sanborn, R.; Natale, R.; Palmero, R.; Bidoli, P.; Bondarenko, I.; Germonpre, P.; Ghizdavescu, D.; et al. Randomized phase III study of docetaxel plus bavituximab in previously treated advanced non-squamous non-small-cell lung cancer. Ann. Oncol. 2018, 29, 1548–1553. [Google Scholar] [CrossRef]
- Freimark, B.D.; Gong, J.; Ye, D.; Gray, M.J.; Nguyen, V.; Yin, S.; Hatch, M.M.; Hughes, C.C.; Schroit, A.J.; Hutchins, J.T.; et al. Antibody-Mediated Phosphatidylserine Blockade Enhances the Antitumor Responses to CTLA-4 and PD-1 Antibodies in Melanoma. Cancer Immunol. Res. 2016, 4, 531–540. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.J.; Gong, J.; Hatch, M.M.; Nguyen, V.; Hughes, C.C.; Hutchins, J.T.; Freimark, B.D. Phosphatidylserine-targeting antibodies augment the anti-tumorigenic activity of anti-PD-1 therapy by enhancing immune activation and downregulating pro-oncogenic factors induced by T-cell checkpoint inhibition in murine triple-negative breast cancers. Breast Cancer Res. 2016, 18, 50. [Google Scholar] [CrossRef]
- Kallinteris, N.; Horn, L.; Tang, M.; Guennel, T.; Yin, S.; Lai, J.; Shan, J.; Sanborn, R.E. Abstract CT159: IFN-γ analysis in blood and tissue as a potential prognostic and/or predictive biomarker. In Proceedings of the AACR Annual Meeting, Washington, DC, USA, 1–5 April 2017. [Google Scholar]
- Desai, T.J.; Toombs, J.E.; Minna, J.D.; Brekken, R.A.; Udugamasooriya, D.G. Identification of lipid-phosphatidylserine (PS) as the target of unbiasedly selected cancer specific peptide-peptoid hybrid PPS1. Oncotarget 2016, 7, 30678–30690. [Google Scholar] [CrossRef]
- Blanco, V.M.; Chu, Z.; Vallabhapurapu, S.D.; Sulaiman, M.K.; Kendler, A.; Rixe, O.; Warnick, R.E.; Franco, R.S.; Qi, X. Phosphatidylserine-selective targeting and anticancer effects of SapC-DOPS nanovesicles on brain tumors. Oncotarget 2014, 5, 7105–7118. [Google Scholar] [CrossRef]
- N’Guessan, K.F.; Patel, P.H.; Qi, X. SapC-DOPS-a Phosphatidylserine-targeted Nanovesicle for selective Cancer therapy. Cell Commun. Signal. 2020, 18, 6. [Google Scholar] [CrossRef]
- Graham, D.K.; DeRyckere, D.; Davies, K.D.; Earp, H.S. The TAM family: Phosphatidylserine sensing receptor tyrosine kinases gone awry in cancer. Nat. Rev. Cancer 2014, 14, 769–785. [Google Scholar] [CrossRef]
- Lemke, G.; Rothlin, C.V. Immunobiology of the TAM receptors. Nat. Rev. Immunol. 2008, 8, 327–336. [Google Scholar] [CrossRef]
- Zeng, M.Y.; Pham, D.; Bagaitkar, J.; Liu, J.; Otero, K.; Shan, M.; Wynn, T.A.; Brombacher, F.; Brutkiewicz, R.R.; Kaplan, M.H.; et al. An efferocytosis-induced, IL-4-dependent macrophage-iNKT cell circuit suppresses sterile inflammation and is defective in murine CGD. Blood 2013, 121, 3473–3483. [Google Scholar] [CrossRef] [PubMed]
- Paolino, M.; Choidas, A.; Wallner, S.; Pranjic, B.; Uribesalgo, I.; Loeser, S.; Jamieson, A.M.; Langdon, W.Y.; Ikeda, F.; Fededa, J.P.; et al. The E3 ligase Cbl-b and TAM receptors regulate cancer metastasis via natural killer cells. Nature 2014, 507, 508–512. [Google Scholar] [CrossRef]
- Cook, R.S.; Jacobsen, K.M.; Wofford, A.M.; DeRyckere, D.; Stanford, J.; Prieto, A.L.; Redente, E.; Sandahl, M.; Hunter, D.M.; Strunk, K.E.; et al. MerTK inhibition in tumor leukocytes decreases tumor growth and metastasis. J. Clin. Investig. 2013, 123, 3231–3242. [Google Scholar] [CrossRef]
- Holtzhausen, A.; Harris, W.; Ubil, E.; Hunter, D.M.; Zhao, J.; Zhang, Y.; Zhang, D.; Liu, Q.; Wang, X.; Graham, D.K.; et al. TAM Family Receptor Kinase Inhibition Reverses MDSC-Mediated Suppression and Augments Anti-PD-1 Therapy in Melanoma. Cancer Immunol. Res. 2019, 7, 1672–1686. [Google Scholar] [CrossRef] [PubMed]
- Linger, R.M.; Keating, A.K.; Earp, H.S.; Graham, D.K. TAM receptor tyrosine kinases: Biologic functions, signaling, and potential therapeutic targeting in human cancer. Adv. Cancer Res. 2008, 100, 35–83. [Google Scholar] [CrossRef] [PubMed]
- Stanford, J.C.; Young, C.; Hicks, D.; Owens, P.; Williams, A.; Vaught, D.B.; Morrison, M.M.; Lim, J.; Williams, M.; Brantley-Sieders, D.M.; et al. Efferocytosis produces a prometastatic landscape during postpartum mammary gland involution. J. Clin. Investig. 2014, 124, 4737–4752. [Google Scholar] [CrossRef] [PubMed]
- Crittenden, M.R.; Baird, J.; Friedman, D.; Savage, T.; Uhde, L.; Alice, A.; Cottam, B.; Young, K.; Newell, P.; Nguyen, C.; et al. Mertk on tumor macrophages is a therapeutic target to prevent tumor recurrence following radiation therapy. Oncotarget 2016, 7, 78653–78666. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Fei, M.; Zhang, G.; Liang, W.C.; Lin, W.; Wu, Y.; Piskol, R.; Ridgway, J.; McNamara, E.; Huang, H.; et al. Blockade of the Phagocytic Receptor MerTK on Tumor-Associated Macrophages Enhances P2X7R-Dependent STING Activation by Tumor-Derived cGAMP. Immunity 2020, 52, 357–373. [Google Scholar] [CrossRef]
- Caetano, M.S.; Younes, A.I.; Barsoumian, H.B.; Quigley, M.; Menon, H.; Gao, C.; Spires, T.; Reilly, T.P.; Cadena, A.P.; Cushman, T.R.; et al. Triple Therapy with MerTK and PD1 Inhibition Plus Radiotherapy Promotes Abscopal Antitumor Immune Responses. Clin. Cancer Res. 2019, 25, 7576–7584. [Google Scholar] [CrossRef] [PubMed]
- Chao, M.P.; Alizadeh, A.A.; Tang, C.; Myklebust, J.H.; Varghese, B.; Gill, S.; Jan, M.; Cha, A.C.; Chan, C.K.; Tan, B.T.; et al. Anti-CD47 antibody synergizes with rituximab to promote phagocytosis and eradicate non-Hodgkin lymphoma. Cell 2010, 142, 699–713. [Google Scholar] [CrossRef]
- Goto, H.; Kojima, Y.; Matsuda, K.; Kariya, R.; Taura, M.; Kuwahara, K.; Nagai, H.; Katano, H.; Okada, S. Efficacy of anti-CD47 antibody-mediated phagocytosis with macrophages against primary effusion lymphoma. Eur. J. Cancer 2014, 50, 1836–1846. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, M.; Wang, X.; Liu, W.; Wang, H.; Yang, Y.G. Vaccination with CD47 deficient tumor cells elicits an antitumor immune response in mice. Nat. Commun. 2020, 11, 581. [Google Scholar] [CrossRef]
- Michaels, A.D.; Newhook, T.E.; Adair, S.J.; Morioka, S.; Goudreau, B.J.; Nagdas, S.; Mullen, M.G.; Persily, J.B.; Bullock, T.N.J.; Slingluff, C.L., Jr.; et al. CD47 Blockade as an Adjuvant Immunotherapy for Resectable Pancreatic Cancer. Clin. Cancer Res. 2018, 24, 1415–1425. [Google Scholar] [CrossRef]
- Tao, H.; Qian, P.; Wang, F.; Yu, H.; Guo, Y. Targeting CD47 Enhances the Efficacy of Anti-PD-1 and CTLA-4 in an Esophageal Squamous Cell Cancer Preclinical Model. Oncol. Res. 2017, 25, 1579–1587. [Google Scholar] [CrossRef]
- Willingham, S.B.; Volkmer, J.P.; Gentles, A.J.; Sahoo, D.; Dalerba, P.; Mitra, S.S.; Wang, J.; Contreras-Trujillo, H.; Martin, R.; Cohen, J.D.; et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 6662–6667. [Google Scholar] [CrossRef]
- Akalu, Y.T.; Rothlin, C.V.; Ghosh, S. TAM receptor tyrosine kinases as emerging targets of innate immune checkpoint blockade for cancer therapy. Immunol. Rev. 2017, 276, 165–177. [Google Scholar] [CrossRef]
- Bosurgi, L.; Bernink, J.H.; Delgado Cuevas, V.; Gagliani, N.; Joannas, L.; Schmid, E.T.; Booth, C.J.; Ghosh, S.; Rothlin, C.V. Paradoxical role of the proto-oncogene Axl and Mer receptor tyrosine kinases in colon cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 13091–13096. [Google Scholar] [CrossRef]
- Suzuki, J.; Fujii, T.; Imao, T.; Ishihara, K.; Kuba, H.; Nagata, S. Calcium-dependent phospholipid scramblase activity of TMEM16 protein family members. J. Biol. Chem. 2013, 288, 13305–13316. [Google Scholar] [CrossRef]
- Segawa, K.; Suzuki, J.; Nagata, S. Constitutive exposure of phosphatidylserine on viable cells. Proc. Natl. Acad. Sci. USA 2011, 108, 19246–19251. [Google Scholar] [CrossRef]
- Geng, K.; Kumar, S.; Kimani, S.G.; Kholodovych, V.; Kasikara, C.; Mizuno, K.; Sandiford, O.; Rameshwar, P.; Kotenko, S.V.; Birge, R.B. Requirement of Gamma-Carboxyglutamic Acid Modification and Phosphatidylserine Binding for the Activation of Tyro3, Axl, and Mertk Receptors by Growth Arrest-Specific 6. Front. Immunol. 2017, 8, 1521. [Google Scholar] [CrossRef]
- Ran, S. Antitumor Effects of a Monoclonal Antibody that Binds Anionic Phospholipids on the Surface of Tumor Blood Vessels in Mice. Clin. Cancer Res. 2005, 11, 1551–1562. [Google Scholar] [CrossRef]
- Ran, S.; Thorpe, P.E. Phosphatidylserine is a marker of tumor vasculature and a potential target for cancer imaging and therapy. Int. J. Radiat. Oncol. 2002, 54, 1479–1484. [Google Scholar] [CrossRef]
- Cabezón, R.; Carrera-Silva, E.A.; Flórez-Grau, G.; Errasti, A.E.; Gómez, E.C.; Lozano, J.J.; Espan, A.C.; Ricart, E.; Panes, J.; Rothlin, C.V.; et al. MERTK as negative regulator of human T cell activation. J. Leukoc. Boil. 2015, 97, 751–760. [Google Scholar] [CrossRef]
- Lea, J.; Sharma, R.; Yang, F.; Zhu, H.; Ward, E.S.; Schroit, A.J. Detection of phosphatidylserine-positive exosomes as a diagnostic marker for ovarian malignancies: A proof of concept study. Oncotarget 2017, 8, 14395–14407. [Google Scholar] [CrossRef]
- Sharma, R.; Huang, X.; Brekken, R.A.; Schroit, A.J. Detection of phosphatidylserine-positive exosomes for the diagnosis of early-stage malignancies. Br. J. Cancer 2017, 117, 545–552. [Google Scholar] [CrossRef]
- Shlomovitz, I.; Speir, M.; Gerlic, M. Flipping the dogma—Phosphatidylserine in non-apoptotic cell death. Cell Commun. Signal. 2019, 17, 1–12. [Google Scholar] [CrossRef]
- Zargarian, S.; Shlomovitz, I.; Erlich, Z.; Hourizadeh, A.; Ofir-Birin, Y.; Croker, B.A.; Regev-Rudzki, N.; Edry-Botzer, L.; Gerlic, M. Phosphatidylserine externalization, “necroptotic bodies” release, and phagocytosis during necroptosis. PLoS Boil. 2017, 15, e2002711. [Google Scholar] [CrossRef]
- Gong, Y.-N.; Guy, C.; Olauson, H.; Becker, J.U.; Yang, M.; Fitzgerald, P.; Linkermann, A.; Green, D.R. ESCRT-III Acts Downstream of MLKL to Regulate Necroptotic Cell Death and Its Consequences. Cell 2017, 169, 286–300. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gadiyar, V.; Lahey, K.C.; Calianese, D.; Devoe, C.; Mehta, D.; Bono, K.; Desind, S.; Davra, V.; Birge, R.B. Cell Death in the Tumor Microenvironment: Implications for Cancer Immunotherapy. Cells 2020, 9, 2207. https://doi.org/10.3390/cells9102207
Gadiyar V, Lahey KC, Calianese D, Devoe C, Mehta D, Bono K, Desind S, Davra V, Birge RB. Cell Death in the Tumor Microenvironment: Implications for Cancer Immunotherapy. Cells. 2020; 9(10):2207. https://doi.org/10.3390/cells9102207
Chicago/Turabian StyleGadiyar, Varsha, Kevin C. Lahey, David Calianese, Connor Devoe, Dhriti Mehta, Kristy Bono, Samuel Desind, Viralkumar Davra, and Raymond B. Birge. 2020. "Cell Death in the Tumor Microenvironment: Implications for Cancer Immunotherapy" Cells 9, no. 10: 2207. https://doi.org/10.3390/cells9102207
APA StyleGadiyar, V., Lahey, K. C., Calianese, D., Devoe, C., Mehta, D., Bono, K., Desind, S., Davra, V., & Birge, R. B. (2020). Cell Death in the Tumor Microenvironment: Implications for Cancer Immunotherapy. Cells, 9(10), 2207. https://doi.org/10.3390/cells9102207