TNF Production and Release from Microglia via Extracellular Vesicles: Impact on Brain Functions



Abstract
{kind=link}
{kind=link}
{kind=link}
1. Introduction
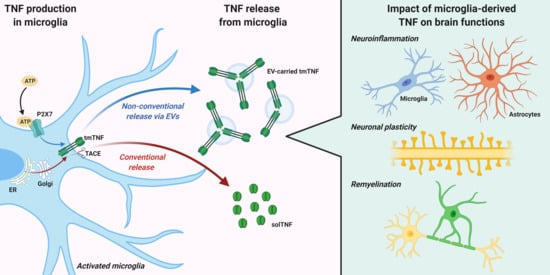
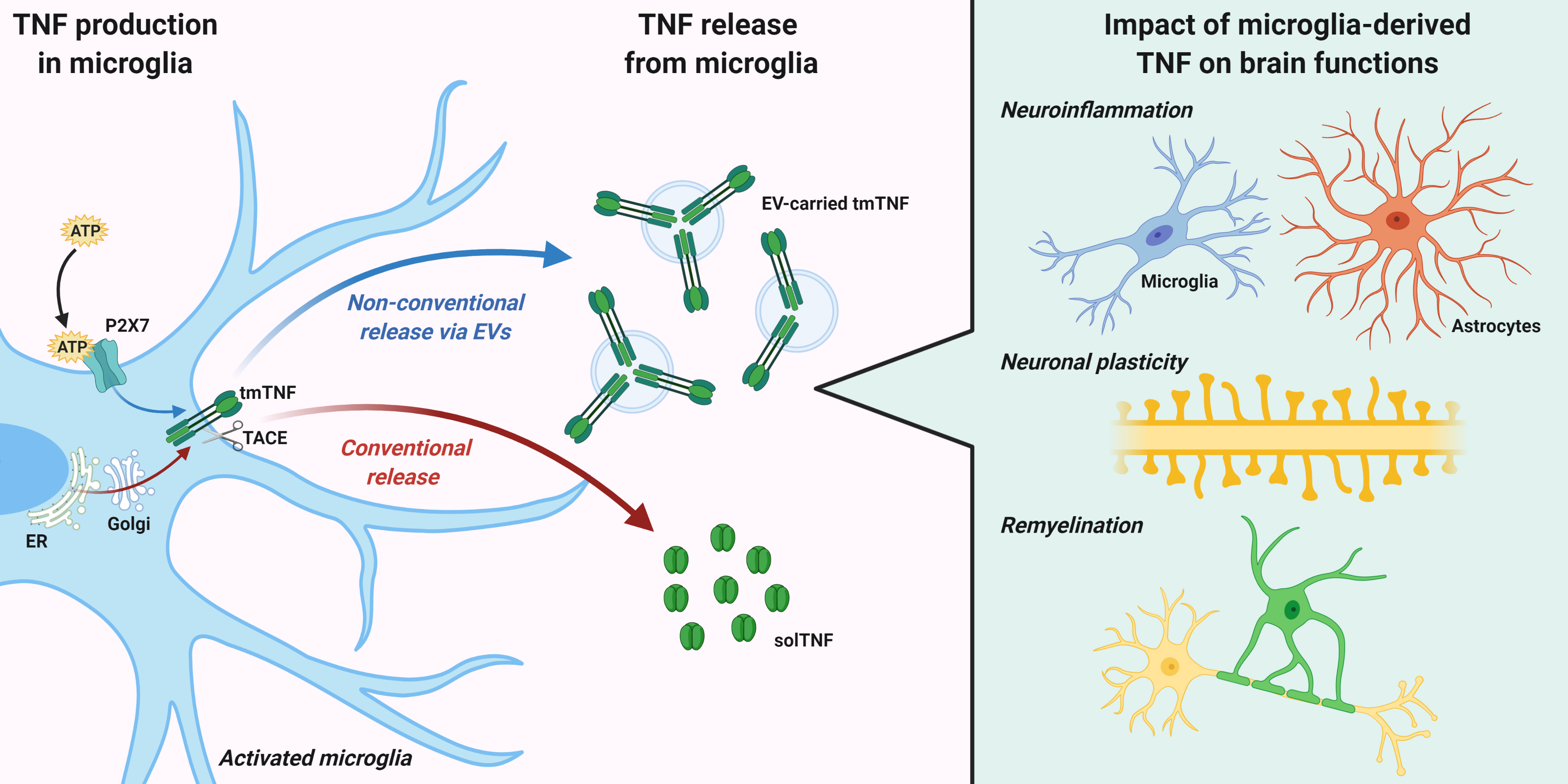
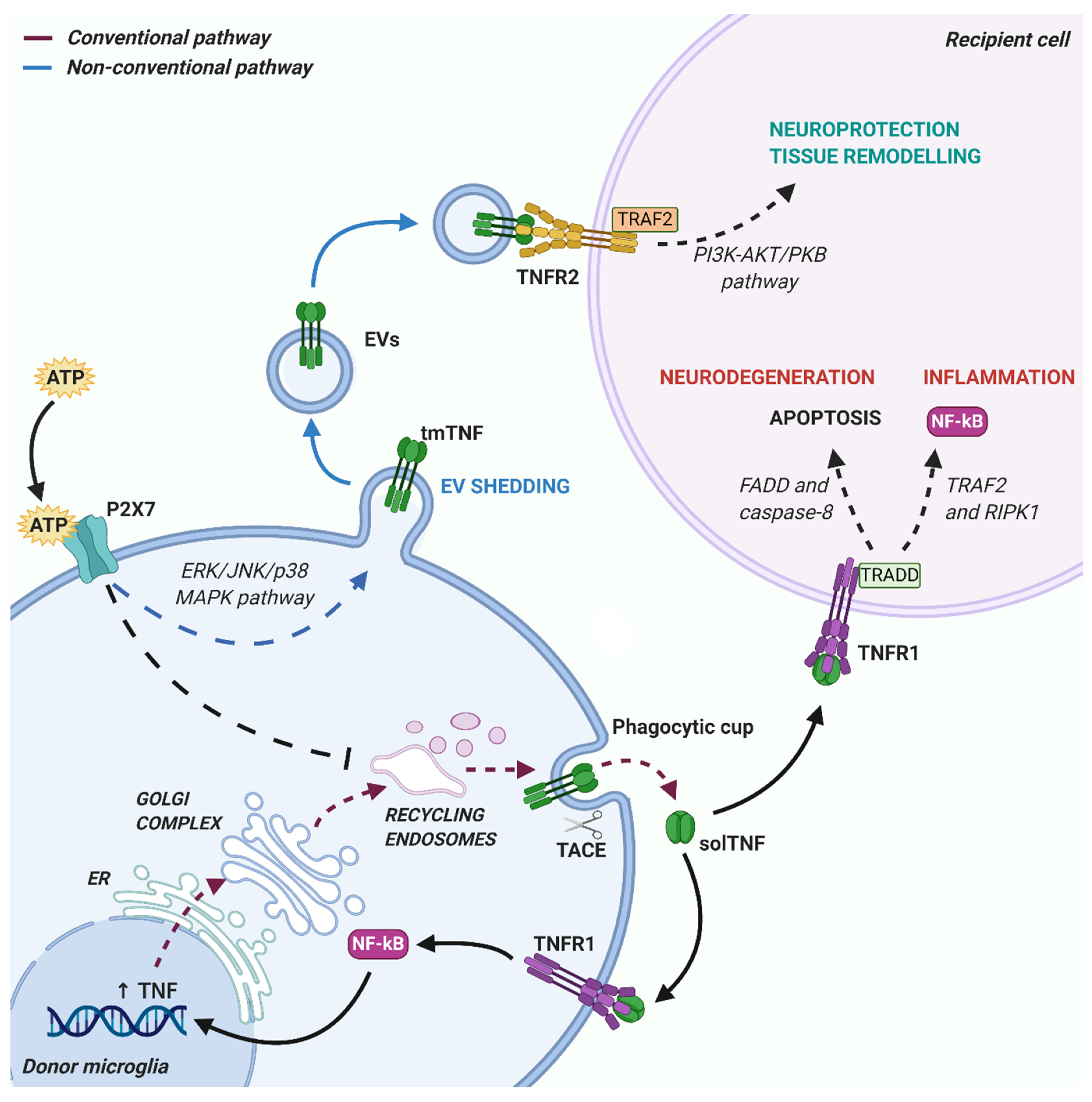
2. TNF Expression, Signaling, and Release from Microglia
3. TNF Release via Extracellular Vesicles (EVs)
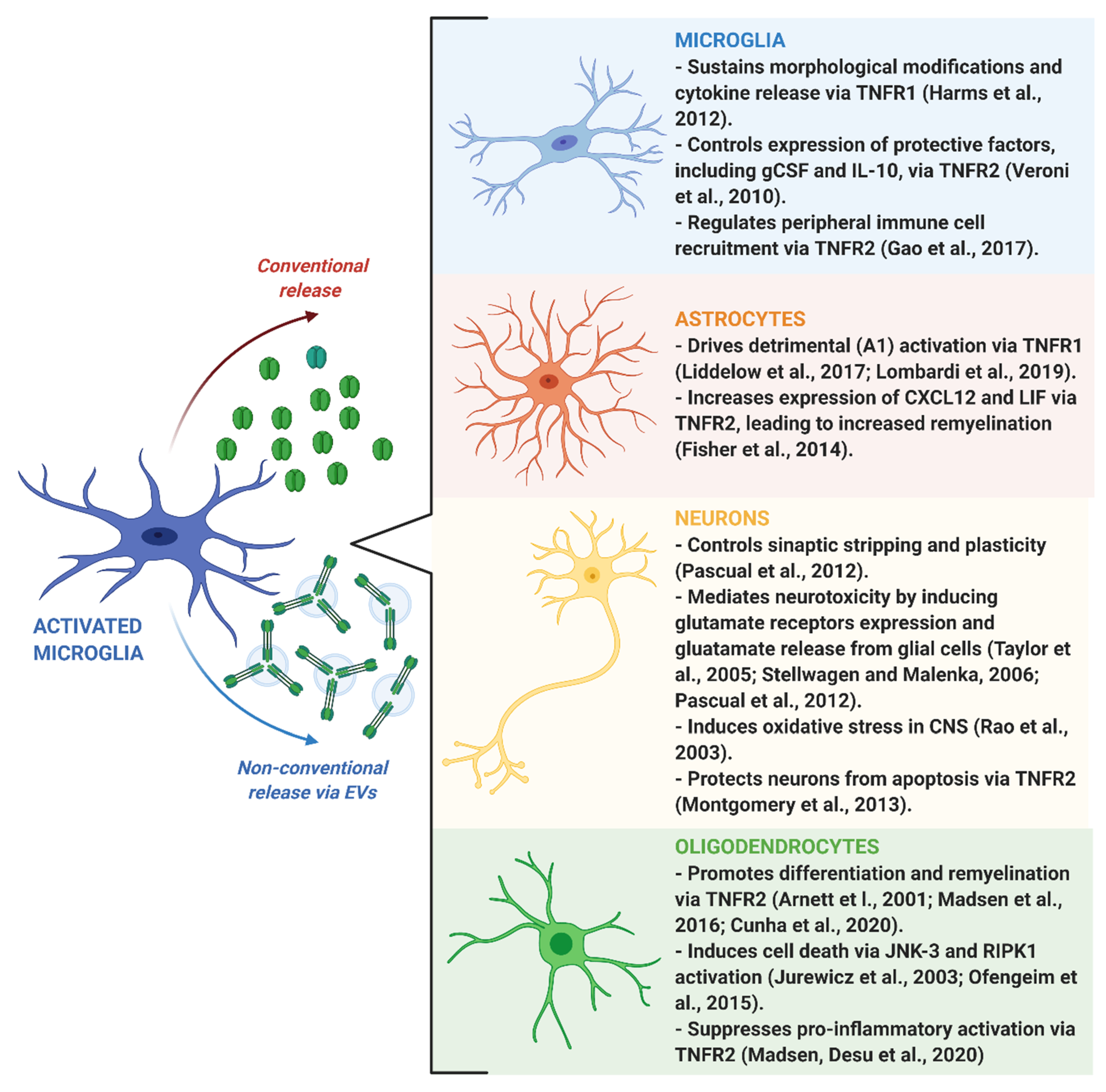
4. Impact of Microglia-Derived TNF on Brain Functions
4.1. Neuroinflammation
4.2. Neuronal Plasticity
4.3. Remyelination
5. Concluding Remarks and Future Perspectives
Funding
Conflicts of Interest
References
- Hanisch, U.K.; Kettenmann, H. Microglia: Active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 2007, 10, 1387–1394. [Google Scholar] [CrossRef] [PubMed]
- Prinz, M.; Jung, S.; Priller, J. Microglia Biology: One Century of Evolving Concepts. Cell 2019, 179, 292–311. [Google Scholar] [CrossRef] [PubMed]
- Fu, R.; Shen, Q.; Xu, P.; Luo, J.J.; Tang, Y. Phagocytosis of microglia in the central nervous system diseases. Mol. Neurobiol. 2014, 49, 1422–1434. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, J.; Wang, J.; Yang, B.; Weng, Q.; He, Q. Targeting microglia and macrophages: A potential treatment strategy for multiple sclerosis. Front. Pharmacol. 2019, 10, 286. [Google Scholar] [CrossRef]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef] [PubMed]
- Stratoulias, V.; Venero, J.L.; Tremblay, M.; Joseph, B. Microglial subtypes: Diversity within the microglial community. EMBO J. 2019, 38. [Google Scholar] [CrossRef]
- Masuda, T.; Sankowski, R.; Staszewski, O.; Böttcher, C.; Amann, L.; Scheiwe, C.; Nessler, S.; Kunz, P.; van Loo, G.; Coenen, V.A.; et al. Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature 2019, 566, 388–392. [Google Scholar] [CrossRef]
- Jordão, M.J.C.; Sankowski, R.; Brendecke, S.M.; Sagar; Locatelli, G.; Tai, Y.H.; Tay, T.L.; Schramm, E.; Armbruster, S.; Hagemeyer, N.; et al. Single-cell profiling identifies myeloid cell subsets with distinct fates during neuroinflammation. Science 2019, 363. [Google Scholar] [CrossRef]
- Mathys, H.; Adaikkan, C.; Gao, F.; Young, J.Z.; Manet, E.; Hemberg, M.; De Jager, P.L.; Ransohoff, R.M.; Regev, A.; Tsai, L.H. Temporal Tracking of Microglia Activation in Neurodegeneration at Single-Cell Resolution. Cell Rep. 2017, 21, 366–380. [Google Scholar] [CrossRef]
- Hammond, T.R.; Dufort, C.; Dissing-Olesen, L.; Giera, S.; Young, A.; Wysoker, A.; Walker, A.J.; Gergits, F.; Segel, M.; Nemesh, J.; et al. Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity 2019, 50, 253–271.e6. [Google Scholar] [CrossRef]
- Probert, L. TNF and its receptors in the CNS: The essential, the desirable and the deleterious effects. Neuroscience 2015, 302, 2–22. [Google Scholar] [CrossRef]
- Horiuchi, T.; Mitoma, H.; Harashima, S.I.; Tsukamoto, H.; Shimoda, T. Transmembrane TNF-α: Structure, function and interaction with anti-TNF agents. Rheumatology 2010, 49, 1215–1228. [Google Scholar] [CrossRef]
- Holtmann, M.; Neurath, M. Differential TNF-Signaling in Chronic Inflammatory Disorders. Curr. Mol. Med. 2005, 4, 439–444. [Google Scholar] [CrossRef]
- Arnett, H.A.; Mason, J.; Marino, M.; Suzuki, K.; Matsushima, G.K.; Ting, J.P.Y. TNFα promotes proliferation of oligodendrocyte progenitors and remyelination. Nat. Neurosci. 2001, 4, 1116–1122. [Google Scholar] [CrossRef] [PubMed]
- Madsen, P.M.; Motti, D.; Karmally, S.; Szymkowski, D.E.; Lambertsen, K.L.; Bethea, J.R.; Brambilla, R. Oligodendroglial TNFR2 mediates membrane TNF-dependent repair in experimental autoimmune encephalomyelitis by promoting oligodendrocyte differentiation and remyelination. J. Neurosci. 2016, 36, 5128–5143. [Google Scholar] [CrossRef] [PubMed]
- Bradley, J.R. TNF-mediated inflammatory disease. J. Pathol. 2008, 214, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Chu, W.M. Tumor necrosis factor. Cancer Lett. 2013, 328, 222–225. [Google Scholar] [CrossRef] [PubMed]
- Micheau, O.; Tschopp, J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 2003, 114, 181–190. [Google Scholar] [CrossRef]
- Walczak, H. TNF and ubiquitin at the crossroads of gene activation, cell death, inflammation, and cancer. Immunol. Rev. 2011, 244, 9–28. [Google Scholar] [CrossRef]
- Medvedev, A.E.; Sundan, A.; Espevik, T. Involvement of the tumor necrosis factor receptor p75 in mediating cytotoxicity and gene regulating activities. Eur. J. Immunol. 1994, 24, 2842–2849. [Google Scholar] [CrossRef]
- Rao, P.; Hsu, K.C.; Chao, M.V. Upregulation of NF-κB-Dependent Gene Expression Mediated by the p75 Tumor Necrosis Factor Receptor. J. Interf. Cytokine Res. 1995, 15, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Fischer, R.; Wajant, H.; Kontermann, R.; Pfizenmaier, K.; Maier, O. Astrocyte-specific activation of TNFR2 promotes oligodendrocyte maturation by secretion of leukemia inhibitory factor. Glia 2014, 62, 272–283. [Google Scholar] [CrossRef] [PubMed]
- Chiaruttini, G.; Piperno, G.M.; Jouve, M.; De Nardi, F.; Larghi, P.; Peden, A.A.; Baj, G.; Müller, S.; Valitutti, S.; Galli, T.; et al. The SNARE VAMP7 Regulates Exocytic Trafficking of Interleukin-12 in Dendritic Cells. Cell Rep. 2016, 14, 2624–2636. [Google Scholar] [CrossRef] [PubMed]
- Hanisch, U.K. Microglia as a source and target of cytokines. Glia 2002, 40, 140–155. [Google Scholar] [CrossRef]
- Kong, X.; Zhang, Z.; Fu, T.; Ji, J.; Yang, J.; Gu, Z. TNF-α regulates microglial activation via the NF-κB signaling pathway in systemic lupus erythematosus with depression. Int. J. Biol. Macromol. 2019, 125, 892–900. [Google Scholar] [CrossRef]
- Wang, J.; Yang, C.; Zhao, Q.; Zhu, Z.; Li, Y.; Yang, P. Microglia activation induced by serum of SLE patients. J. Neuroimmunol. 2017, 310, 135–142. [Google Scholar] [CrossRef]
- McCoy, M.K.; Martinez, T.N.; Ruhn, K.A.; Szymkowski, D.E.; Smith, C.G.; Botterman, B.R.; Tansey, K.E.; Tansey, M.G. Blocking soluble tumor necrosis factor signaling with dominant-negative tumor necrosis factor inhibitor attenuates loss of dopaminergic neurons in models of Parkinson’s disease. J. Neurosci. 2006, 26, 9365–9375. [Google Scholar] [CrossRef]
- McAlpine, F.E.; Lee, J.K.; Harms, A.S.; Ruhn, K.A.; Blurton-Jones, M.; Hong, J.; Das, P.; Golde, T.E.; LaFerla, F.M.; Oddo, S.; et al. Inhibition of soluble TNF signaling in a mouse model of Alzheimer’s disease prevents pre-plaque amyloid-associated neuropathology. Neurobiol. Dis. 2009, 34, 163–177. [Google Scholar] [CrossRef]
- Ajami, B.; Samusik, N.; Wieghofer, P.; Ho, P.P.; Crotti, A.; Bjornson, Z.; Prinz, M.; Fantl, W.J.; Nolan, G.P.; Steinman, L. Single-cell mass cytometry reveals distinct populations of brain myeloid cells in mouse neuroinflammation and neurodegeneration models. Nat. Neurosci. 2018, 21, 541–551. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Brás, J.P.; Bravo, J.; Freitas, J.; Barbosa, M.A.; Santos, S.G.; Summavielle, T.; Almeida, M.I. TNF-alpha-induced microglia activation requires miR-342: Impact on NF-kB signaling and neurotoxicity. Cell Death Dis. 2020, 11, 415. [Google Scholar] [CrossRef] [PubMed]
- Tarkowski, E.; Rosengren, L.; Blomstrand, C.; Wikkelsö, C.; Jensen, C.; Ekholm, S.; Tarkowski, A. Intrathecal release of pro- and anti-inflammatory cytokines during stroke. Clin. Exp. Immunol. 1997, 110, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Sotgiu, S.; Zanda, B.; Marchetti, B.; Fois, M.L.; Arru, G.; Pes, G.M.; Salaris, F.S.; Arru, A.; Pirisi, A.; Rosati, G. Inflammatory biomarkers in blood of patients with acute brain ischemia. Eur. J. Neurol. 2006, 13, 505–513. [Google Scholar] [CrossRef]
- Frank-Cannon, T.C.; Alto, L.T.; McAlpine, F.E.; Tansey, M.G. Does neuroinflammation fan the flame in neurodegenerative diseases? Mol. Neurodegener. 2009, 4, 47. [Google Scholar] [CrossRef]
- Whitton, P.S. Inflammation as a causative factor in the aetiology of Parkinson’s disease. Br. J. Pharmacol. 2007, 150, 963–976. [Google Scholar] [CrossRef]
- Kappelmann, N.; Lewis, G.; Dantzer, R.; Jones, P.B.; Khandaker, G.M. Antidepressant activity of anti-cytokine treatment: A systematic review and meta-analysis of clinical trials of chronic inflammatory conditions. Mol. Psychiatry 2018, 23, 335–343. [Google Scholar] [CrossRef]
- Ménard, C.; Pfau, M.L.; Hodes, G.E.; Russo, S.J. Immune and Neuroendocrine Mechanisms of Stress Vulnerability and Resilience. Neuropsychopharmacology 2017, 42, 62–80. [Google Scholar] [CrossRef]
- Monaco, C.; Nanchahal, J.; Taylor, P.; Feldmann, M. Anti-TNF therapy: Past, present and future. Int. Immunol. 2015, 27, 55–62. [Google Scholar] [CrossRef]
- Inoue, K.; Nakajima, K.; Morimoto, T.; Kikuchi, Y.; Koizumi, S.; Illes, P.; Kohsaka, S. ATP stimulation of Ca 2+ -dependent plasminogen release from cultured microglia. Br. J. Pharmacol. 1998, 123, 1304–1310. [Google Scholar] [CrossRef]
- Abbracchio, M.P.; Burnstock, G.; Verkhratsky, A.; Zimmermann, H. Purinergic signalling in the nervous system: An overview. Trends Neurosci. 2009, 32, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Bianco, F.; Pravettoni, E.; Colombo, A.; Schenk, U.; Möller, T.; Matteoli, M.; Verderio, C. Astrocyte-Derived ATP Induces Vesicle Shedding and IL-1β Release from Microglia. J. Immunol. 2005, 174, 7268–7277. [Google Scholar] [CrossRef]
- Koizumi, S.; Ohsawa, K.; Inoue, K.; Kohsaka, S. Purinergic receptors in microglia: Functional modal shifts of microglia mediated by P2 and P1 receptors. Glia 2013, 61, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Hide, I.; Tanaka, M.; Inoue, A.; Nakajima, K.; Kohsaka, S.; Inoue, K.; Nakata, Y. Extracellular ATP triggers tumor necrosis factor-α release from rat microglia. J. Neurochem. 2000, 75, 965–972. [Google Scholar] [CrossRef] [PubMed]
- Shieh, C.H.; Heinrich, A.; Serchov, T.; van Calker, D.; Biber, K. P2X7-dependent, but differentially regulated release of IL-6, CCL2, and TNF-α in cultured mouse microglia. Glia 2014, 62, 592–607. [Google Scholar] [CrossRef]
- Robinson, M.J.; Cobb, M.H. Mitogen-activated protein kinase pathways. Curr. Opin. Cell Biol. 1997, 9, 180–186. [Google Scholar] [CrossRef]
- Ferrari, R. The role of TNF in cardiovascular disease. Pharmacol. Res. 1999, 40, 97–105. [Google Scholar] [CrossRef]
- Suzuki, T.; Hide, I.; Ido, K.; Kohsaka, S.; Inoue, K.; Nakata, Y. Production and Release of Neuroprotective Tumor Necrosis Factor by P2X 7 Receptor-Activated Microglia. J. Neurosci. 2004, 24, 1–7. [Google Scholar] [CrossRef]
- Murray, R.Z.; Stow, J.L. Cytokine secretion in macrophages: SNAREs, Rabs, and membrane trafficking. Front. Immunol. 2014, 5, 538. [Google Scholar] [CrossRef]
- De Matteis, M.A.; Luini, A. Exiting the Golgi complex. Nat. Rev. Mol. Cell Biol. 2008, 9, 273–284. [Google Scholar] [CrossRef]
- Kay, J.G.; Murray, R.Z.; Pagan, J.K.; Stow, J.L. Cytokine secretion via cholesterol-rich lipid raft-associated SNAREs at the phagocytic cup. J. Biol. Chem. 2006, 281, 11949–11954. [Google Scholar] [CrossRef]
- Tellier, E.; Canault, M.; Rebsomen, L.; Bonardo, B.; Juhan-Vague, I.; Nalbone, G.; Peiretti, F. The shedding activity of ADAM17 is sequestered in lipid rafts. Exp. Cell Res. 2006, 312, 3969–3980. [Google Scholar] [CrossRef]
- Revelo, N.H.; Ter Beest, M.; van den Bogaart, G. Membrane trafficking as an active regulator of constitutively secreted cytokines. J. Cell Sci. 2019, 133. [Google Scholar] [CrossRef] [PubMed]
- Daniels, M.J.D.; Brough, D. Unconventional pathways of secretion contribute to inflammation. Int. J. Mol. Sci. 2017, 18, 102. [Google Scholar] [CrossRef] [PubMed]
- Flieger, O.; Engling, A.; Bucala, R.; Lue, H.; Nickel, W.; Bernhagen, J. Regulated secretion of macrophage migration inhibitory factor is mediated by a non-classical pathway involving an ABC transporter. FEBS Lett. 2003, 551, 78–86. [Google Scholar] [CrossRef]
- Bergsbaken, T.; Fink, S.L.; Cookson, B.T. Pyroptosis: Host cell death and inflammation. Nat. Rev. Microbiol. 2009, 7, 99–109. [Google Scholar] [CrossRef]
- Afonina, I.S.; Tynan, G.A.; Logue, S.E.; Cullen, S.P.; Bots, M.; Lüthi, A.U.; Reeves, E.P.; McElvaney, N.G.; Medema, J.P.; Lavelle, E.C.; et al. Granzyme B-dependent proteolysis acts as a switch to enhance the proinflammatory activity of IL-1α. Mol. Cell 2011, 44, 265–278. [Google Scholar] [CrossRef]
- Soni, S.; O’Dea, K.P.; Tan, Y.Y.; Cho, K.; Abe, E.; Romano, R.; Cui, J.; Ma, D.; Sarathchandra, P.; Wilson, M.R.; et al. ATP redirects cytokine trafficking and promotes novel membrane TNF signaling via microvesicles. FASEB J. 2019, 33, 6442–6455. [Google Scholar] [CrossRef]
- Ikeda, M.; Tsuno, S.; Sugiyama, T.; Hashimoto, A.; Yamoto, K.; Takeuchi, K.; Kishi, H.; Mizuguchi, H.; Kohsaka, S.i.; Yoshioka, T. Ca2+ spiking activity caused by the activation of store-operated Ca2+ channels mediates TNF-α release from microglial cells under chronic purinergic stimulation. Biochim. Biophys. Acta Mol. Cell Res. 2013, 1833, 2573–2585. [Google Scholar] [CrossRef][Green Version]
- Bianco, F.; Perrotta, C.; Novellino, L.; Francolini, M.; Riganti, L.; Menna, E.; Saglietti, L.; Schuchman, E.H.; Furlan, R.; Clementi, E.; et al. Acid sphingomyelinase activity triggers microparticle release from glial cells. EMBO J. 2009, 28, 1043–1054. [Google Scholar] [CrossRef]
- Budnik, V.; Ruiz-Cañada, C.; Wendler, F. Extracellular vesicles round off communication in the nervous system. Nat. Rev. Neurosci. 2016, 17, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, W.; Freeman, M.L.; Lederman, M.M.; Vasilieva, E.; Romero, R.; Margolis, L. A System of Cytokines Encapsulated in ExtraCellular Vesicles. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7. [Google Scholar] [CrossRef]
- Garzetti, L.; Menon, R.; Finardi, A.; Bergami, A.; Sica, A.; Martino, G.; Comi, G.; Verderio, C.; Farina, C.; Furlan, R. Activated macrophages release microvesicles containing polarized M1 or M2 mRNAs. J. Leukoc. Biol. 2014, 95, 817–825. [Google Scholar] [CrossRef]
- Nigro, A.; Colombo, F.; Casella, G.; Finardi, A.; Verderio, C.; Furlan, R. Myeloid Extracellular Vesicles: Messengers from the Demented Brain. Front. Immunol. 2016, 7, 17. [Google Scholar] [CrossRef]
- Joshi, P.; Turola, E.; Ruiz, A.; Bergami, A.; Libera, D.D.; Benussi, L.; Giussani, P.; Magnani, G.; Comi, G.; Legname, G.; et al. Microglia convert aggregated amyloid-β into neurotoxic forms through the shedding of microvesicles. Cell Death Differ. 2014, 21, 582–593. [Google Scholar] [CrossRef]
- Crotti, A.; Sait, H.R.; McAvoy, K.M.; Estrada, K.; Ergun, A.; Szak, S.; Marsh, G.; Jandreski, L.; Peterson, M.; Reynolds, T.L.; et al. BIN1 favors the spreading of Tau via extracellular vesicles. Sci. Rep. 2019, 9, 1–20. [Google Scholar] [CrossRef]
- Delpech, J.C.; Herron, S.; Botros, M.B.; Ikezu, T. Neuroimmune Crosstalk through Extracellular Vesicles in Health and Disease. Trends Neurosci. 2019, 42, 361–372. [Google Scholar] [CrossRef]
- Pérez, M.; Avila, J.; Hernández, F. Propagation of tau via extracellular vesicles. Front. Neurosci. 2019, 13, 698. [Google Scholar] [CrossRef]
- Prada, I.; Gabrielli, M.; Turola, E.; Iorio, A.; D’Arrigo, G.; Parolisi, R.; De Luca, M.; Pacifici, M.; Bastoni, M.; Lombardi, M.; et al. Glia-to-neuron transfer of miRNAs via extracellular vesicles: A new mechanism underlying inflammation-induced synaptic alterations. Acta Neuropathol. 2018, 135, 529–550. [Google Scholar] [CrossRef]
- Lombardi, M.; Parolisi, R.; Scaroni, F.; Bonfanti, E.; Gualerzi, A.; Gabrielli, M.; Kerlero de Rosbo, N.; Uccelli, A.; Giussani, P.; Viani, P.; et al. Detrimental and protective action of microglial extracellular vesicles on myelin lesions: Astrocyte involvement in remyelination failure. Acta Neuropathol. 2019, 138, 987–1012. [Google Scholar] [CrossRef]
- Casella, G.; Colombo, F.; Finardi, A.; Descamps, H.; Ill-Raga, G.; Spinelli, A.; Podini, P.; Bastoni, M.; Martino, G.; Muzio, L.; et al. Extracellular Vesicles Containing IL-4 Modulate Neuroinflammation in a Mouse Model of Multiple Sclerosis. Mol. Ther. 2018, 26, 2107–2118. [Google Scholar] [CrossRef]
- Lim, C.Z.J.; Natalia, A.; Sundah, N.R.; Shao, H. Biomarker Organization in Circulating Extracellular Vesicles: New Applications in Detecting Neurodegenerative Diseases. Adv. Biosyst. 2020, 1900309. [Google Scholar] [CrossRef] [PubMed]
- Wiklander, O.P.B.; Brennan, M.; Lötvall, J.; Breakefield, X.O.; Andaloussi, S.E.L. Advances in therapeutic applications of extracellular vesicles. Sci. Transl. Med. 2019, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Boza-Serrano, A.; Dunning, C.J.R.; Clausen, B.H.; Lambertsen, K.L.; Deierborg, T. Inflammation leads to distinct populations of extracellular vesicles from microglia. J. Neuroinflamm. 2018, 15, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Verderio, C.; Muzio, L.; Turola, E.; Bergami, A.; Novellino, L.; Ruffini, F.; Riganti, L.; Corradini, I.; Francolini, M.; Garzetti, L.; et al. Myeloid microvesicles are a marker and therapeutic target for neuroinflammation. Ann. Neurol. 2012, 72, 610–624. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.; Lang, H.; Geng, N.; Wang, J.; Li, N.; Wang, X. Exosomes of BV-2 cells induced by alpha-synuclein: Important mediator of neurodegeneration in PD. Neurosci. Lett. 2013, 548, 190–195. [Google Scholar] [CrossRef]
- Pegoretti, V.; Baron, W.; Laman, J.D.; Eisel, U.L.M. Selective modulation of TNF-TNFRs signaling: Insights for multiple sclerosis treatment. Front. Immunol. 2018, 9, 925. [Google Scholar] [CrossRef]
- Islam, A.; Jones, H.; Hiroi, T.; Lam, J.; Zhang, J.; Moss, J.; Vaughan, M.; Levine, S.J. cAMP-dependent protein kinase A (PKA) signaling induces TNFR1 exosome-like vesicle release via anchoring of PKA regulatory subunit RIIβ to BIG2. J. Biol. Chem. 2008, 283, 25364–25371. [Google Scholar] [CrossRef]
- Sohda, M.; Misumi, Y.; Oda, K. TNFα triggers release of extracellular vesicles containing TNFR1 and TRADD, which can modulate TNFα responses of the parental cells. Arch. Biochem. Biophys. 2015, 587, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Hawari, F.I.; Shamburek, R.D.; Adamik, B.; Kaler, M.; Islam, A.; Liao, D.W.; Rouhani, F.N.; Ingham, M.; Levine, S.J. Circulating TNFR1 exosome-like vesicles partition with the LDL fraction of human plasma. Biochem. Biophys. Res. Commun. 2008, 366, 579–584. [Google Scholar] [CrossRef]
- Qu, Y.; Zhao, G.; Li, H. Forward and reverse signaling mediated by transmembrane tumor necrosis factor-alpha and TNF receptor 2: Potential roles in an immunosuppressive tumor microenvironment. Front. Immunol. 2017, 8, 1675. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.H.; Seo, D.; Lim, S.G.; Suk, K. Reverse Signaling of Tumor Necrosis Factor Superfamily Proteins in Factor Superfamily Proteins in Macrophages and microgia: Superfamily portrait in the neuroimmune interface. Front. Immunol. 2019, 10, 262. [Google Scholar] [CrossRef] [PubMed]
- Pallai, A.; Kiss, B.; Vereb, G.; Armaka, M.; Kollias, G.; Szekanecz, Z.; Szondy, Z. Transmembrane TNF-α Reverse Signaling Inhibits Lipopolysaccharide-Induced Proinflammatory Cytokine Formation in Macrophages by Inducing TGF-β: Therapeutic Implications. J. Immunol. 2016, 196, 1146–1157. [Google Scholar] [CrossRef] [PubMed]
- Szondy, Z.; Pallai, A. Transmembrane TNF-alpha reverse signaling leading to TGF-beta production is selectively activated by TNF targeting molecules: Therapeutic implications. Pharmacol. Res. 2017, 115, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Schierer, S.; Blume, K.; Dindorf, J.; Wittki, S.; Xiang, W.; Ostalecki, C.; Koliha, N.; Wild, S.; Schuler, G.; et al. HIV-Nef and ADAM17-Containing Plasma Extracellular Vesicles Induce and Correlate with Immune Pathogenesis in Chronic HIV Infection. EBioMedicine 2016, 6, 103–113. [Google Scholar] [CrossRef]
- Zhao, Z.; Kesti, T.; Uğurlu, H.; Baur, A.S.; Fagerlund, R.; Saksela, K. Tyrosine phosphorylation directs TACE into extracellular vesicles via unconventional secretion. Traffic 2019, 20, 202–212. [Google Scholar] [CrossRef]
- Edrissi, H.; Schock, S.C.; Hakim, A.M.; Thompson, C.S. Microparticles generated during chronic cerebral ischemia increase the permeability of microvascular endothelial barriers in vitro. Brain Res. 2016, 1634, 83–93. [Google Scholar] [CrossRef]
- Pieragostino, D.; Lanuti, P.; Cicalini, I.; Cufaro, M.C.; Ciccocioppo, F.; Ronci, M.; Simeone, P.; Onofrj, M.; van der Pol, E.; Fontana, A.; et al. Proteomics characterization of extracellular vesicles sorted by flow cytometry reveals a disease-specific molecular cross-talk from cerebrospinal fluid and tears in multiple sclerosis. J. Proteomics 2019, 204, 103403. [Google Scholar] [CrossRef]
- Kawata, K.; Mitsuhashi, M.; Aldret, R. A Preliminary Report on Brain-Derived Extracellular Vesicle as Novel Blood Biomarkers for Sport-Related Concussions. Front. Neurol. 2018, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- Mansur, R.B.; Delgado-Peraza, F.; Subramaniapillai, M.; Lee, Y.; Iacobucci, M.; Rodrigues, N.; Rosenblat, J.D.; Brietzke, E.; Cosgrove, V.E.; Kramer, N.E.; et al. Extracellular Vesicle Biomarkers Reveal Inhibition of Neuroinflammation by Infliximab in Association with Antidepressant Response in Adults with Bipolar Depression. Cells 2020, 9, 895. [Google Scholar] [CrossRef] [PubMed]
- Croese, T.; Furlan, R. Extracellular vesicles in neurodegenerative diseases. Mol. Aspects Med. 2018, 60, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.Q.; Zhou, J.W. Neuroinflammation in the central nervous system: Symphony of glial cells. Glia 2019, 67, 1017–1035. [Google Scholar] [CrossRef]
- Kuno, R.; Wang, J.; Kawanokuchi, J.; Takeuchi, H.; Mizuno, T.; Suzumura, A. Autocrine activation of microglia by tumor necrosis factor-α. J. Neuroimmunol. 2005, 162, 89–96. [Google Scholar] [CrossRef]
- Harms, A.S.; Lee, J.K.; Nguyen, T.A.; Chang, J.; Ruhn, K.M.; Treviño, I.; Tansey, M.G. Regulation of microglia effector functions by tumor necrosis factor signaling. Glia 2012, 60, 189–202. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Trindade, P.; Loiola, E.C.; Gasparotto, J.; Ribeiro, C.T.; Cardozo, P.L.; Devalle, S.; Salerno, J.A.; Ornelas, I.M.; Ledur, P.F.; Ribeiro, F.M.; et al. Short and long TNF-alpha exposure recapitulates canonical astrogliosis events in human-induced pluripotent stem cells-derived astrocytes. Glia 2020, 68, 1396–1409. [Google Scholar] [CrossRef]
- Clarke, L.E.; Liddelow, S.A.; Chakraborty, C.; Münch, A.E.; Heiman, M.; Barres, B.A. Normal aging induces A1-like astrocyte reactivity. Proc. Natl. Acad. Sci. USA 2018, 115, E1896–E1905. [Google Scholar] [CrossRef]
- Sierra, A.; Gottfried-Blackmore, A.C.; Mcewen, B.S.; Bulloch, K. Microglia derived from aging mice exhibit an altered inflammatory profile. Glia 2007, 55, 412–424. [Google Scholar] [CrossRef]
- Hu, W.T.; Howell, J.C.; Ozturk, T.; Gangishetti, U.; Kollhoff, A.L.; Hatcher-Martin, J.M.; Anderson, A.M.; Tyor, W.R. CSF cytokines in aging, multiple sclerosis, and dementia. Front. Immunol. 2019, 10, 480. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, K.R.; Lin, Y.; Langan, T.J.; Iwakura, Y.; Chou, R.C. Innate Immune Functions of Astrocytes are Dependent Upon Tumor Necrosis Factor-Alpha. Sci. Rep. 2020, 10, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.H.; Oyarzabal, E.A.; Sung, Y.F.; Chu, C.H.; Wang, Q.; Chen, S.L.; Lu, R.B.; Hong, J.S. Microglial regulation of immunological and neuroprotective functions of astroglia. Glia 2015, 63, 118–131. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Tang, Y.; Feng, J. Cross talk between activation of microglia and astrocytes in pathological conditions in the central nervous system. Life Sci. 2011, 89, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Jha, M.K.; Jo, M.; Kim, J.H.; Suk, K. Microglia-Astrocyte Crosstalk: An Intimate Molecular Conversation. Neuroscientist 2019, 25, 227–240. [Google Scholar] [CrossRef]
- Marchetti, B.; Abbracchio, M.P. To be or not to be (inflamed)—Is that the question in anti-inflammatory drug therapy of neurodegenerative disorders? Trends Pharmacol. Sci. 2005, 26, 517–525. [Google Scholar] [CrossRef]
- Gao, H.; Danzi, M.C.; Choi, C.S.; Taherian, M.; Dalby-Hansen, C.; Ellman, D.G.; Madsen, P.M.; Bixby, J.L.; Lemmon, V.P.; Lambertsen, K.L.; et al. Opposing Functions of Microglial and Macrophagic TNFR2 in the Pathogenesis of Experimental Autoimmune Encephalomyelitis. Cell Rep. 2017, 18, 198–212. [Google Scholar] [CrossRef]
- Veroni, C.; Gabriele, L.; Canini, I.; Castiello, L.; Coccia, E.; Remoli, M.E.; Columba-Cabezas, S.; Aricò, E.; Aloisi, F.; Agresti, C. Activation of TNF receptor 2 in microglia promotes induction of anti-inflammatory pathways. Mol. Cell. Neurosci. 2010, 45, 234–244. [Google Scholar] [CrossRef]
- Kettenmann, H.; Kirchhoff, F.; Verkhratsky, A. Microglia: New Roles for the Synaptic Stripper. Neuron 2013, 77, 10–18. [Google Scholar] [CrossRef]
- Pascual, O.; Achour, S.B.; Rostaing, P.; Triller, A.; Bessis, A. Microglia activation triggers astrocyte-mediated modulation of excitatory neurotransmission. Proc. Natl. Acad. Sci. USA 2012, 109, 197–205. [Google Scholar] [CrossRef]
- Stellwagen, D.; Malenka, R.C. Synaptic scaling mediated by glial TNF-α. Nature 2006, 440, 1054–1059. [Google Scholar] [CrossRef] [PubMed]
- Beattie, M.S.; Hermann, G.E.; Rogers, R.C.; Bresnahan, J.C. Cell death in models of spinal cord injury. Prog. Brain Res. 2002, 137, 37–47. [Google Scholar] [PubMed]
- Aizenman, C.D.; Pratt, K.G. There’s More Than One Way to Scale a Synapse. Neuron 2008, 58, 651–653. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ogoshi, F.; Yin, H.Z.; Kuppumbatti, Y.; Song, B.; Amindari, S.; Weiss, J.H. Tumor necrosis-factor-alpha (TNF-α) induces rapid insertion of Ca2+-permeable α-amino-3-hydroxyl-5-methyl-4-isoxazole- propionate (AMPA)/kainate (Ca-A/K) channels in a subset of hippocampal pyramidal neurons. Exp. Neurol. 2005, 193, 384–393. [Google Scholar] [CrossRef]
- Yu, Z.F.; Cheng, G.; Wen, X.; Wu, G.D.; Lee, W.T.; Pleasure, D. Tumor necrosis factor α increases neuronal vulnerability to excitotoxic necrosis by inducing expression of the AMPA-glutamate receptor subunit GluR1 via an acid sphingomyelinase-and NF-κB-dependent mechanism. Neurobiol. Dis. 2002, 11, 199–213. [Google Scholar] [CrossRef]
- Sitcheran, R.; Gupta, P.; Fisher, P.B.; Baldwin, A.S. Positive and negative regulation of EAAT2 by NF-κB: A role for N-myc in TNFα-controlled repression. EMBO J. 2005, 24, 510–520. [Google Scholar] [CrossRef]
- Wheeler, D.; Knapp, E.; Bandaru, V.V.R.; Wang, Y.; Knorr, D.; Poirier, C.; Mattson, M.P.; Geiger, J.D.; Haughey, N.J. Tumor necrosis factor-α-induced neutral sphingomyelinase-2 modulates synaptic plasticity by controlling the membrane insertion of NMDA receptors. J. Neurochem. 2009, 109, 1237–1249. [Google Scholar] [CrossRef]
- Chao, C.C.; Hu, S. Tumor necrosis factor-alpha potentiates glutamate neurotoxicity in human fetal brain cell cultures. Dev. Neurosci. 1994, 16, 172–179. [Google Scholar] [CrossRef]
- Taylor, D.L.; Jones, F.; Chen Seho Kubota, E.S.F.; Pocock, J.M. Stimulation of microglial metabotropic glutamate receptor mGlu2 triggers tumor necrosis factor α-induced neurotoxicity in concert with microglial-derived Fas ligand. J. Neurosci. 2005, 25, 2952–2964. [Google Scholar] [CrossRef]
- Feng, W.; Wang, Y.; Liu, Z.Q.; Zhang, X.; Han, R.; Miao, Y.Z.; Qin, Z.H. Microglia activation contributes to quinolinic acid-induced neuronal excitotoxicity through TNF-α. Apoptosis 2017, 22, 696–709. [Google Scholar] [CrossRef]
- Rao, S.D.; Yin, H.Z.; Weiss, J.H. Disruption of glial glutamate transport by reactive oxygen species produced in motor neurons. J. Neurosci. 2003, 23, 2627–2633. [Google Scholar] [CrossRef] [PubMed]
- Bernardino, L.; Xapelli, S.; Silva, A.P.; Jakobsen, B.; Poulsen, F.R.; Oliveira, C.R.; Vezzani, A.; Malva, J.O.; Zimmer, J. Modulator effects of interleukin-1β and tumor necrosis factor-α on AMPA-induced excitotoxicity in mouse organotypic hippocampal slice cultures. J. Neurosci. 2005, 25, 6734–6744. [Google Scholar] [CrossRef]
- Stellwagen, D.; Beattie, E.C.; Seo, J.Y.; Malenka, R.C. Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-α. J. Neurosci. 2005, 25, 3219–3228. [Google Scholar] [CrossRef] [PubMed]
- Pribiag, H.; Stellwagen, D. Tnf-α downregulates inhibitory neurotransmission through protein phosphatase 1-dependent trafficking of GABAA receptors. J. Neurosci. 2013, 33, 15879–15893. [Google Scholar] [CrossRef] [PubMed]
- Riazi, K.; Galic, M.A.; Kuzmiski, J.B.; Ho, W.; Sharkey, K.A.; Pittman, Q.J. Microglial activation and TNFα production mediate altered CNS excitability following peripheral inflammation. Proc. Natl. Acad. Sci. USA 2008, 105, 17151–17156. [Google Scholar] [CrossRef]
- Antonucci, F.; Turola, E.; Riganti, L.; Caleo, M.; Gabrielli, M.; Perrotta, C.; Novellino, L.; Clementi, E.; Giussani, P.; Viani, P.; et al. Microvesicles released from microglia stimulate synaptic activity via enhanced sphingolipid metabolism. EMBO J. 2012, 31, 1231–1240. [Google Scholar] [CrossRef]
- Marrone, M.C.; Morabito, A.; Giustizieri, M.; Chiurchiù, V.; Leuti, A.; Mattioli, M.; Marinelli, S.; Riganti, L.; Lombardi, M.; Murana, E.; et al. TRPV1 channels are critical brain inflammation detectors and neuropathic pain biomarkers in mice. Nat. Commun. 2017, 8, 15292. [Google Scholar] [CrossRef]
- Riganti, L.; Antonucci, F.; Gabrielli, M.; Prada, I.; Giussani, P.; Viani, P.; Valtorta, F.; Menna, E.; Matteoli, M.; Verderio, C. Sphingosine-1-phosphate (S1P) impacts presynaptic functions by regulating synapsin i localization in the presynaptic compartment. J. Neurosci. 2016, 36, 4624–4634. [Google Scholar] [CrossRef]
- Gabrielli, M.; Battista, N.; Riganti, L.; Prada, I.; Antonucci, F.; Cantone, L.; Matteoli, M.; Maccarrone, M.; Verderio, C. Active endocannabinoids are secreted on extracellular membrane vesicles. EMBO Rep. 2015, 16, 213–220. [Google Scholar] [CrossRef]
- Boka, G.; Anglade, P.; Wallach, D.; Javoy-Agid, F.; Agid, Y.; Hirsch, E.C. Immunocytochemical analysis of tumor necrosis factor and its receptors in Parkinson’s disease. Neurosci. Lett. 1994, 172, 151–154. [Google Scholar] [CrossRef]
- Sriram, K.; Miller, D.B.; O’Callaghan, J.P. Minocycline attenuates microglial activation but fails to mitigate striatal dopaminergic neurotoxicity: Role of tumor necrosis factor-α. J. Neurochem. 2006, 96, 706–718. [Google Scholar] [CrossRef] [PubMed]
- Rousselet, E.; Callebert, J.; Parain, K.; Joubert, C.; Hunot, S.; Hartmann, A.; Jacque, C.; Perez-Diaz, F.; Cohen-Salmon, C.; Launay, J.M.; et al. Role of TNF-α receptors in mice intoxicated with the parkinsonian toxin MPTP. Exp. Neurol. 2002, 177, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Morgan, J.A.; Singhal, G.; Corrigan, F.; Jaehne, E.J.; Jawahar, M.C.; Baune, B.T. Exercise related anxiety-like behaviours are mediated by TNF receptor signaling, but not depression-like behaviours. Brain Res. 2018, 1695, 10–17. [Google Scholar] [CrossRef]
- Wang, W.Y.; Tan, M.S.; Yu, J.T.; Tan, L. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann. Transl. Med. 2015, 3, 136. [Google Scholar] [PubMed]
- Shin, J.W.; Cheong, Y.J.; Koo, Y.M.; Kim, S.; Noh, C.K.; Son, Y.H.; Kang, C.; Sohn, N.W. α-Asarone ameliorates memory deficit in lipopolysaccharide-treated mice via suppression of pro-inflammatory cytokines and microglial activation. Biomol. Ther. 2014, 22, 17–26. [Google Scholar] [CrossRef]
- Tweedie, D.; Ferguson, R.A.; Fishman, K.; Frankola, K.A.; Van Praag, H.; Holloway, H.W.; Luo, W.; Li, Y.; Caracciolo, L.; Russo, I.; et al. Tumor necrosis factor-α synthesis inhibitor 3,6′-dithiothalidomide attenuates markers of inflammation, Alzheimer pathology and behavioral deficits in animal models of neuroinflammation and Alzheimer’s disease. J. Neuroinflamm. 2012, 9, 106. [Google Scholar] [CrossRef]
- He, P.; Cheng, X.; Staufenbiel, M.; Li, R.; Shen, Y. Long-Term Treatment of Thalidomide Ameliorates Amyloid-Like Pathology through Inhibition of β-Secretase in a Mouse Model of Alzheimer’s Disease. PLoS ONE 2013, 8, e55091. [Google Scholar] [CrossRef]
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L.; et al. Inflammation and Alzheimer’s disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar] [CrossRef]
- Montgomery, S.L.; Narrow, W.C.; Mastrangelo, M.A.; Olschowka, J.A.; O’Banion, M.K.; Bowers, W.J. Chronic neuron- and age-selective down-regulation of TNF receptor expression in triple-transgenic alzheimer disease mice leads to significant modulation of amyloid- and Tau-related pathologies. Am. J. Pathol. 2013, 182, 2285–2297. [Google Scholar] [CrossRef]
- Tarkowski, E.; Blennow, K.; Wallin, A.; Tarkowski, A. Intracerebral production of tumor necrosis factor-α, a local neuroprotective agent, in Alzheimer disease and vascular dementia. J. Clin. Immunol. 1999, 19, 223–230. [Google Scholar] [CrossRef]
- Ohgidani, M.; Kato, T.A.; Sagata, N.; Hayakawa, K.; Shimokawa, N.; Sato-Kasai, M.; Kanba, S. TNF-α from hippocampal microglia induces working memory deficits by acute stress in mice. Brain Behav. Immun. 2016, 55, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Botchkina, G.I.; Meistrell, M.E.; Botchkina, I.L.; Tracey, K.J. Expression of TNF and TNF receptors (p55 and p75) in the rat brain after focal cerebral ischemia. Mol. Med. 1997, 3, 765–781. [Google Scholar] [CrossRef] [PubMed]
- Botchkina, G.I.; Geimonen, E.; Bilof, M.L.; Villarreal, O.; Tracey, K.J. Loss of NF-κB activity during cerebral ischemia and TNF cytotoxicity. Mol. Med. 1999, 5, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Lambertsen, K.L.; Clausen, B.H.; Babcock, A.A.; Gregersen, R.; Fenger, C.; Nielsen, H.H.; Haugaard, L.S.; Wirenfeldt, M.; Nielsen, M.; Dagnaes-Hansen, F.; et al. Microglia protect neurons against ischemia by synthesis of tumor necrosis factor. J. Neurosci. 2009, 29, 1319–1330. [Google Scholar] [CrossRef] [PubMed]
- Harry, G.J.; Lefebvre D’Hellencourt, C.; McPherson, C.A.; Funk, J.A.; Aoyama, M.; Wine, R.N. Tumor necrosis factor p55 and p75 receptors are involved in chemical-induced apoptosis of dentate granule neurons. J. Neurochem. 2008, 106, 281–298. [Google Scholar] [CrossRef]
- Sairanen, T.R.; Lindsberg, P.J.; Brenner, M.; Carpén, O.; Sirén, A.L. Differential cellular expression of tumor necrosis factor-α and Type I tumor necrosis factor receptor after transient global forebrain ischemia. J. Neurol. Sci. 2001, 186, 87–99. [Google Scholar] [CrossRef]
- Nawashiro, H.; Tasaki, K.; Ruetzler, C.A.; Hallenbeck, J.M. TNF-α pretreatment induces protective effects against focal cerebral ischemia in mice. J. Cereb. Blood Flow Metab. 1997, 17, 483–490. [Google Scholar] [CrossRef]
- Pradillo, J.M.; Romera, C.; Hurtado, O.; Cárdenas, A.; Moro, M.A.; Leza, J.C.; Dávalos, A.; Castillo, J.; Lorenzo, P.; Lizasoain, I. TNFR1 upregulation mediates tolerance after brain ischemic preconditioning. J. Cereb. Blood Flow Metab. 2005, 25, 193–203. [Google Scholar] [CrossRef]
- Morgan, J.A.; Singhal, G.; Corrigan, F.; Jaehne, E.J.; Jawahar, M.C.; Baune, B.T. TNF signalling via the TNF receptors mediates the effects of exercise on cognition-like behaviours. Behav. Brain Res. 2018, 353, 74–82. [Google Scholar] [CrossRef]
- Nave, K.A. Myelination and support of axonal integrity by glia. Nature 2010, 468, 244–252. [Google Scholar] [CrossRef]
- Kremer, D.; Göttle, P.; Hartung, H.-P.; Küry, P. Pushing Forward: Remyelination as the New Frontier in CNS Diseases. Trends Neurosci. 2016, 39, 246–263. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, M.; Lecca, D.; Abbracchio, M.P. CNS remyelination as a novel reparative approach to neurodegenerative diseases: The roles of purinergic signaling and the P2Y-like receptor GPR17. Neuropharmacology 2016, 104, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Franklin, R.J.M.; Ffrench-Constant, C. Regenerating CNS myelin—From mechanisms to experimental medicines. Nat. Rev. Neurosci. 2017, 18, 753–769. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, A.F.; Miron, V.E. The pro-remyelination properties of microglia in the central nervous system. Nat. Rev. Neurol. 2019, 29–34. [Google Scholar] [CrossRef]
- Lampron, A.; Larochelle, A.; Laflamme, N.; Préfontaine, P.; Plante, M.M.; Sánchez, M.G.; Wee Yong, V.; Stys, P.K.; Tremblay, M.È.; Rivest, S. Inefficient clearance of myelin debris by microglia impairs remyelinating processes. J. Exp. Med. 2015, 212, 481–495. [Google Scholar] [CrossRef]
- Cantuti-Castelvetri, L.; Fitzner, D.; Bosch-Queralt, M.; Weil, M.T.; Su, M.; Sen, P.; Ruhwedel, T.; Mitkovski, M.; Trendelenburg, G.; Lütjohann, D.; et al. Defective cholesterol clearance limits remyelination in the aged central nervous system. Science 2018, 359, 684–688. [Google Scholar] [CrossRef]
- Gruchot, J.; Weyers, V.; Göttle, P.; Förster, M.; Hartung, H.P.; Küry, P.; Kremer, D. The Molecular Basis for Remyelination Failure in Multiple Sclerosis. Cells 2019, 8, 825. [Google Scholar] [CrossRef]
- Karamita, M.; Barnum, C.; Möbius, W.; Tansey, M.G.; Szymkowski, D.E.; Lassmann, H.; Probert, L. Therapeutic inhibition of soluble brain TNF promotes remyelination by increasing myelin phagocytosis by microglia. JCI Insight 2017, 2, e87455. [Google Scholar] [CrossRef]
- Cunha, M.I.; Su, M.; Cantuti-Castelvetri, L.; Müller, S.A.; Schifferer, M.; Djannatian, M.; Alexopoulos, I.; van der Meer, F.; Winkler, A.; van Ham, T.J.; et al. Pro-inflammatory activation following demyelination is required for myelin clearance and oligodendrogenesis. J. Exp. Med. 2020, 217, e20191390. [Google Scholar] [CrossRef]
- Jurewicz, A.; Matysiak, M.; Tybor, K.; Selmaj, K. TNF-induced death of adult human oligodendrocytes is mediated by c-jun NH2-terminal kinase-3. Brain 2003, 126, 1358–1370. [Google Scholar] [CrossRef]
- Ofengeim, D.; Ito, Y.; Najafov, A.; Zhang, Y.; Shan, B.; DeWitt, J.P.; Ye, J.; Zhang, X.; Chang, A.; Vakifahmetoglu-Norberg, H.; et al. Activation of necroptosis in multiple sclerosis. Cell Rep. 2015, 10, 1836–1849. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Marino, M.W.; Wong, G.; Grail, D.; Dunn, A.; Bettadapura, J.; Slavin, A.J.; Old, L.; Bernard, C.C.A. TNF is a potent anti-inflammatory cytokine in autoimmune-mediated demyelination. Nat. Med. 1998, 4, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Hövelmeyer, N.; Hao, Z.; Kranidioti, K.; Kassiotis, G.; Buch, T.; Frommer, F.; von Hoch, L.; Kramer, D.; Minichiello, L.; Kollias, G.; et al. Apoptosis of Oligodendrocytes via Fas and TNF-R1 Is a Key Event in the Induction of Experimental Autoimmune Encephalomyelitis. J. Immunol. 2005, 175, 5875–5884. [Google Scholar]
- Kim, S.; Steelman, A.J.; Koito, H.; Li, J. Astrocytes promote TNF-mediated toxicity to oligodendrocyte precursors. J. Neurochem. 2011, 116, 53–66. [Google Scholar] [CrossRef] [PubMed]
- Magliozzi, R.; Howell, O.W.; Durrenberger, P.; Aricò, E.; James, R.; Cruciani, C.; Reeves, C.; Roncaroli, F.; Nicholas, R.; Reynolds, R. Meningeal inflammation changes the balance of TNF signalling in cortical grey matter in multiple sclerosis. J. Neuroinflamm. 2019, 16, 259. [Google Scholar] [CrossRef] [PubMed]
- Madsen, P.M.; Desu, H.L.; Vaccari, J.P.d.R.; Florimon, Y.; Ellman, D.G.; Keane, R.W.; Clausen, B.H.; Lambertsen, K.L.; Brambilla, R. Oligodendrocytes modulate the immune-inflammatory response in EAE via TNFR2 signaling. Brain Behav. Immun. 2020, 84, 132–146. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Yuan, Y.; Chen, J.; Zhu, Y.; Qiu, Y.; Zhu, F.; Huang, A.; He, C. Reactive astrocytes inhibit the survival and differentiation of oligodendrocyte precursor cells by secreted TNF-α. J. Neurotrauma 2011, 28, 1089–1100. [Google Scholar] [CrossRef]
- Arnason, B.G.W. TNF neutralization in MS: Results of a randomized, placebo-controlled multicenter study. Neurology 1999, 53, 457–465. [Google Scholar]
- Kemanetzoglou, E.; Andreadou, E. CNS Demyelination with TNF-α Blockers. Curr. Neurol. Neurosci. Rep. 1910, 17, 36. [Google Scholar] [CrossRef]
- Brambilla, R.; Ashbaugh, J.J.; Magliozzi, R.; Dellarole, A.; Karmally, S.; Szymkowski, D.E.; Bethea, J.R. Inhibition of soluble tumour necrosis factor is therapeutic in experimental autoimmune encephalomyelitis and promotes axon preservation and remyelination. Brain 2011, 134, 2736–2754. [Google Scholar] [CrossRef]
- Madsen, P.M.; Clausen, B.H.; Degn, M.; Thyssen, S.; Kristensen, L.K.; Svensson, M.; Ditzel, N.; Finsen, B.; Deierborg, T.; Brambilla, R.; et al. Genetic ablation of soluble tumor necrosis factor with preservation of membrane tumor necrosis factor is associated with neuroprotection after focal cerebral ischemia. J. Cereb. Blood Flow Metab. 2016, 36, 1553–1569. [Google Scholar] [CrossRef] [PubMed]
- Yli-Karjanmaa, M.; Clausen, B.H.; Degn, M.; Novrup, H.G.; Ellman, D.G.; Toft-Jensen, P.; Szymkowski, D.E.; Stensballe, A.; Meyer, M.; Brambilla, R.; et al. Topical administration of a soluble TNF inhibitor reduces infarct volume after focal cerebral ischemia in mice. Front. Neurosci. 2019, 13, 781. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Fischer, R.; Naudé, P.J.W.; Maier, O.; Nyakas, C.; Duffey, M.; Van Der Zee, E.A.; Dekens, D.; Douwenga, W.; Herrmann, A.; et al. Essential protective role of tumor necrosis factor receptor 2 in neurodegeneration. Proc. Natl. Acad. Sci. USA 2016, 113, 12304–12309. [Google Scholar] [CrossRef] [PubMed]
- Fischer, R.; Padutsch, T.; Bracchi-Ricard, V.; Murphy, K.L.; Martinez, G.F.; Delguercio, N.; Elmer, N.; Sendetski, M.; Diem, R.; Eisel, U.L.M.; et al. Exogenous activation of tumor necrosis factor receptor 2 promotes recovery from sensory and motor disease in a model of multiple sclerosis. Brain Behav. Immun. 2019, 81, 247–259. [Google Scholar] [CrossRef] [PubMed]
- Catalano, M.; O’Driscoll, L. Inhibiting extracellular vesicles formation and release: A review of EV inhibitors. J. Extracell. Vesicles 2020, 9, 1703244. [Google Scholar] [CrossRef]
- Fumagalli, M.; Lombardi, M.; Gressens, P.; Verderio, C. How to reprogram microglia toward beneficial functions. Glia 2018, 66, 2531–2549. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raffaele, S.; Lombardi, M.; Verderio, C.; Fumagalli, M. TNF Production and Release from Microglia via Extracellular Vesicles: Impact on Brain Functions. Cells 2020, 9, 2145. https://doi.org/10.3390/cells9102145
Raffaele S, Lombardi M, Verderio C, Fumagalli M. TNF Production and Release from Microglia via Extracellular Vesicles: Impact on Brain Functions. Cells. 2020; 9(10):2145. https://doi.org/10.3390/cells9102145
Chicago/Turabian StyleRaffaele, Stefano, Marta Lombardi, Claudia Verderio, and Marta Fumagalli. 2020. "TNF Production and Release from Microglia via Extracellular Vesicles: Impact on Brain Functions" Cells 9, no. 10: 2145. https://doi.org/10.3390/cells9102145
APA StyleRaffaele, S., Lombardi, M., Verderio, C., & Fumagalli, M. (2020). TNF Production and Release from Microglia via Extracellular Vesicles: Impact on Brain Functions. Cells, 9(10), 2145. https://doi.org/10.3390/cells9102145