Distinct Role of CD11b+Ly6G−Ly6C− Myeloid-Derived Cells on the Progression of the Primary Tumor and Therapy-Associated Recurrent Brain Tumor


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Cell Line Cultures
2.3. Plasmids and Cell Transfection
2.4. Flow Cytometry Analysis
2.5. Orthotopic Intracranial Tumor Model
2.6. Immunohistochemistry
2.7. Drug Preparation
2.8. Statistics
3. Results
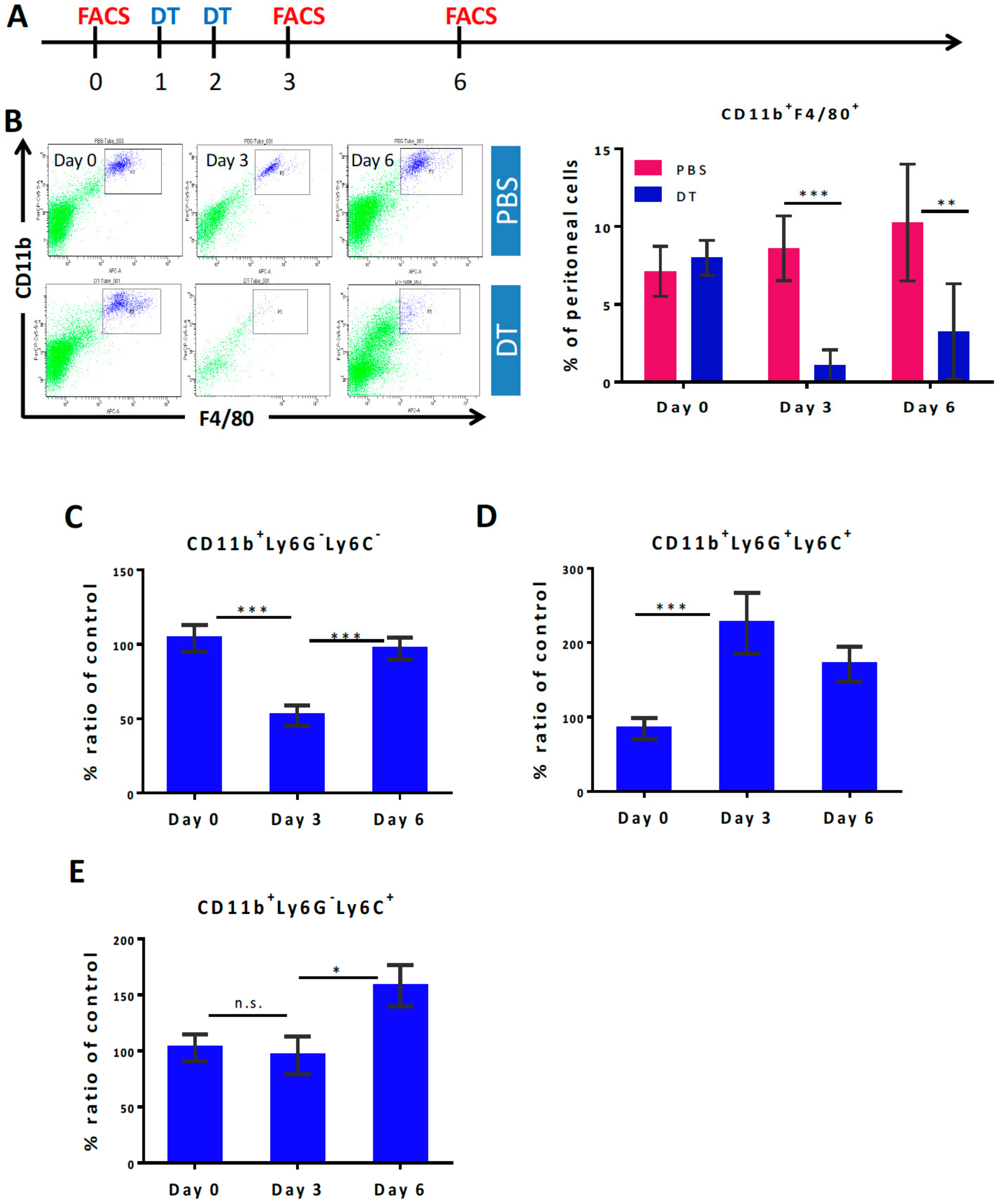
3.1. Selective Myeloid Cells Depletion in CD11b-DTR Transgenic Mice
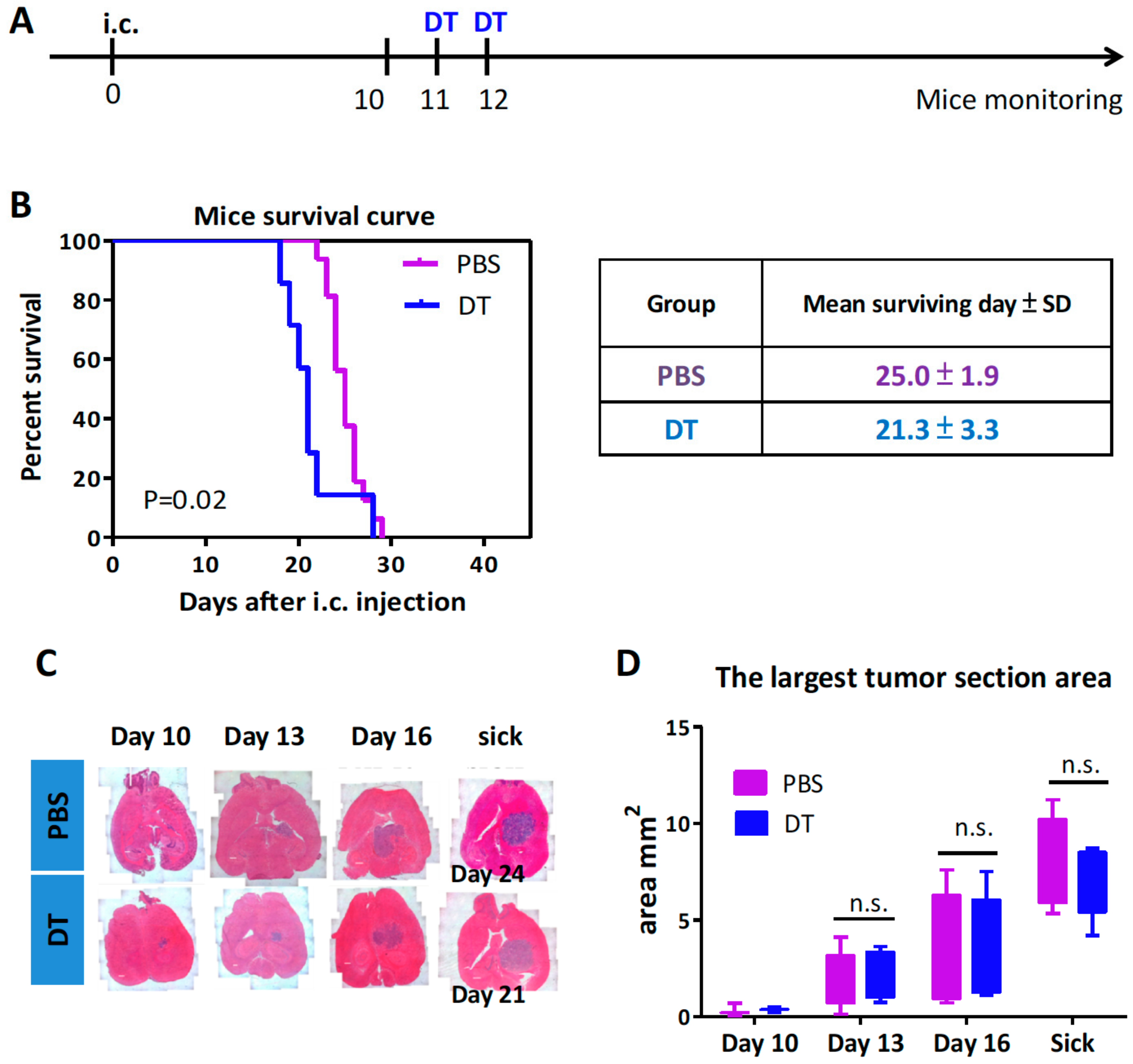
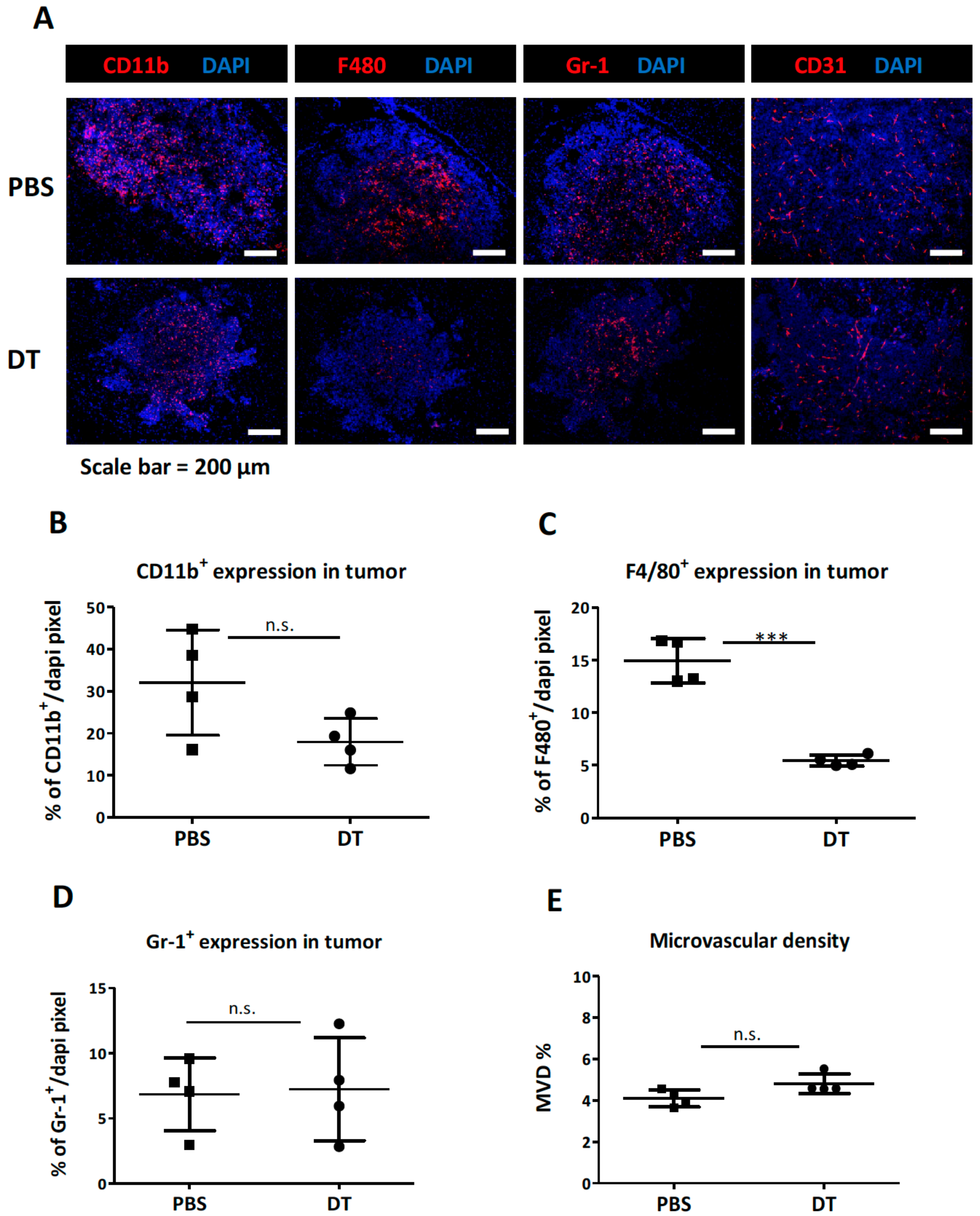
3.2. Depletion of the CD11b+Ly6G−Ly6C− MDCs Alone Did Not Impede Tumor Growth
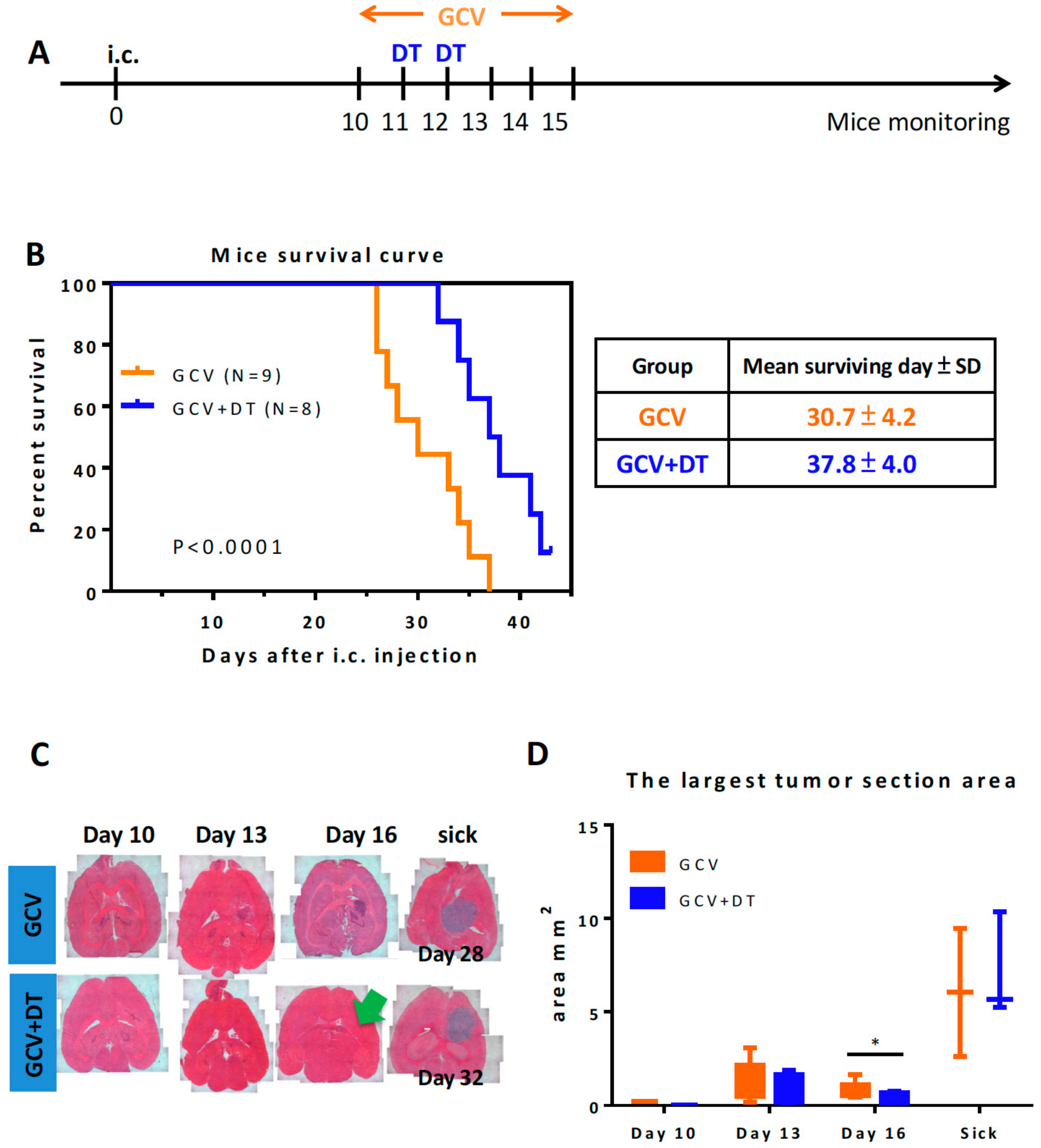
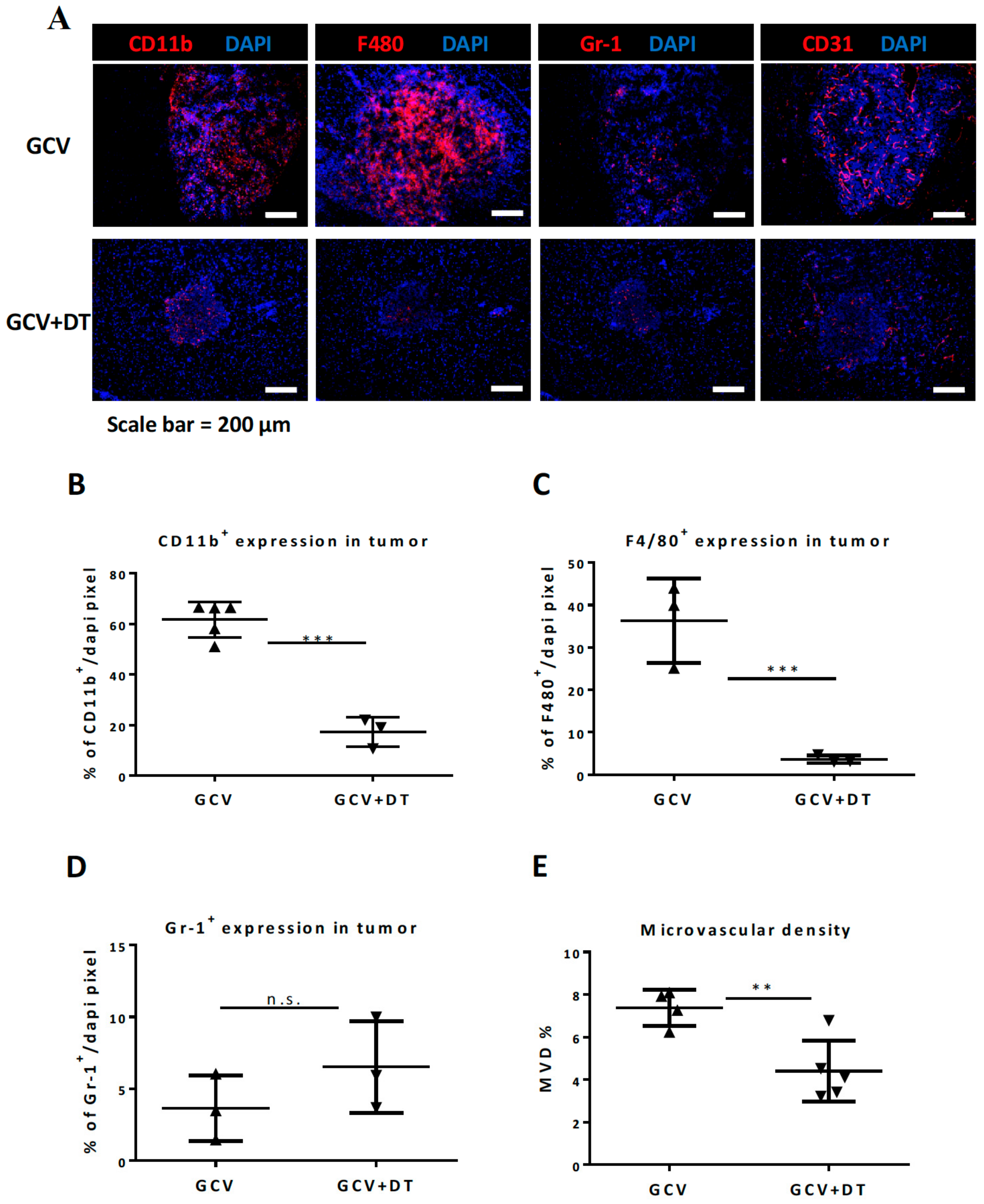
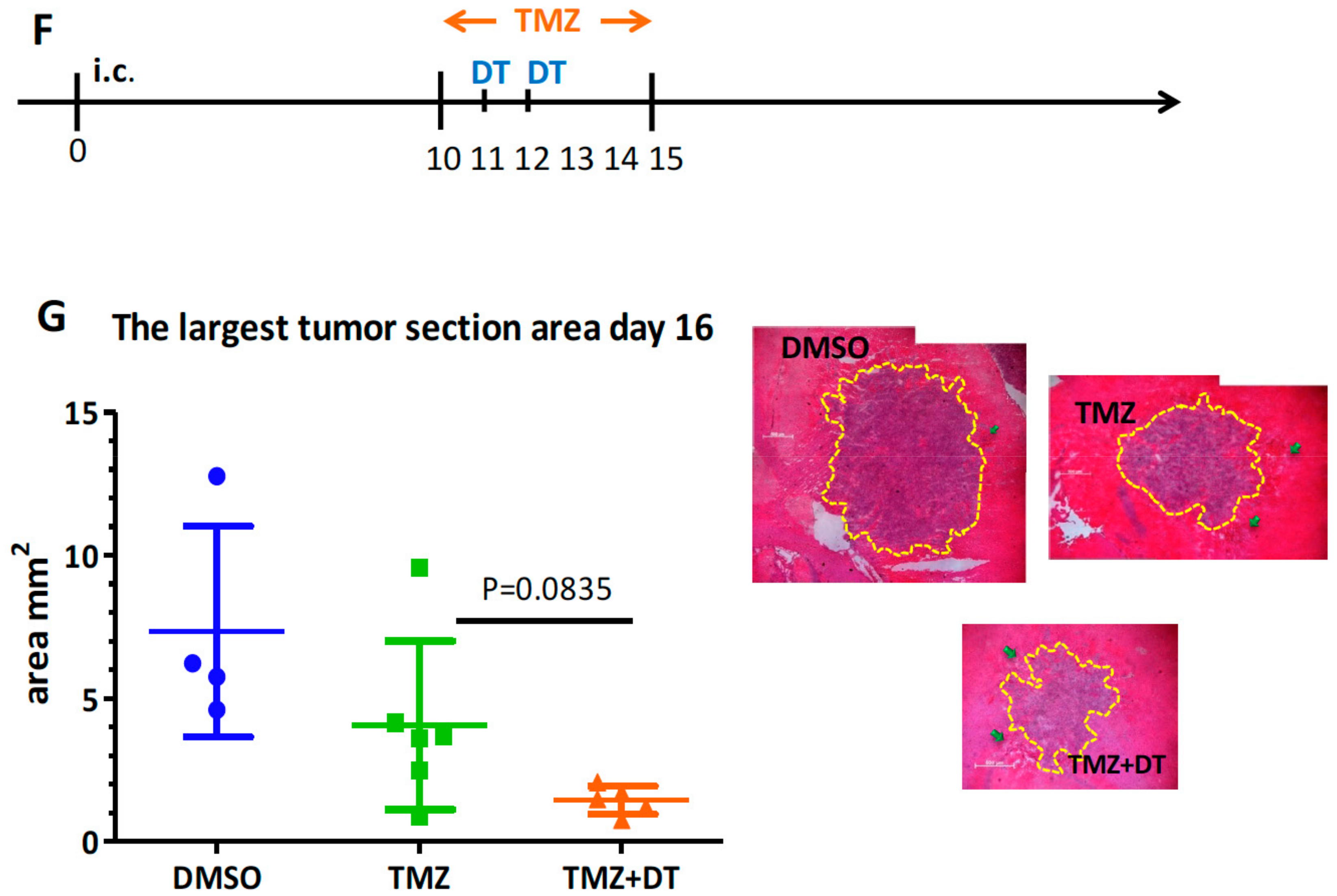
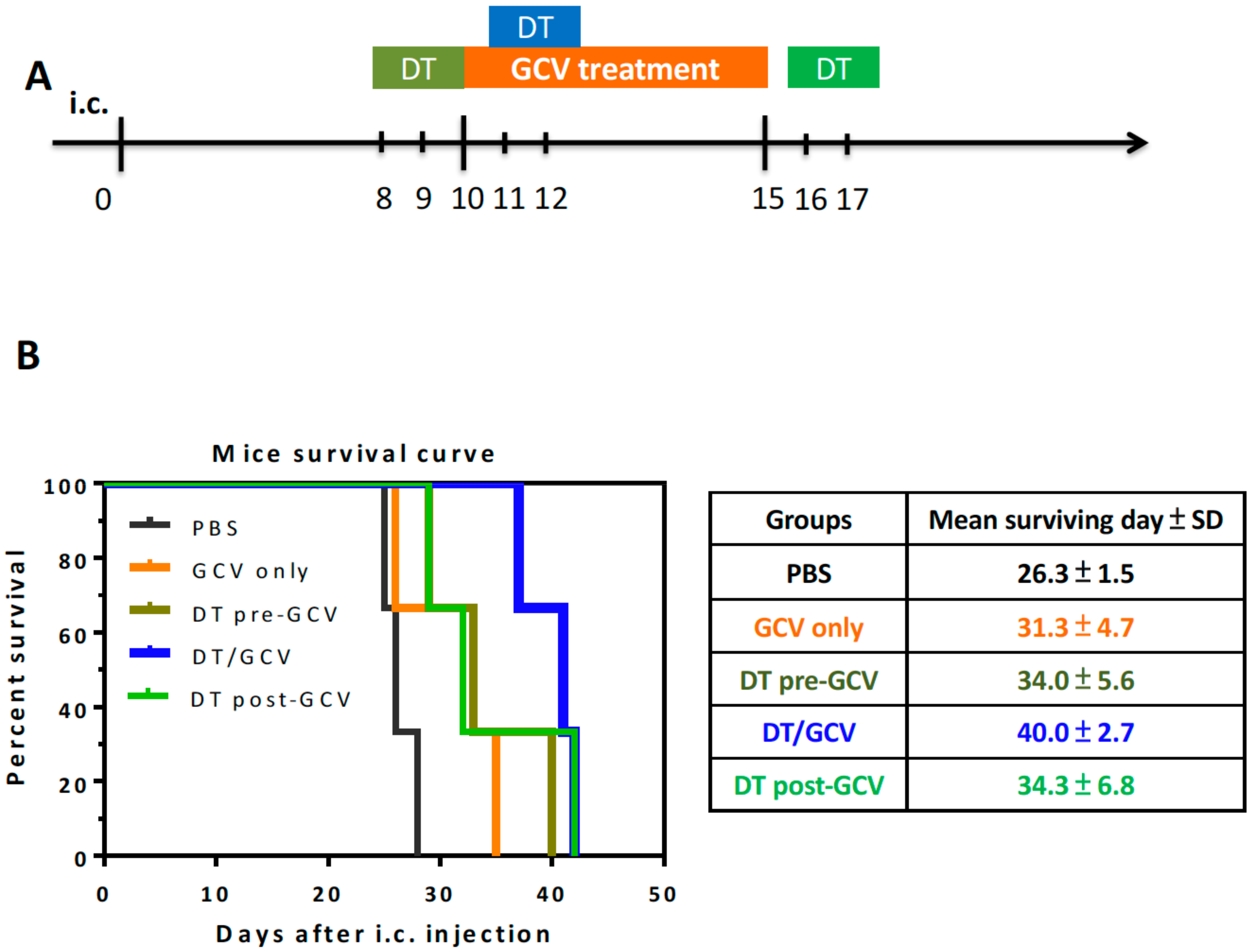
3.3. Selective MDCs Depletion Benefits tk/GCV Therapy
3.4. The Time-Dependent Effect of DT Administration
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Bauchet, L.; Davis, F.G.; Deltour, I.; Fisher, J.L.; Langer, C.E.; Pekmezci, M.; Schwartzbaum, J.A.; Turner, M.C.; Walsh, K.M.; et al. The epidemiology of glioma in adults: A “state of the science” review. Neuro Oncol. 2014, 16, 896–913. [Google Scholar] [CrossRef]
- Wong, E.T.; Hess, K.R.; Gleason, M.J.; Jaeckle, K.A.; Kyritsis, A.P.; Prados, M.D.; Levin, V.A.; Yung, W.K. Outcomes and prognostic factors in recurrent glioma patients enrolled onto phase II clinical trials. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 1999, 17, 2572–2578. [Google Scholar] [CrossRef] [PubMed]
- Ballman, K.V.; Buckner, J.C.; Brown, P.D.; Giannini, C.; Flynn, P.J.; LaPlant, B.R.; Jaeckle, K.A. The relationship between six-month progression-free survival and 12-month overall survival end points for phase II trials in patients with glioblastoma multiforme. Neuro Oncol. 2007, 9, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Chae, M.; Peterson, T.E.; Balgeman, A.; Chen, S.; Zhang, L.; Renner, D.N.; Johnson, A.J.; Parney, I.F. Increasing glioma-associated monocytes leads to increased intratumoral and systemic myeloid-derived suppressor cells in a murine model. Neuro Oncol. 2015, 17, 978–991. [Google Scholar] [CrossRef] [PubMed]
- Brandenburg, S.; Muller, A.; Turkowski, K.; Radev, Y.T.; Rot, S.; Schmidt, C.; Bungert, A.D.; Acker, G.; Schorr, A.; Hippe, A.; et al. Resident microglia rather than peripheral macrophages promote vascularization in brain tumors and are source of alternative pro-angiogenic factors. Acta Neuropathol. 2016, 131, 365–378. [Google Scholar] [CrossRef]
- Bronte, V.; Chappell, D.B.; Apolloni, E.; Cabrelle, A.; Wang, M.; Hwu, P.; Restifo, N.P. Unopposed production of granulocyte-macrophage colony-stimulating factor by tumors inhibits CD8+ T cell responses by dysregulating antigen-presenting cell maturation. J. Immunol. 1999, 162, 5728–5737. [Google Scholar]
- Gabrilovich, D.I.; Bronte, V.; Chen, S.H.; Colombo, M.P.; Ochoa, A.; Ostrand-Rosenberg, S.; Schreiber, H. The terminology issue for myeloid-derived suppressor cells. Cancer Res. 2007, 67, 425. [Google Scholar] [CrossRef]
- Gabrilovich, D.I. Myeloid-Derived Suppressor Cells. Cancer Immunol. Res. 2017, 5, 3–8. [Google Scholar] [CrossRef]
- Kallberg, E.; Stenstrom, M.; Liberg, D.; Ivars, F.; Leanderson, T. CD11b+Ly6C++Ly6G- cells show distinct function in mice with chronic inflammation or tumor burden. BMC Immunol. 2012, 13, 69. [Google Scholar] [CrossRef] [PubMed]
- Ouzounova, M.; Lee, E.; Piranlioglu, R.; El Andaloussi, A.; Kolhe, R.; Demirci, M.F.; Marasco, D.; Asm, I.; Chadli, A.; Hassan, K.A.; et al. Monocytic and granulocytic myeloid derived suppressor cells differentially regulate spatiotemporal tumour plasticity during metastatic cascade. Nat. Commun. 2017, 8, 14979. [Google Scholar] [CrossRef] [PubMed]
- Gieryng, A.; Kaminska, B. Myeloid-derived suppressor cells in gliomas. Contemp. Oncol. (Pozn.) 2016, 20, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Safarzadeh, E.; Orangi, M.; Mohammadi, H.; Babaie, F.; Baradaran, B. Myeloid-derived suppressor cells: Important contributors to tumor progression and metastasis. J. Cell Physiol. 2018, 233, 3024–3036. [Google Scholar] [CrossRef]
- Hey, Y.Y.; Tan, J.K.; O’Neill, H.C. Redefining Myeloid Cell Subsets in Murine Spleen. Front. Immunol. 2015, 6, 652. [Google Scholar] [CrossRef]
- Bingle, L.; Lewis, C.E.; Corke, K.P.; Reed, M.W.; Brown, N.J. Macrophages promote angiogenesis in human breast tumour spheroids in vivo. Br. J. Cancer 2006, 94, 101–107. [Google Scholar] [CrossRef]
- Laoui, D.; Movahedi, K.; Van Overmeire, E.; Van den Bossche, J.; Schouppe, E.; Mommer, C.; Nikolaou, A.; Morias, Y.; De Baetselier, P.; Van Ginderachter, J.A. Tumor-associated macrophages in breast cancer: Distinct subsets, distinct functions. Int. J. Dev. Biol. 2011, 55, 861–867. [Google Scholar] [CrossRef]
- Liu, C.Y.; Xu, J.Y.; Shi, X.Y.; Huang, W.; Ruan, T.Y.; Xie, P.; Ding, J.L. M2-polarized tumor-associated macrophages promoted epithelial-mesenchymal transition in pancreatic cancer cells, partially through TLR4/IL-10 signaling pathway. Lab. Investig. A J. Tech. Methods Pathol. 2013, 93, 844–854. [Google Scholar] [CrossRef]
- Lanciotti, M.; Masieri, L.; Raspollini, M.R.; Minervini, A.; Mari, A.; Comito, G.; Giannoni, E.; Carini, M.; Chiarugi, P.; Serni, S. The role of M1 and M2 macrophages in prostate cancer in relation to extracapsular tumor extension and biochemical recurrence after radical prostatectomy. BioMed Res. Int. 2014, 2014, 486798. [Google Scholar] [CrossRef]
- Yuan, A.; Hsiao, Y.J.; Chen, H.Y.; Chen, H.W.; Ho, C.C.; Chen, Y.Y.; Liu, Y.C.; Hong, T.H.; Yu, S.L.; Chen, J.J.; et al. Opposite Effects of M1 and M2 Macrophage Subtypes on Lung Cancer Progression. Sci Rep. 2015, 5, 14273. [Google Scholar] [CrossRef]
- Morantz, R.A.; Wood, G.W.; Foster, M.; Clark, M.; Gollahon, K. Macrophages in experimental and human brain tumors. Part 2: Studies of the macrophage content of human brain tumors. J. Neurosurg. 1979, 50, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.L.; Cruz Sanchez, F.; Hughes, J.T.; Esiri, M.M.; Coakham, H.B. Immunocytochemical study of the cellular immune response in meningiomas. J. Clin. Pathol. 1988, 41, 314–319. [Google Scholar] [CrossRef] [PubMed]
- Simmons, G.W.; Pong, W.W.; Emnett, R.J.; White, C.R.; Gianino, S.M.; Rodriguez, F.J.; Gutmann, D.H. Neurofibromatosis-1 heterozygosity increases microglia in a spatially and temporally restricted pattern relevant to mouse optic glioma formation and growth. J. Neuropathol. Exp. Neurol. 2011, 70, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Arcuri, C.; Fioretti, B.; Bianchi, R.; Mecca, C.; Tubaro, C.; Beccari, T.; Franciolini, F.; Giambanco, I.; Donato, R. Microglia-glioma cross-talk: A two way approach to new strategies against glioma. Front. Biosci. (Landmark Ed.) 2017, 22, 268–309. [Google Scholar] [CrossRef]
- Honjo, T.; Nishizuka, Y.; Hayaishi, O. Diphtheria toxin-dependent adenosine diphosphate ribosylation of aminoacyl transferase II and inhibition of protein synthesis. J. Biol. Chem. 1968, 243, 3553–3555. [Google Scholar]
- Van Ness, B.G.; Howard, J.B.; Bodley, J.W. ADP-ribosylation of elongation factor 2 by diphtheria toxin. NMR spectra and proposed structures of ribosyl-diphthamide and its hydrolysis products. J. Biol. Chem. 1980, 255, 10710–10716. [Google Scholar]
- Saito, M.; Iwawaki, T.; Taya, C.; Yonekawa, H.; Noda, M.; Inui, Y.; Mekada, E.; Kimata, Y.; Tsuru, A.; Kohno, K. Diphtheria toxin receptor-mediated conditional and targeted cell ablation in transgenic mice. Nat. Biotechnol. 2001, 19, 746–750. [Google Scholar] [CrossRef]
- Robinson, E.A.; Henriksen, O.; Maxwell, E.S. Elongation Factor 2: Amino acid sequence at the site of adenosine diphosphate ribosylation. J. Biol. Chem. 1974, 249, 5088–5093. [Google Scholar]
- Duffield, J.S.; Tipping, P.G.; Kipari, T.; Cailhier, J.F.; Clay, S.; Lang, R.; Bonventre, J.V.; Hughes, J. Conditional ablation of macrophages halts progression of crescentic glomerulonephritis. Am. J. Pathol. 2005, 167, 1207–1219. [Google Scholar] [CrossRef]
- Stoneman, V.; Braganza, D.; Figg, N.; Mercer, J.; Lang, R.; Goddard, M.; Bennett, M. Monocyte/macrophage suppression in CD11b diphtheria toxin receptor transgenic mice differentially affects atherogenesis and established plaques. Circ. Res. 2007, 100, 884–893. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, N.; Li, Q.; Zhang, W.; Ke, F.; Leng, Q.; Wang, H.; Chen, J.; Wang, H. Tumor-associated macrophages recruit CCR6+ regulatory T cells and promote the development of colorectal cancer via enhancing CCL20 production in mice. PLoS ONE 2011, 6, e19495. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Lim, S.Y.; Gordon-Weeks, A.N.; Tapmeier, T.T.; Im, J.H.; Cao, Y.; Beech, J.; Allen, D.; Smart, S.; Muschel, R.J. Recruitment of a myeloid cell subset (CD11b/Gr1 mid) via CCL2/CCR2 promotes the development of colorectal cancer liver metastasis. Hepatology 2013, 57, 829–839. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Velez-Delgado, A.; Mathew, E.; Li, D.; Mendez, F.M.; Flannagan, K.; Rhim, A.D.; Simeone, D.M.; Beatty, G.L.; Pasca di Magliano, M. Myeloid cells are required for PD-1/PD-L1 checkpoint activation and the establishment of an immunosuppressive environment in pancreatic cancer. Gut 2017, 66, 124–136. [Google Scholar] [CrossRef] [PubMed]
- Moolten, F.L. Tumor chemosensitivity conferred by inserted herpes thymidine kinase genes: Paradigm for a prospective cancer control strategy. Cancer Res. 1986, 46, 5276–5281. [Google Scholar]
- Fillat, C.; Carrio, M.; Cascante, A.; Sangro, B. Suicide gene therapy mediated by the Herpes Simplex virus thymidine kinase gene/Ganciclovir system: Fifteen years of application. Curr. Gene 2003, 3, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Duarte, S.; Carle, G.; Faneca, H.; de Lima, M.C.; Pierrefite-Carle, V. Suicide gene therapy in cancer: Where do we stand now? Cancer Lett. 2012, 324, 160–170. [Google Scholar] [CrossRef]
- Yu, C.F.; Hong, J.H.; Chiang, C.S. The roles of macrophages and nitric oxide in interleukin-3-enhanced HSV-Sr39tk-mediated prodrug therapy. PLoS ONE 2013, 8, e56508. [Google Scholar] [CrossRef]
- Wang, S.C.; Hong, J.H.; Hsueh, C.; Chiang, C.S. Tumor-secreted SDF-1 promotes glioma invasiveness and TAM tropism toward hypoxia in a murine astrocytoma model. Lab. Investig. J. Tech. Methods Pathol. 2012, 92, 151–162. [Google Scholar] [CrossRef]
- Chiang, C.S.; Fu, S.Y.; Wang, S.C.; Yu, C.F.; Chen, F.H.; Lin, C.M.; Hong, J.H. Irradiation promotes an M2 macrophage phenotype in tumor hypoxia. Front. Oncol. 2012, 2, 89. [Google Scholar] [CrossRef]
- Kioi, M.; Vogel, H.; Schultz, G.; Hoffman, R.M.; Harsh, G.R.; Brown, J.M. Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. J. Clin. Investig. 2010, 120, 694–705. [Google Scholar] [CrossRef]
- Kozin, S.V.; Kamoun, W.S.; Huang, Y.; Dawson, M.R.; Jain, R.K.; Duda, D.G. Recruitment of myeloid but not endothelial precursor cells facilitates tumor regrowth after local irradiation. Cancer Res. 2010, 70, 5679–5685. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.C.; Yu, C.F.; Hong, J.H.; Tsai, C.S.; Chiang, C.S. Radiation therapy-induced tumor invasiveness is associated with SDF-1-regulated macrophage mobilization and vasculogenesis. PLoS ONE 2013, 8, e69182. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.E.; Harney, A.S.; Pollard, J.W. The Multifaceted Role of Perivascular Macrophages in Tumors. Cancer Cell 2016, 30, 365. [Google Scholar] [CrossRef] [PubMed]
- Middleton, J.D.; Stover, D.G.; Hai, T. Chemotherapy-Exacerbated Breast Cancer Metastasis: A Paradox Explainable by Dysregulated Adaptive-Response. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- DeNardo, D.G.; Brennan, D.J.; Rexhepaj, E.; Ruffell, B.; Shiao, S.L.; Madden, S.F.; Gallagher, W.M.; Wadhwani, N.; Keil, S.D.; Junaid, S.A.; et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 2011, 1, 54–67. [Google Scholar] [CrossRef]
- Shree, T.; Olson, O.C.; Elie, B.T.; Kester, J.C.; Garfall, A.L.; Simpson, K.; Bell-McGuinn, K.M.; Zabor, E.C.; Brogi, E.; Joyce, J.A. Macrophages and cathepsin proteases blunt chemotherapeutic response in breast cancer. Genes Dev. 2011, 25, 2465–2479. [Google Scholar] [CrossRef]
- Dijkgraaf, E.M.; Heusinkveld, M.; Tummers, B.; Vogelpoel, L.T.; Goedemans, R.; Jha, V.; Nortier, J.W.; Welters, M.J.; Kroep, J.R.; van der Burg, S.H. Chemotherapy alters monocyte differentiation to favor generation of cancer-supporting M2 macrophages in the tumor microenvironment. Cancer Res. 2013, 73, 2480–2492. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P. The interaction of anticancer therapies with tumor-associated macrophages. J. Exp. Med. 2015, 212, 435–445. [Google Scholar] [CrossRef]
- Leek, R.D.; Lewis, C.E.; Whitehouse, R.; Greenall, M.; Clarke, J.; Harris, A.L. Association of macrophage infiltration with angiogenesis and prognosis in invasive breast carcinoma. Cancer Res. 1996, 56, 4625–4629. [Google Scholar]
- Lamagna, C.; Aurrand-Lions, M.; Imhof, B.A. Dual role of macrophages in tumor growth and angiogenesis. J. Leukoc. Biol. 2006, 80, 705–713. [Google Scholar] [CrossRef]
- Coffelt, S.B.; Hughes, R.; Lewis, C.E. Tumor-associated macrophages: Effectors of angiogenesis and tumor progression. Biochim. Et Biophys. Acta 2009, 1796, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Shiao, S.L.; Ruffell, B.; DeNardo, D.G.; Faddegon, B.A.; Park, C.C.; Coussens, L.M. TH2-Polarized CD4(+) T Cells and Macrophages Limit Efficacy of Radiotherapy. Cancer Immunol Res. 2015, 3, 518–525. [Google Scholar] [CrossRef] [PubMed]
- Kalbasi, A.; Komar, C.; Tooker, G.M.; Liu, M.; Lee, J.W.; Gladney, W.L.; Ben-Josef, E.; Beatty, G.L. Tumor-Derived CCL2 Mediates Resistance to Radiotherapy in Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res. 2017, 23, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wang, Y.; Nelson, D.; Tian, S.; Mulvey, E.; Patel, B.; Conti, I.; Jaen, J.; Rollins, B.J. CCL2/CCR2 Regulates the Tumor Microenvironment in HER-2/neu-Driven Mammary Carcinomas in Mice. PLoS ONE 2016, 11, e0165595. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.B.; Zhang, X.; Paul, D.; Kays, L.M.; Ye, M.; Vaillancourt, P.; Dowless, M.; Stancato, L.F.; Stewart, J.; Uhlik, M.T.; et al. Inhibition of CXCR4 by LY2624587, a Fully Humanized Anti-CXCR4 Antibody Induces Apoptosis of Hematologic Malignancies. PLoS ONE 2016, 11, e0150585. [Google Scholar] [CrossRef] [PubMed]
- Prinz, M.; Priller, J. Microglia and brain macrophages in the molecular age: From origin to neuropsychiatric disease. Nat. Rev. Neurosci. 2014, 15, 300–312. [Google Scholar] [CrossRef] [PubMed]
- Frieler, R.A.; Nadimpalli, S.; Boland, L.K.; Xie, A.; Kooistra, L.J.; Song, J.; Chung, Y.; Cho, K.W.; Lumeng, C.N.; Wang, M.M.; et al. Depletion of macrophages in CD11b diphtheria toxin receptor mice induces brain inflammation and enhances inflammatory signaling during traumatic brain injury. Brain Res. 2015, 1624, 103–112. [Google Scholar] [CrossRef]
- Ueno, M.; Fujita, Y.; Tanaka, T.; Nakamura, Y.; Kikuta, J.; Ishii, M.; Yamashita, T. Layer V cortical neurons require microglial support for survival during postnatal development. Nat. Neurosci. 2013, 16, 543–551. [Google Scholar] [CrossRef]
- Satoh, J.; Kino, Y.; Asahina, N.; Takitani, M.; Miyoshi, J.; Ishida, T.; Saito, Y. TMEM119 marks a subset of microglia in the human brain. Neuropathology 2016, 36, 39–49. [Google Scholar] [CrossRef]
- Haage, V.; Semtner, M.; Vidal, R.O.; Hernandez, D.P.; Pong, W.W.; Chen, Z.; Hambardzumyan, D.; Magrini, V.; Ly, A.; Walker, J.; et al. Comprehensive gene expression meta-analysis identifies signature genes that distinguish microglia from peripheral monocytes/macrophages in health and glioma. Acta Neuropathol. Commun. 2019, 7, 20. [Google Scholar] [CrossRef]
- Kolaczkowska, E.; Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 2013, 13, 159–175. [Google Scholar] [CrossRef] [PubMed]
- Nauseef, W.M.; Borregaard, N. Neutrophils at work. Nat. Immunol. 2014, 15, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Kruger, P.; Saffarzadeh, M.; Weber, A.N.; Rieber, N.; Radsak, M.; von Bernuth, H.; Benarafa, C.; Roos, D.; Skokowa, J.; Hartl, D. Neutrophils: Between host defence, immune modulation, and tissue injury. PLoS Pathog. 2015, 11, e1004651. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Melton, D.W.; Porter, L.; Sarwar, Z.U.; McManus, L.M.; Shireman, P.K. Altered macrophage phenotype transition impairs skeletal muscle regeneration. Am. J. Pathol. 2014, 184, 1167–1184. [Google Scholar] [CrossRef] [PubMed]
- Ries, C.H.; Cannarile, M.A.; Hoves, S.; Benz, J.; Wartha, K.; Runza, V.; Rey-Giraud, F.; Pradel, L.P.; Feuerhake, F.; Klaman, I.; et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell 2014, 25, 846–859. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.Y.; Yuzhalin, A.E.; Gordon-Weeks, A.N.; Muschel, R.J. Targeting the CCL2-CCR2 signaling axis in cancer metastasis. Oncotarget 2016, 7, 28697–28710. [Google Scholar] [CrossRef] [PubMed]
- Mollica Poeta, V.; Massara, M.; Capucetti, A.; Bonecchi, R. Chemokines and Chemokine Receptors: New Targets for Cancer Immunotherapy. Front. Immunol. 2019, 10, 379. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, S.-Y.; Chiang, C.-S. Distinct Role of CD11b+Ly6G−Ly6C− Myeloid-Derived Cells on the Progression of the Primary Tumor and Therapy-Associated Recurrent Brain Tumor. Cells 2020, 9, 51. https://doi.org/10.3390/cells9010051
Wu S-Y, Chiang C-S. Distinct Role of CD11b+Ly6G−Ly6C− Myeloid-Derived Cells on the Progression of the Primary Tumor and Therapy-Associated Recurrent Brain Tumor. Cells. 2020; 9(1):51. https://doi.org/10.3390/cells9010051
Chicago/Turabian StyleWu, Sheng-Yan, and Chi-Shiun Chiang. 2020. "Distinct Role of CD11b+Ly6G−Ly6C− Myeloid-Derived Cells on the Progression of the Primary Tumor and Therapy-Associated Recurrent Brain Tumor" Cells 9, no. 1: 51. https://doi.org/10.3390/cells9010051
APA StyleWu, S.-Y., & Chiang, C.-S. (2020). Distinct Role of CD11b+Ly6G−Ly6C− Myeloid-Derived Cells on the Progression of the Primary Tumor and Therapy-Associated Recurrent Brain Tumor. Cells, 9(1), 51. https://doi.org/10.3390/cells9010051