Neutrophil Extracellular Traps and Cardiovascular Diseases: An Update



Abstract
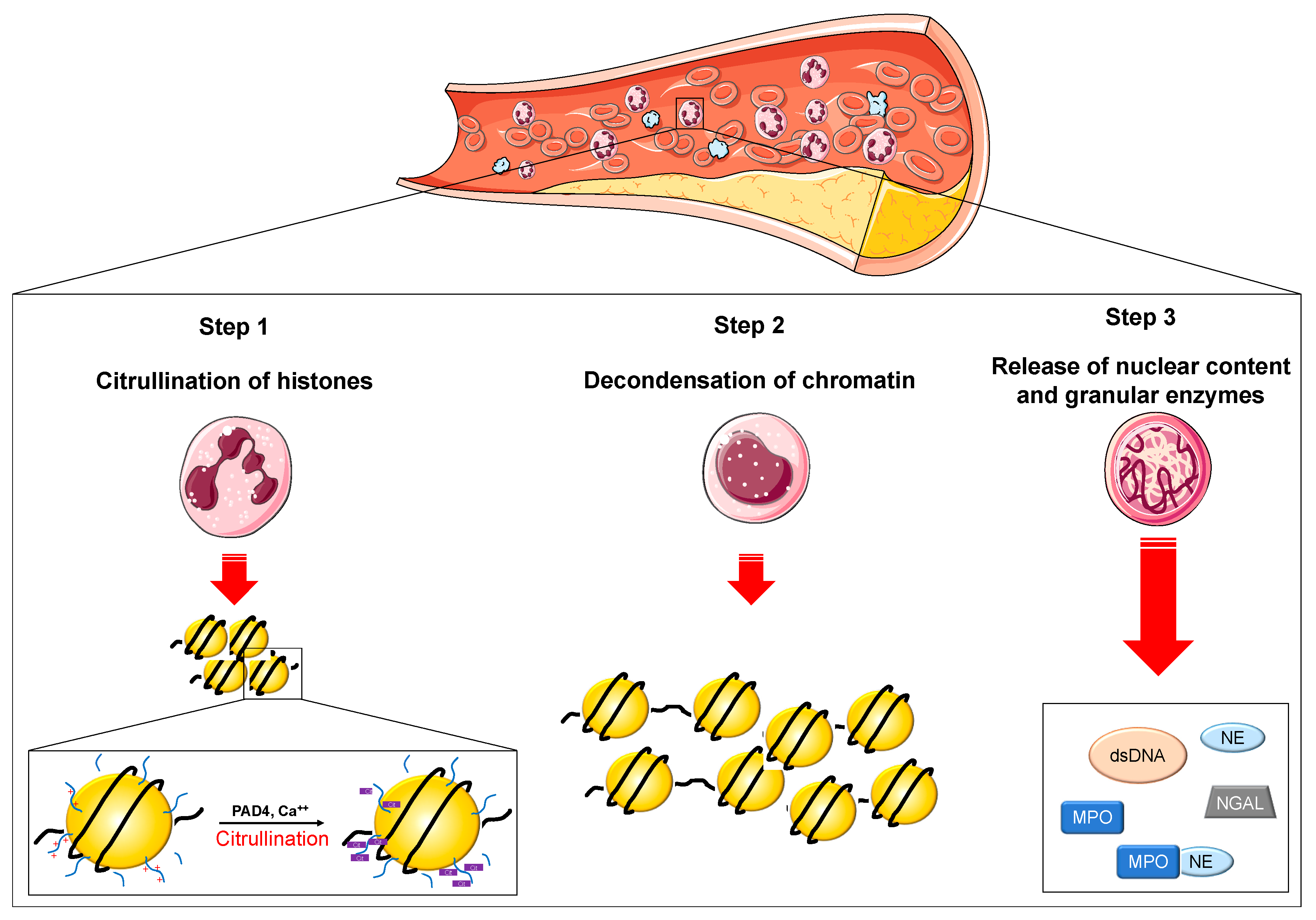
1. Introduction
2. Review Criteria
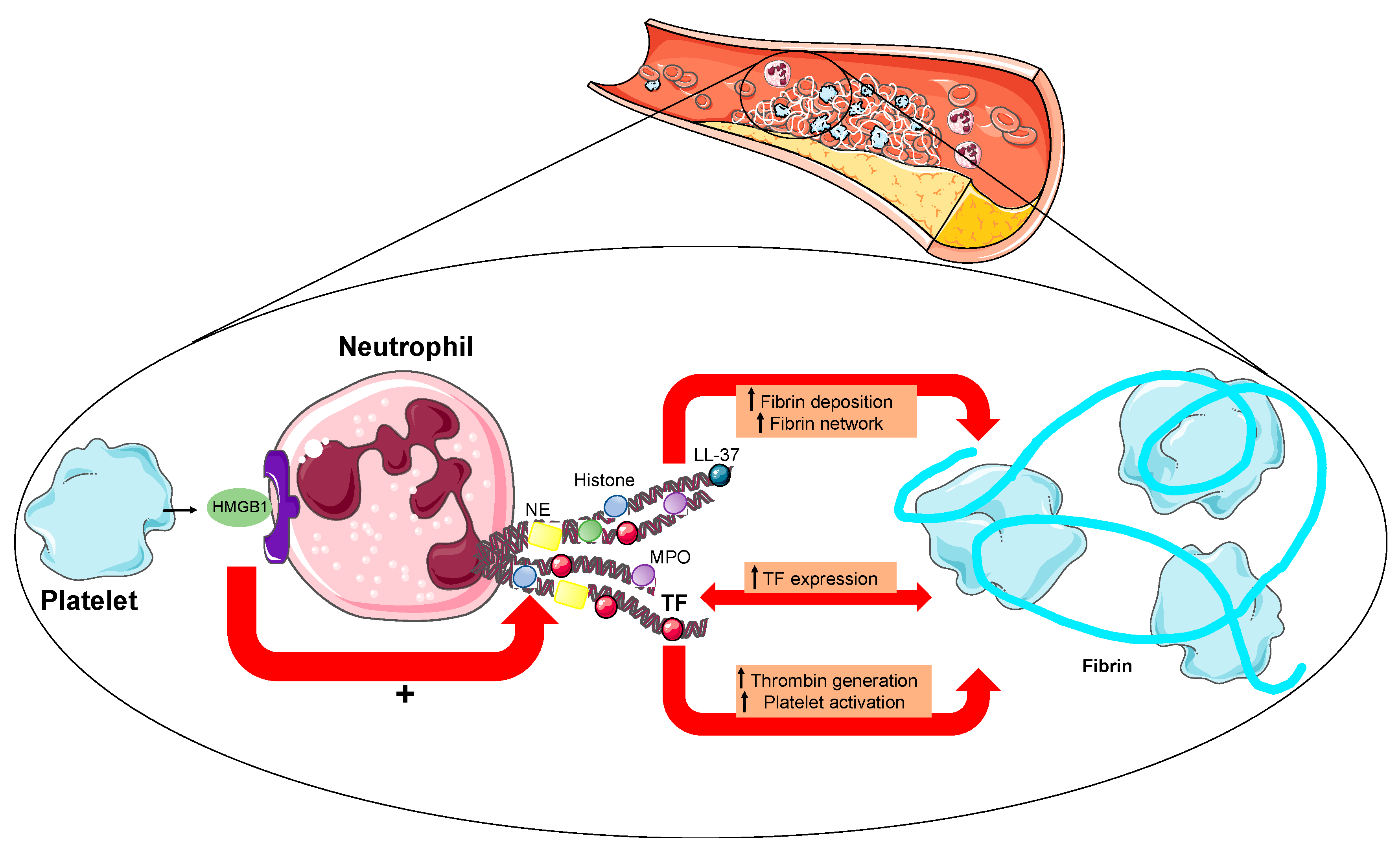
3. NETs in Acute Myocardial Infarction
4. NETs in Diabetes
5. NETs in Obesity
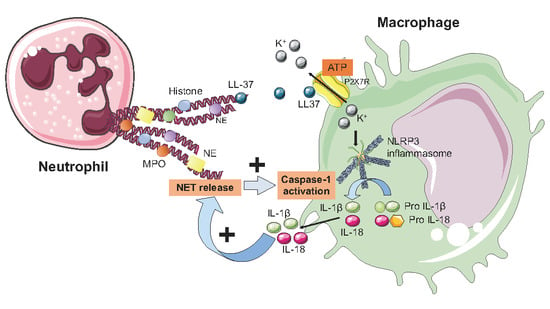
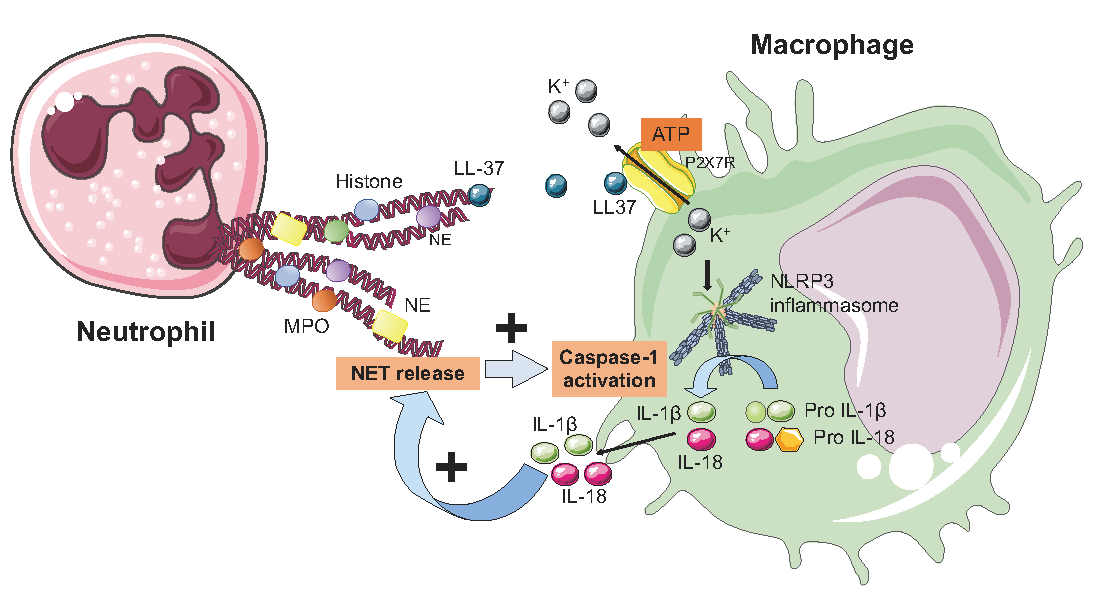
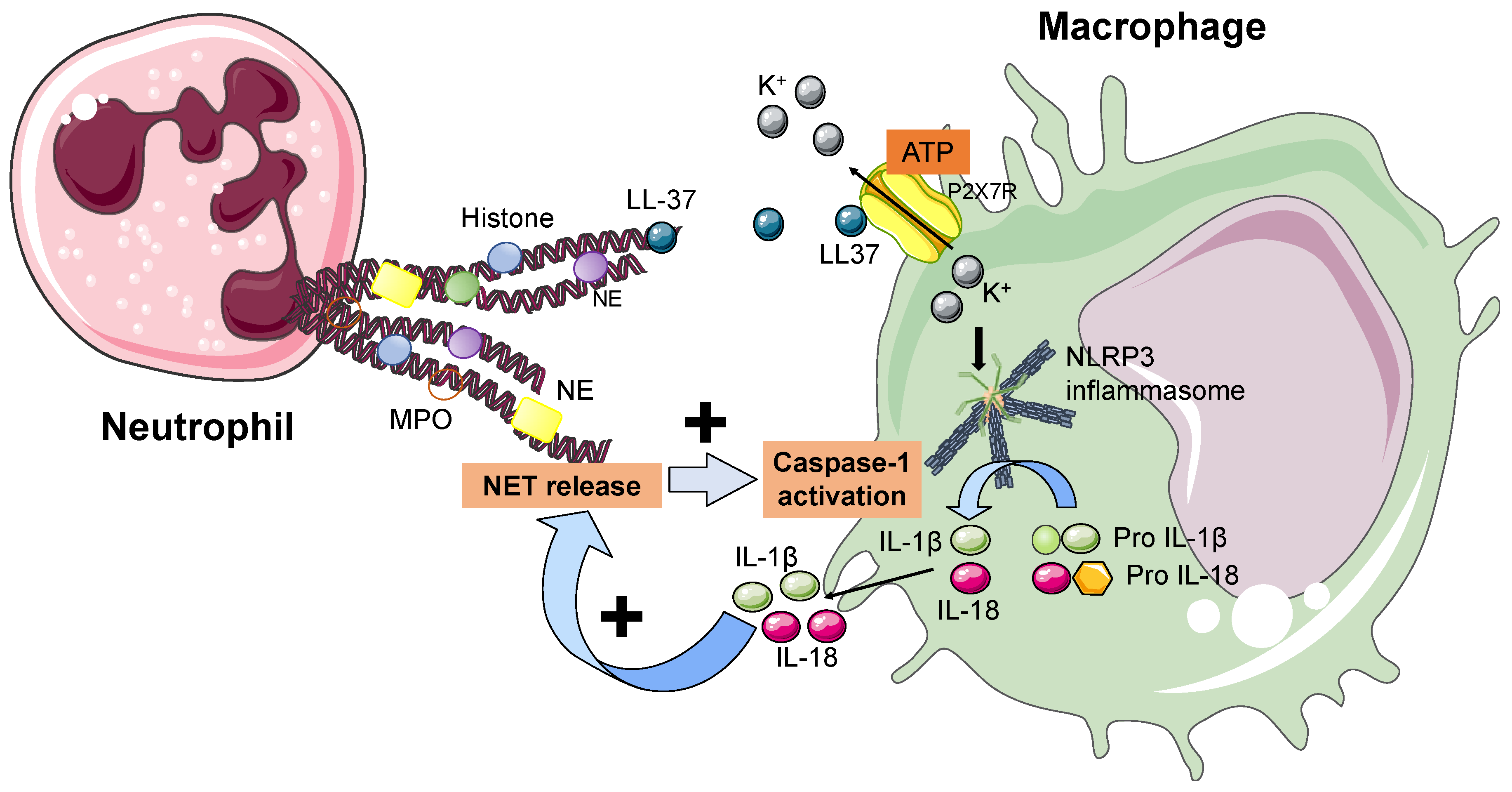
6. NETs and the Inflammasome
7. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Borregaard, N. Neutrophils, from marrow to microbes. Immunity 2010, 33, 657–670. [Google Scholar] [CrossRef] [PubMed]
- Bonaventura, A.; Montecucco, F.; Dallegri, F.; Carbone, F.; Luscher, T.F.; Camici, G.G.; Liberale, L. Novel findings in neutrophil biology and their impact on cardiovascular disease. Cardiovasc. Res. 2019, 115, 1266–1285. [Google Scholar] [CrossRef] [PubMed]
- Cowland, J.B.; Borregaard, N. Granulopoiesis and granules of human neutrophils. Immunol. Rev. 2016, 273, 11–28. [Google Scholar] [CrossRef] [PubMed]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2018, 18, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, O.E.; Borregaard, N. Neutrophil extracellular traps—The dark side of neutrophils. J. Clin. Investig. 2016, 126, 1612–1620. [Google Scholar] [CrossRef] [PubMed]
- Bonaventura, A.; Liberale, L.; Carbone, F.; Vecchie, A.; Diaz-Canestro, C.; Camici, G.G.; Montecucco, F.; Dallegri, F. The Pathophysiological Role of Neutrophil Extracellular Traps in Inflammatory Diseases. Thromb. Haemost. 2018, 118, 6–27. [Google Scholar] [CrossRef]
- Mohanan, S.; Cherrington, B.D.; Horibata, S.; McElwee, J.L.; Thompson, P.R.; Coonrod, S.A. Potential role of peptidylarginine deiminase enzymes and protein citrullination in cancer pathogenesis. Biochem. Res. Int. 2012, 2012, 895343. [Google Scholar] [CrossRef]
- Papayannopoulos, V.; Metzler, K.D.; Hakkim, A.; Zychlinsky, A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J. Cell Biol. 2010, 191, 677–691. [Google Scholar] [CrossRef]
- Metzler, K.D.; Fuchs, T.A.; Nauseef, W.M.; Reumaux, D.; Roesler, J.; Schulze, I.; Wahn, V.; Papayannopoulos, V.; Zychlinsky, A. Myeloperoxidase is required for neutrophil extracellular trap formation: Implications for innate immunity. Blood 2011, 117, 953–959. [Google Scholar] [CrossRef]
- Belaaouaj, A.; McCarthy, R.; Baumann, M.; Gao, Z.; Ley, T.J.; Abraham, S.N.; Shapiro, S.D. Mice lacking neutrophil elastase reveal impaired host defense against gram negative bacterial sepsis. Nat. Med. 1998, 4, 615–618. [Google Scholar] [CrossRef]
- Doring, Y.; Soehnlein, O.; Weber, C. Neutrophils cast NETs in atherosclerosis: Employing peptidylarginine deiminase as a therapeutic target. Circ. Res. 2014, 114, 931–934. [Google Scholar] [CrossRef] [PubMed]
- Castanheira, F.V.S.; Kubes, P. Neutrophils and NETs in modulating acute and chronic inflammation. Blood 2019, 133, 2178–2185. [Google Scholar] [CrossRef]
- Toldo, S.; Abbate, A. The NLRP3 inflammasome in acute myocardial infarction. Nat. Rev. Cardiol. 2018, 15, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Jorch, S.K.; Kubes, P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat. Med. 2017, 23, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Crea, F.; Libby, P. Acute Coronary Syndromes: The Way Forward from Mechanisms to Precision Treatment. Circulation 2017, 136, 1155–1166. [Google Scholar] [CrossRef] [PubMed]
- Bonaventura, A.; Montecucco, F.; Dallegri, F. Cellular recruitment in myocardial ischaemia/reperfusion injury. Eur. J. Clin. Investig. 2016, 46, 590–601. [Google Scholar] [CrossRef] [PubMed]
- Montecucco, F.; Liberale, L.; Bonaventura, A.; Vecchie, A.; Dallegri, F.; Carbone, F. The Role of Inflammation in Cardiovascular Outcome. Curr. Atheroscler. Rep. 2017, 19, 11. [Google Scholar] [CrossRef]
- Distelmaier, K.; Adlbrecht, C.; Jakowitsch, J.; Winkler, S.; Dunkler, D.; Gerner, C.; Wagner, O.; Lang, I.M.; Kubicek, M. Local complement activation triggers neutrophil recruitment to the site of thrombus formation in acute myocardial infarction. Thromb. Haemost. 2009, 102, 564–572. [Google Scholar] [CrossRef]
- Horne, B.D.; Anderson, J.L.; John, J.M.; Weaver, A.; Bair, T.L.; Jensen, K.R.; Renlund, D.G.; Muhlestein, J.B. Which white blood cell subtypes predict increased cardiovascular risk? J. Am. Coll. Cardiol. 2005, 45, 1638–1643. [Google Scholar] [CrossRef]
- Distelmaier, K.; Winter, M.P.; Dragschitz, F.; Redwan, B.; Mangold, A.; Gleiss, A.; Perkmann, T.; Maurer, G.; Adlbrecht, C.; Lang, I.M. Prognostic value of culprit site neutrophils in acute coronary syndrome. Eur. J. Clin. Investig. 2014, 44, 257–265. [Google Scholar] [CrossRef]
- Ge, L.; Zhou, X.; Ji, W.J.; Lu, R.Y.; Zhang, Y.; Zhang, Y.D.; Ma, Y.Q.; Zhao, J.H.; Li, Y.M. Neutrophil extracellular traps in ischemia-reperfusion injury-induced myocardial no-reflow: Therapeutic potential of DNase-based reperfusion strategy. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H500–H509. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, T.A.; Brill, A.; Duerschmied, D.; Schatzberg, D.; Monestier, M.; Myers, D.D., Jr.; Wrobleski, S.K.; Wakefield, T.W.; Hartwig, J.H.; Wagner, D.D. Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. USA 2010, 107, 15880–15885. [Google Scholar] [CrossRef] [PubMed]
- Stakos, D.A.; Kambas, K.; Konstantinidis, T.; Mitroulis, I.; Apostolidou, E.; Arelaki, S.; Tsironidou, V.; Giatromanolaki, A.; Skendros, P.; Konstantinides, S.; et al. Expression of functional tissue factor by neutrophil extracellular traps in culprit artery of acute myocardial infarction. Eur. Heart J. 2015, 36, 1405–1414. [Google Scholar] [CrossRef] [PubMed]
- Noubouossie, D.F.; Whelihan, M.F.; Yu, Y.B.; Sparkenbaugh, E.; Pawlinski, R.; Monroe, D.M.; Key, N.S. In vitro activation of coagulation by human neutrophil DNA and histone proteins but not neutrophil extracellular traps. Blood 2017, 129, 1021–1029. [Google Scholar] [CrossRef]
- Maugeri, N.; Campana, L.; Gavina, M.; Covino, C.; De Metrio, M.; Panciroli, C.; Maiuri, L.; Maseri, A.; D’Angelo, A.; Bianchi, M.E.; et al. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J. Thromb. Haemost. 2014, 12, 2074–2088. [Google Scholar] [CrossRef]
- Quillard, T.; Araujo, H.A.; Franck, G.; Shvartz, E.; Sukhova, G.; Libby, P. TLR2 and neutrophils potentiate endothelial stress, apoptosis and detachment: Implications for superficial erosion. Eur. Heart J. 2015, 36, 1394–1404. [Google Scholar] [CrossRef]
- De Boer, O.J.; Li, X.; Teeling, P.; Mackaay, C.; Ploegmakers, H.J.; van der Loos, C.M.; Daemen, M.J.; de Winter, R.J.; van der Wal, A.C. Neutrophils, neutrophil extracellular traps and interleukin-17 associate with the organisation of thrombi in acute myocardial infarction. Thromb. Haemost. 2013, 109, 290–297. [Google Scholar]
- Mangold, A.; Alias, S.; Scherz, T.; Hofbauer, T.; Jakowitsch, J.; Panzenbock, A.; Simon, D.; Laimer, D.; Bangert, C.; Kammerlander, A.; et al. Coronary neutrophil extracellular trap burden and deoxyribonuclease activity in ST-elevation acute coronary syndrome are predictors of ST-segment resolution and infarct size. Circ. Res. 2015, 116, 1182–1192. [Google Scholar] [CrossRef]
- Borissoff, J.I.; Joosen, I.A.; Versteylen, M.O.; Brill, A.; Fuchs, T.A.; Savchenko, A.S.; Gallant, M.; Martinod, K.; Ten Cate, H.; Hofstra, L.; et al. Elevated levels of circulating DNA and chromatin are independently associated with severe coronary atherosclerosis and a prothrombotic state. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2032–2040. [Google Scholar] [CrossRef]
- Cui, M.; Fan, M.; Jing, R.; Wang, H.; Qin, J.; Sheng, H.; Wang, Y.; Wu, X.; Zhang, L.; Zhu, J.; et al. Cell-Free circulating DNA: A new biomarker for the acute coronary syndrome. Cardiology 2013, 124, 76–84. [Google Scholar] [CrossRef]
- Helseth, R.; Solheim, S.; Arnesen, H.; Seljeflot, I.; Opstad, T.B. The Time Course of Markers of Neutrophil Extracellular Traps in Patients Undergoing Revascularisation for Acute Myocardial Infarction or Stable Angina Pectoris. Mediat. Inflamm. 2016, 2016, 2182358. [Google Scholar] [CrossRef] [PubMed]
- Hofbauer, T.M.; Mangold, A.; Scherz, T.; Seidl, V.; Panzenbock, A.; Ondracek, A.S.; Muller, J.; Schneider, M.; Binder, T.; Hell, L.; et al. Neutrophil extracellular traps and fibrocytes in ST-segment elevation myocardial infarction. Basic Res. Cardiol. 2019, 114, 33. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yang, D.; Wang, X.; Zhu, Z.; Wang, T.; Ma, A.; Liu, P. Neutrophil extracellular traps and dsDNA predict outcomes among patients with ST-elevation myocardial infarction. Sci. Rep. 2019, 9, 11599. [Google Scholar] [CrossRef] [PubMed]
- Helseth, R.; Knudsen, E.C.; Eritsland, J.; Opstad, T.B.; Arnesen, H.; Andersen, G.O.; Seljeflot, I. Glucose associated NETosis in patients with ST-elevation myocardial infarction: An observational study. BMC Cardiovasc. Disord. 2019, 19, 221. [Google Scholar] [CrossRef] [PubMed]
- Mangold, A.; Hofbauer, T.M.; Ondracek, A.S.; Artner, T.; Scherz, T.; Speidl, W.S.; Krychtiuk, K.A.; Sadushi-Kolici, R.; Jakowitsch, J.; Lang, I.M. Neutrophil extracellular traps and monocyte subsets at the culprit lesion site of myocardial infarction patients. Sci. Rep. 2019, 9, 16304. [Google Scholar] [CrossRef] [PubMed]
- Liberale, L.; Holy, E.W.; Akhmedov, A.; Bonetti, N.R.; Nietlispach, F.; Matter, C.M.; Mach, F.; Montecucco, F.; Beer, J.H.; Paneni, F.; et al. Interleukin-1β Mediates Arterial Thrombus Formation via NET-Associated Tissue Factor. J. Clin. Med. 2019, 8, 2072. [Google Scholar] [CrossRef]
- Rodriguez-Espinosa, O.; Rojas-Espinosa, O.; Moreno-Altamirano, M.M.; Lopez-Villegas, E.O.; Sanchez-Garcia, F.J. Metabolic requirements for neutrophil extracellular traps formation. Immunology 2015, 145, 213–224. [Google Scholar] [CrossRef]
- Langseth, M.S.; Opstad, T.B.; Bratseth, V.; Solheim, S.; Arnesen, H.; Pettersen, A.A.; Seljeflot, I.; Helseth, R. Markers of neutrophil extracellular traps are associated with adverse clinical outcome in stable coronary artery disease. Eur. J. Prev. Cardiol. 2018, 25, 762–769. [Google Scholar] [CrossRef]
- Wellen, K.E.; Hotamisligil, G.S. Inflammation, stress, and diabetes. J. Clin. Investig. 2005, 115, 1111–1119. [Google Scholar] [CrossRef]
- Bonaventura, A.; Liberale, L.; Montecucco, F. Aspirin in primary prevention for patients with diabetes: Still a matter of debate. Eur. J. Clin. Investig. 2018, 48, e13001. [Google Scholar] [CrossRef]
- Vecchie, A.; Montecucco, F.; Vecchie, F.; Dallegri, F.; Bonaventura, A. Diabetes and Vascular Disease: Is it all about Glycemia? Curr. Pharm. Des. 2019, 25, 3112–3127. [Google Scholar] [CrossRef] [PubMed]
- Stegenga, M.E.; van der Crabben, S.N.; Blumer, R.M.; Levi, M.; Meijers, J.C.; Serlie, M.J.; Tanck, M.W.; Sauerwein, H.P.; van der Poll, T. Hyperglycemia enhances coagulation and reduces neutrophil degranulation, whereas hyperinsulinemia inhibits fibrinolysis during human endotoxemia. Blood 2008, 112, 82–89. [Google Scholar] [CrossRef] [PubMed]
- De Souza Ferreira, C.; Araujo, T.H.; Angelo, M.L.; Pennacchi, P.C.; Okada, S.S.; de Araujo Paula, F.B.; Migliorini, S.; Rodrigues, M.R. Neutrophil dysfunction induced by hyperglycemia: Modulation of myeloperoxidase activity. Cell Biochem. Funct. 2012, 30, 604–610. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, M. Neutrophils and type 1 autoimmune diabetes. Curr. Opin. Hematol. 2014, 21, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yang, Z.; Zhang, W.; Niu, Y.; Li, X.; Qin, L.; Su, Q. White blood cell subtypes and risk of type 2 diabetes. J. Diabetes Complicat. 2017, 31, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Bonaventura, A.; Liberale, L.; Carbone, F.; Vecchie, A.; Bonomi, A.; Scopinaro, N.; Camerini, G.B.; Papadia, F.S.; Maggi, D.; Cordera, R.; et al. Baseline neutrophil-to-lymphocyte ratio is associated with long-term T2D remission after metabolic surgery. Acta Diabetol. 2019, 56, 741–748. [Google Scholar] [CrossRef]
- Milosevic, D.; Panin, V.L. Relationship Between Hematological Parameters and Glycemic Control in Type 2 Diabetes Mellitus Patients. J. Med. Biochem. 2019, 38, 164–171. [Google Scholar] [CrossRef]
- Wong, S.L.; Demers, M.; Martinod, K.; Gallant, M.; Wang, Y.; Goldfine, A.B.; Kahn, C.R.; Wagner, D.D. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat. Med. 2015, 21, 815–819. [Google Scholar] [CrossRef]
- Valle, A.; Giamporcaro, G.M.; Scavini, M.; Stabilini, A.; Grogan, P.; Bianconi, E.; Sebastiani, G.; Masini, M.; Maugeri, N.; Porretti, L.; et al. Reduction of circulating neutrophils precedes and accompanies type 1 diabetes. Diabetes 2013, 62, 2072–2077. [Google Scholar] [CrossRef]
- Harsunen, M.H.; Puff, R.; D’Orlando, O.; Giannopoulou, E.; Lachmann, L.; Beyerlein, A.; von Meyer, A.; Ziegler, A.G. Reduced blood leukocyte and neutrophil numbers in the pathogenesis of type 1 diabetes. Horm. Metab. Res. 2013, 45, 467–470. [Google Scholar] [CrossRef]
- Vecchio, F.; Lo Buono, N.; Stabilini, A.; Nigi, L.; Dufort, M.J.; Geyer, S.; Rancoita, P.M.; Cugnata, F.; Mandelli, A.; Valle, A.; et al. Abnormal neutrophil signature in the blood and pancreas of presymptomatic and symptomatic type 1 diabetes. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Wang, Y.; Xiao, Y.; Zhong, L.; Ye, D.; Zhang, J.; Tu, Y.; Bornstein, S.R.; Zhou, Z.; Lam, K.S.; Xu, A. Increased neutrophil elastase and proteinase 3 and augmented NETosis are closely associated with beta-cell autoimmunity in patients with type 1 diabetes. Diabetes 2014, 63, 4239–4248. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Fu, S.; Speake, C.; Greenbaum, C.J.; Odegard, J.M. NETosis-associated serum biomarkers are reduced in type 1 diabetes in association with neutrophil count. Clin. Exp. Immunol. 2016, 184, 318–322. [Google Scholar] [CrossRef] [PubMed]
- Menegazzo, L.; Ciciliot, S.; Poncina, N.; Mazzucato, M.; Persano, M.; Bonora, B.; Albiero, M.; Vigili de Kreutzenberg, S.; Avogaro, A.; Fadini, G.P. NETosis is induced by high glucose and associated with type 2 diabetes. Acta Diabetol. 2015, 52, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Kim, J.E.; Gu, J.Y.; Yoo, H.J.; Park, S.H.; Kim, Y.I.; Nam-Goong, I.S.; Kim, E.S.; Kim, H.K. Evaluation of Circulating Markers of Neutrophil Extracellular Trap (NET) Formation as Risk Factors for Diabetic Retinopathy in a Case-Control Association Study. Exp. Clin. Endocrinol. Diabetes 2016, 124, 557–561. [Google Scholar] [CrossRef] [PubMed]
- Joshi, M.B.; Lad, A.; Bharath Prasad, A.S.; Balakrishnan, A.; Ramachandra, L.; Satyamoorthy, K. High glucose modulates IL-6 mediated immune homeostasis through impeding neutrophil extracellular trap formation. FEBS Lett. 2013, 587, 2241–2246. [Google Scholar] [CrossRef] [PubMed]
- Carestia, A.; Frechtel, G.; Cerrone, G.; Linari, M.A.; Gonzalez, C.D.; Casais, P.; Schattner, M. NETosis before and after Hyperglycemic Control in Type 2 Diabetes Mellitus Patients. PLoS ONE 2016, 11, e0168647. [Google Scholar] [CrossRef]
- Menegazzo, L.; Scattolini, V.; Cappellari, R.; Bonora, B.M.; Albiero, M.; Bortolozzi, M.; Romanato, F.; Ceolotto, G.; Vigili de Kreutzeberg, S.; Avogaro, A.; et al. The antidiabetic drug metformin blunts NETosis in vitro and reduces circulating NETosis biomarkers in vivo. Acta Diabetol. 2018, 55, 593–601. [Google Scholar] [CrossRef]
- Wang, L.; Zhou, X.; Yin, Y.; Mai, Y.; Wang, D.; Zhang, X. Hyperglycemia Induces Neutrophil Extracellular Traps Formation Through an NADPH Oxidase-Dependent Pathway in Diabetic Retinopathy. Front. Immunol. 2018, 9, 3076. [Google Scholar] [CrossRef]
- Fadini, G.P.; Menegazzo, L.; Rigato, M.; Scattolini, V.; Poncina, N.; Bruttocao, A.; Ciciliot, S.; Mammano, F.; Ciubotaru, C.D.; Brocco, E.; et al. NETosis Delays Diabetic Wound Healing in Mice and Humans. Diabetes 2016, 65, 1061–1071. [Google Scholar] [CrossRef]
- Vecchie, A.; Dallegri, F.; Carbone, F.; Bonaventura, A.; Liberale, L.; Portincasa, P.; Fruhbeck, G.; Montecucco, F. Obesity phenotypes and their paradoxical association with cardiovascular diseases. Eur. J. Intern. Med. 2018, 48, 6–17. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.C.; Lee, J. Cellular and molecular players in adipose tissue inflammation in the development of obesity-induced insulin resistance. Biochim. Biophys. Acta 2014, 1842, 446–462. [Google Scholar] [CrossRef] [PubMed]
- Liberale, L.; Bertolotto, M.; Carbone, F.; Contini, P.; Wust, P.; Spinella, G.; Pane, B.; Palombo, D.; Bonaventura, A.; Pende, A.; et al. Resistin exerts a beneficial role in atherosclerotic plaque inflammation by inhibiting neutrophil migration. Int. J. Cardiol. 2018, 272, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Liberale, L.; Bonaventura, A.; Carbone, F.; Bertolotto, M.; Contini, P.; Scopinaro, N.; Camerini, G.B.; Papadia, F.S.; Cordera, R.; Camici, G.G.; et al. Early reduction of matrix metalloproteinase-8 serum levels is associated with leptin drop and predicts diabetes remission after bariatric surgery. Int. J. Cardiol. 2017, 245, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, Q.; Venugopal, J.; Wang, J.; Kleiman, K.; Guo, C.; Eitzman, D.T. Obesity-induced Endothelial Dysfunction is Prevented by Neutrophil Extracellular Trap Inhibition. Sci. Rep. 2018, 8, 4881. [Google Scholar] [CrossRef] [PubMed]
- Braster, Q.; Silvestre Roig, C.; Hartwig, H.; Beckers, L.; den Toom, M.; Doring, Y.; Daemen, M.J.; Lutgens, E.; Soehnlein, O. Inhibition of NET Release Fails to Reduce Adipose Tissue Inflammation in Mice. PLoS ONE 2016, 11, e0163922. [Google Scholar] [CrossRef]
- D’Abbondanza, M.; Martorelli, E.E.; Ricci, M.A.; De Vuono, S.; Migliola, E.N.; Godino, C.; Corradetti, S.; Siepi, D.; Paganelli, M.T.; Maugeri, N.; et al. Increased plasmatic NETs by-products in patients in severe obesity. Sci. Rep. 2019, 9, 14678. [Google Scholar] [CrossRef]
- Toldo, S.; Mezzaroma, E.; Mauro, A.G.; Salloum, F.; Van Tassell, B.W.; Abbate, A. The inflammasome in myocardial injury and cardiac remodeling. Antioxid. Redox Signal. 2015, 22, 1146–1161. [Google Scholar] [CrossRef]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef]
- Franchi, L.; Eigenbrod, T.; Munoz-Planillo, R.; Nunez, G. The inflammasome: A caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat. Immunol. 2009, 10, 241–247. [Google Scholar] [CrossRef]
- Jin, Y.; Fu, J. Novel Insights into the NLRP 3 Inflammasome in Atherosclerosis. J. Am. Heart Assoc. 2019, 8, e012219. [Google Scholar] [CrossRef] [PubMed]
- Abbate, A.; Van Tassell, B.W.; Biondi-Zoccai, G.G. Blocking interleukin-1 as a novel therapeutic strategy for secondary prevention of cardiovascular events. BioDrugs 2012, 26, 217–233. [Google Scholar] [CrossRef] [PubMed]
- Zuurbier, C.J.; Abbate, A.; Cabrera-Fuentes, H.A.; Cohen, M.V.; Collino, M.; De Kleijn, D.P.V.; Downey, J.M.; Pagliaro, P.; Preissner, K.T.; Takahashi, M.; et al. Innate immunity as a target for acute cardioprotection. Cardiovasc. Res. 2019, 115, 1131–1142. [Google Scholar] [CrossRef] [PubMed]
- Bonaventura, A.; Montecucco, F. Inflammation and pericarditis: Are neutrophils actors behind the scenes? J. Cell. Physiol. 2019, 234, 5390–5398. [Google Scholar] [CrossRef] [PubMed]
- Mauro, A.G.; Bonaventura, A.; Mezzaroma, E.; Quader, M.; Toldo, S. NLRP3 Inflammasome in Acute Myocardial Infarction. J. Cardiovasc. Pharmacol. 2019, 74, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Bonaventura, A.; Cannata, A.; Mezzaroma, E.; Sinagra, G.; Bussani, R.; Toldo, S.; Gandhi, R.; Kim, M.; Paolini, J.; Montecucco, F.; et al. Abstract 13363: Intensification of the Inflammasome Formation in the Pericardium of Patients with Chronic Severe Pericarditis. Circulation 2019, 140 (Suppl. 1), A13363. [Google Scholar]
- Dinarello, C.A. The IL-1 family of cytokines and receptors in rheumatic diseases. Nat. Rev. Rheumatol. 2019, 15, 612–632. [Google Scholar] [CrossRef]
- Elssner, A.; Duncan, M.; Gavrilin, M.; Wewers, M.D. A novel P2X7 receptor activator, the human cathelicidin-derived peptide LL37, induces IL-1 beta processing and release. J. Immunol. 2004, 172, 4987–4994. [Google Scholar] [CrossRef]
- Kahlenberg, J.M.; Carmona-Rivera, C.; Smith, C.K.; Kaplan, M.J. Neutrophil extracellular trap-associated protein activation of the NLRP3 inflammasome is enhanced in lupus macrophages. J. Immunol. 2013, 190, 1217–1226. [Google Scholar] [CrossRef]
- Mitroulis, I.; Kambas, K.; Chrysanthopoulou, A.; Skendros, P.; Apostolidou, E.; Kourtzelis, I.; Drosos, G.I.; Boumpas, D.T.; Ritis, K. Neutrophil extracellular trap formation is associated with IL-1beta and autophagy-related signaling in gout. PLoS ONE 2011, 6, e29318. [Google Scholar] [CrossRef]
- Hu, Q.; Shi, H.; Zeng, T.; Liu, H.; Su, Y.; Cheng, X.; Ye, J.; Yin, Y.; Liu, M.; Zheng, H.; et al. Increased neutrophil extracellular traps activate NLRP3 and inflammatory macrophages in adult-onset Still’s disease. Arthritis Res. Ther. 2019, 21, 9. [Google Scholar] [CrossRef] [PubMed]
- Lachowicz-Scroggins, M.E.; Dunican, E.M.; Charbit, A.R.; Raymond, W.; Looney, M.R.; Peters, M.C.; Gordon, E.D.; Woodruff, P.G.; Lefrancais, E.; Phillips, B.R.; et al. Extracellular DNA, Neutrophil Extracellular Traps, and Inflammasome Activation in Severe Asthma. Am. J. Respir. Crit. Care Med. 2019, 199, 1076–1085. [Google Scholar] [CrossRef] [PubMed]
- Awad, F.; Assrawi, E.; Jumeau, C.; Georgin-Lavialle, S.; Cobret, L.; Duquesnoy, P.; Piterboth, W.; Thomas, L.; Stankovic-Stojanovic, K.; Louvrier, C.; et al. Impact of human monocyte and macrophage polarization on NLR expression and NLRP3 inflammasome activation. PLoS ONE 2017, 12, e0175336. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, H.N.; Herzig, A.; Zychlinsky, A. Beyond the grave: When is cell death critical for immunity to infection? Curr. Opin. Immunol. 2016, 38, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.K.; Adameova, A.; Hill, J.A.; Baines, C.P.; Kang, P.M.; Downey, J.M.; Narula, J.; Takahashi, M.; Abbate, A.; Piristine, H.C.; et al. Guidelines for evaluating myocardial cell death. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H891–H922. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016, 535, 153–158. [Google Scholar] [CrossRef]
- Shi, J.; Gao, W.; Shao, F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem. Sci. 2017, 42, 245–254. [Google Scholar] [CrossRef]
- Sollberger, G.; Choidas, A.; Burn, G.L.; Habenberger, P.; Di Lucrezia, R.; Kordes, S.; Menninger, S.; Eickhoff, J.; Nussbaumer, P.; Klebl, B.; et al. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci. Immunol. 2018, 3. [Google Scholar] [CrossRef]
- Metzler, K.D.; Goosmann, C.; Lubojemska, A.; Zychlinsky, A.; Papayannopoulos, V. A myeloperoxidase-containing complex regulates neutrophil elastase release and actin dynamics during NETosis. Cell Rep. 2014, 8, 883–896. [Google Scholar] [CrossRef]
- Chen, K.W.; Monteleone, M.; Boucher, D.; Sollberger, G.; Ramnath, D.; Condon, N.D.; von Pein, J.B.; Broz, P.; Sweet, M.J.; Schroder, K. Noncanonical inflammasome signaling elicits gasdermin D-dependent neutrophil extracellular traps. Sci. Immunol. 2018, 3, eaar6676. [Google Scholar] [CrossRef] [PubMed]
- Toldo, S.; Mezzaroma, E.; McGeough, M.D.; Pena, C.A.; Marchetti, C.; Sonnino, C.; Van Tassell, B.W.; Salloum, F.N.; Voelkel, N.F.; Hoffman, H.M.; et al. Independent roles of the priming and the triggering of the NLRP3 inflammasome in the heart. Cardiovasc. Res. 2015, 105, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Toldo, S.; Marchetti, C.; Mauro, A.G.; Chojnacki, J.; Mezzaroma, E.; Carbone, S.; Zhang, S.; Van Tassell, B.; Salloum, F.N.; Abbate, A. Inhibition of the NLRP3 inflammasome limits the inflammatory injury following myocardial ischemia-reperfusion in the mouse. Int. J. Cardiol. 2016, 209, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Toldo, S.; Mauro, A.G.; Cutter, Z.; Abbate, A. Inflammasome, pyroptosis, and cytokines in myocardial ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1553–H1568. [Google Scholar] [CrossRef]
- Kawaguchi, M.; Takahashi, M.; Hata, T.; Kashima, Y.; Usui, F.; Morimoto, H.; Izawa, A.; Takahashi, Y.; Masumoto, J.; Koyama, J.; et al. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation 2011, 123, 594–604. [Google Scholar] [CrossRef]
- Mezzaroma, E.; Toldo, S.; Farkas, D.; Seropian, I.M.; Van Tassell, B.W.; Salloum, F.N.; Kannan, H.R.; Menna, A.C.; Voelkel, N.F.; Abbate, A. The inflammasome promotes adverse cardiac remodeling following acute myocardial infarction in the mouse. Proc. Natl. Acad. Sci. USA 2011, 108, 19725–19730. [Google Scholar] [CrossRef]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nunez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef]
- Tall, A.R.; Westerterp, M. Inflammasomes, neutrophil extracellular traps, and cholesterol. J. Lipid Res. 2019, 60, 721–727. [Google Scholar] [CrossRef]
- Westerterp, M.; Fotakis, P.; Ouimet, M.; Bochem, A.E.; Zhang, H.; Molusky, M.M.; Wang, W.; Abramowicz, S.; la Bastide-van Gemert, S.; Wang, N. Cholesterol Efflux Pathways Suppress Inflammasome Activation, NETosis, and Atherogenesis. Circulation 2018, 138, 898–912. [Google Scholar] [CrossRef]
- Warnatsch, A.; Ioannou, M.; Wang, Q.; Papayannopoulos, V. Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science 2015, 349, 316–320. [Google Scholar] [CrossRef] [PubMed]
- Everett, B.M.; Donath, M.Y.; Pradhan, A.D.; Thuren, T.; Pais, P.; Nicolau, J.C.; Glynn, R.J.; Libby, P.; Ridker, P.M. Anti-Inflammatory Therapy with Canakinumab for the Prevention and Management of Diabetes. J. Am. Coll. Cardiol. 2018, 71, 2392–2401. [Google Scholar] [CrossRef] [PubMed]
- Hahn, J.; Schauer, C.; Czegley, C.; Kling, L.; Petru, L.; Schmid, B.; Weidner, D.; Reinwald, C.; Biermann, M.H.C.; Blunder, S.; et al. Aggregated neutrophil extracellular traps resolve inflammation by proteolysis of cytokines and chemokines and protection from antiproteases. FASEB J. 2019, 33, 1401–1414. [Google Scholar] [CrossRef] [PubMed]
- Schauer, C.; Janko, C.; Munoz, L.E.; Zhao, Y.; Kienhofer, D.; Frey, B.; Lell, M.; Manger, B.; Rech, J.; Naschberger, E.; et al. Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines. Nat. Med. 2014, 20, 511–517. [Google Scholar] [CrossRef]
- Clancy, D.M.; Henry, C.M.; Sullivan, G.P.; Martin, S.J. Neutrophil extracellular traps can serve as platforms for processing and activation of IL-1 family cytokines. FEBS J. 2017, 284, 1712–1725. [Google Scholar] [CrossRef]
- Petretto, A.; Bruschi, M.; Pratesi, F.; Croia, C.; Candiano, G.; Ghiggeri, G.; Migliorini, P. Neutrophil extracellular traps (NET) induced by different stimuli: A comparative proteomic analysis. PLoS ONE 2019, 14, e0218946. [Google Scholar] [CrossRef]
- Sorvillo, N.; Cherpokova, D.; Martinod, K.; Wagner, D.D. Extracellular DNA NET-Works with Dire Consequences for Health. Circ. Res. 2019, 125, 470–488. [Google Scholar] [CrossRef]
- Wong, S.L.; Wagner, D.D. Peptidylarginine deiminase 4: A nuclear button triggering neutrophil extracellular traps in inflammatory diseases and aging. FASEB J. 2018, 32, 6358–6370. [Google Scholar] [CrossRef]
- Wolach, O.; Sellar, R.S.; Martinod, K.; Cherpokova, D.; McConkey, M.; Chappell, R.J.; Silver, A.J.; Adams, D.; Castellano, C.A.; Schneider, R.K.; et al. Increased neutrophil extracellular trap formation promotes thrombosis in myeloproliferative neoplasms. Sci. Transl. Med. 2018, 10, eaan8292. [Google Scholar] [CrossRef]
- Keyel, P.A. Dnases in health and disease. Dev. Biol. 2017, 429, 1–11. [Google Scholar] [CrossRef]
- Boeltz, S.; Amini, P.; Anders, H.J.; Andrade, F.; Bilyy, R.; Chatfield, S.; Cichon, I.; Clancy, D.M.; Desai, J.; Dumych, T.; et al. To NET or not to NET:current opinions and state of the science regarding the formation of neutrophil extracellular traps. Cell Death Differ. 2019, 26, 395–408. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Author | Year | Patients | Biomarkers | Results |
|---|---|---|---|---|
| Borissoff et al. [29] | 2013 | 282 patients with suspected CAD undergoing coronary CTA, grouped based on the presence and severity of CAD | dsDNA, nucleosomes, citH4, and MPO-DNA complexes | dsDNA, nucleosome, and MPO-DNA complex levels were higher in patients with severe CAD (p < 0.05 for all) compared to healthy controls and correlated with the severity of luminal stenosis and the number of diseased coronary artery vessels (p ≤ 0.001 for all). Baseline higher-than-median values of dsDNA (OR 3.12, p = 0.013), nucleosome (OR 2.59, p = 0.030), and MPO–DNA complexes (OR 3.53, p = 0.009) were significantly associated with the occurrence of MACEs. |
| Cui et al. [30] | 2013 | 137 ACS patients (51 UA, 37 NSTEMI, and 49 STEMI), 13 stable AP patients, and 60 healthy controls | dsDNA | ACS patients showed higher dsDNA levels compared to stable AP patients and control group (p < 0.05 for both). Significant differences in dsDNA concentrations were observed among UA, NSTEMI, and STEMI sub-groups (p < 0.05 for all). |
| Mangold et al. [28] | 2015 | 111 patients with STEMI undergoing PCI (TIMI flow 0–1) | Nucleosomes and dsDNA | NE, MPO, nucleosome, and dsDNA concentrations were increased at the CLS compared to the femoral site (p < 0.001 for all). Nucleosome and dsDNA levels positively correlated with coronary thrombus NET burden (p < 0.05 for both), the latter being positively correlated with ST resolution and both enzymatic (CK-MB AUC) and CMR-assessed infarct size (p < 0.01 for all). |
| Helseth et al. [31] | 2016 | 30 patients with CAD undergoing PCI (20 with STEMI and 10 with stable AP) | Nucleosomes and dsDNA | dsDNA and nucleosome levels were higher in patients with STEMI compared to those with AP (p < 0.05 for both). dsDNA significantly correlated with peak TnT and CK-MB at day 5 (p = 0.03 for both) and with CMR-assessed infarct size at days 5 and 7 (p < 0.05 for both), while only nucleosomes correlated with infarct size at day 5 (p = 0.02). |
| Hofbauer et al. [32] | 2019 | 50 patients with STEMI undergoing PCI (TIMI flow 0) | dsDNA and citH3 | dsDNA and citH3 levels were significantly increased at the CLS than at the femoral artery (p < 0.01 for both). This trend was confirmed only for dsDNA when compared to healthy controls (p < 0.0001). Both dsDNA and citH3 were positively correlated with enzymatic infarct size (p < 0.05 for both). dsDNA measured at the CLS at the time of PCI was positively correlated with WMSI at the 24 ± 8-month follow-up (p = 0.039). |
| Liu et al. [33] | 2019 | 83 patients with STEMI undergoing PCI (TIMI 0) | dsDNA and MPO-DNA complexes | A larger number of NETting neutrophils from IRA was found compared to peripheral arteries and healthy controls (p < 0.05 for both). Higher concentrations of dsDNA and MPO-DNA complexes were retrieved within IRA compared to peripheral arteries (p < 0.05 for both). Baseline levels of coronary dsDNA were higher in patients experiencing a MACE (0.70 vs. 0.46 μg/mL, p = 0.002). Additionally, dsDNA was found to independently predict in-hospital MACEs (OR 46.26, p = 0.001). A cutoff of 0.39 μg/mL for dsDNA was reported as a better prognostic marker compared to TnT and CK-MB (sensitivity 78%, specificity 53%). |
| Helseth et al. [34] | 2019 | 224 patients with STEMI undergoing PCI followed for 3 months | dsDNA and MPO-DNA complexes | dsDNA and MPO-DNA levels were correlated to leukocyte count at admission (p < 0.01 for both) and to each other only in the acute phase (p < 0.001), but not after 3 months. dsDNA weakly correlated with glucose in the acute phase and after 3 months (p < 0.05 for both), while MPO-DNA did not. |
| Mangold et al. [35] | 2019 | 91 patients with STEMI receiving thrombectomy during PCI | dsDNA and citH3 | dsDNA and citH3 were significantly elevated at the CLS compared to femoral plasma (p < 0.0001 for both) and correlated with enzymatic infarct size (CK-MB AUC, p < 0.001 and p < 0.01, respectively). High CLS dsDNA correlated with low, non-classical (anti-inflammatory) monocyte percentage at the culprit site (p < 0.05). Low CX3CR1 expression of non-classical monocytes (i.e., anti-inflammatory) negatively correlated with high CLS dsDNA and citH3 levels (p < 0.05 for both). |
| Liberale et al. [36] | 2019 | 66 patients undergoing PCI | MPO-DNA and TF-DNA complexes | MPO-DNA complexes were higher in the high- compared to the low-CRP group (p < 0.01). Patients with high CRP levels showed increased levels of TF-DNA complexes than patients with low CRP levels (p < 0.01). A positive correlation with NETosis markers (MPO-DNA and TF-DNA complexes) was recorded (p < 0.0001). |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bonaventura, A.; Vecchié, A.; Abbate, A.; Montecucco, F. Neutrophil Extracellular Traps and Cardiovascular Diseases: An Update. Cells 2020, 9, 231. https://doi.org/10.3390/cells9010231
Bonaventura A, Vecchié A, Abbate A, Montecucco F. Neutrophil Extracellular Traps and Cardiovascular Diseases: An Update. Cells. 2020; 9(1):231. https://doi.org/10.3390/cells9010231
Chicago/Turabian StyleBonaventura, Aldo, Alessandra Vecchié, Antonio Abbate, and Fabrizio Montecucco. 2020. "Neutrophil Extracellular Traps and Cardiovascular Diseases: An Update" Cells 9, no. 1: 231. https://doi.org/10.3390/cells9010231
APA StyleBonaventura, A., Vecchié, A., Abbate, A., & Montecucco, F. (2020). Neutrophil Extracellular Traps and Cardiovascular Diseases: An Update. Cells, 9(1), 231. https://doi.org/10.3390/cells9010231