ETV6/RUNX1 Fusion Gene Abrogation Decreases the Oncogenicity of Tumour Cells in a Preclinical Model of Acute Lymphoblastic Leukaemia

 , , , and
, , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Background
2. Material and Methods
2.1. Cell Lines and Culture Conditions
2.2. Sgrnas Design and Cloning
2.3. Sgrna Transfections
2.4. Flow Cytometry Analysis and Cell Sorting
2.5. Sequencing of sgrNA Targets Sites
2.6. Off-Target Sequence Analysis
2.7. RT-qPCR
2.8. Transcriptome Sequencing
2.9. Western Blotting
2.10. Cell Viability, Cell Cycle Analysis and Proliferation Assays
2.11. B-ALL-Stromal Cell Co-Culture
2.12. Drugs and Treatments
2.13. Mouse Xenograft Tumourigenesis
2.14. Histopathology and Immunohistochemistry
2.15. Statistical Analysis
3. Results
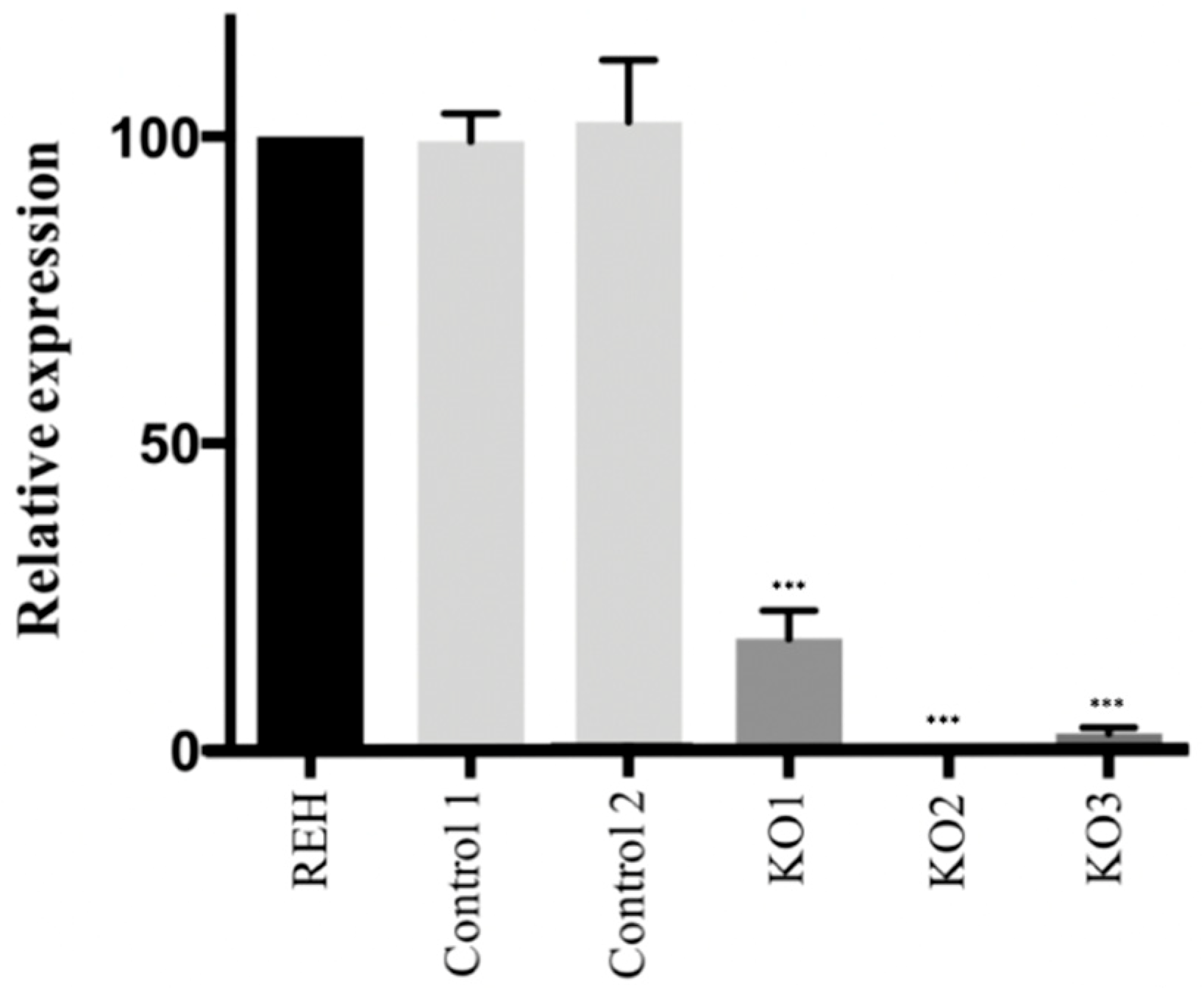
3.1. CRISPR/Cas9 Edited Lymphoid Cell Line Showed a Loss of E/R Functionality
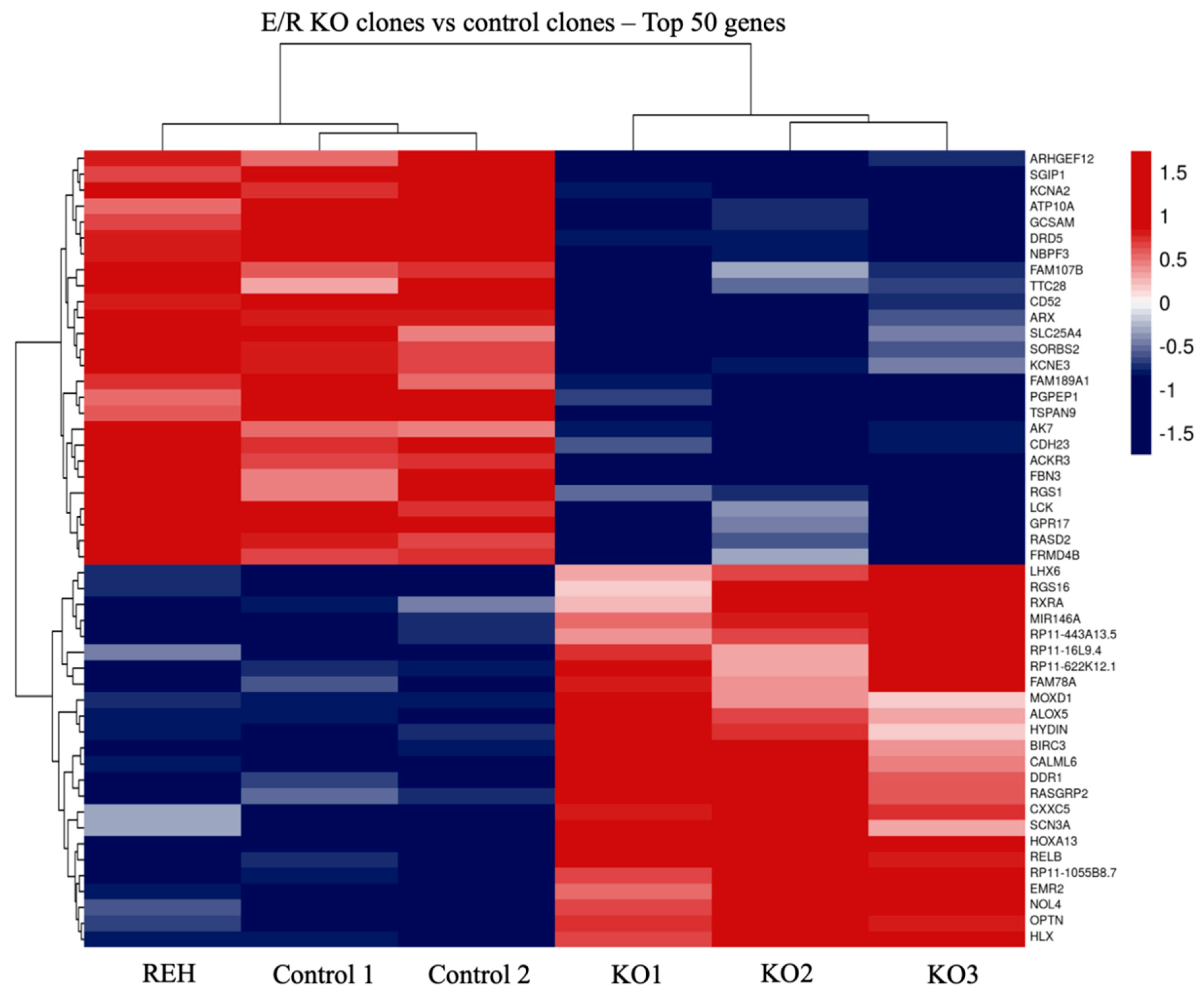
3.2. Transcriptomic Analysis of E/R KO Lymphoid Cell Line Generated by CRISPR/Cas9 Showed a Distinct Expression Signature and a Deregulation of Its Downstream Signalling Genes
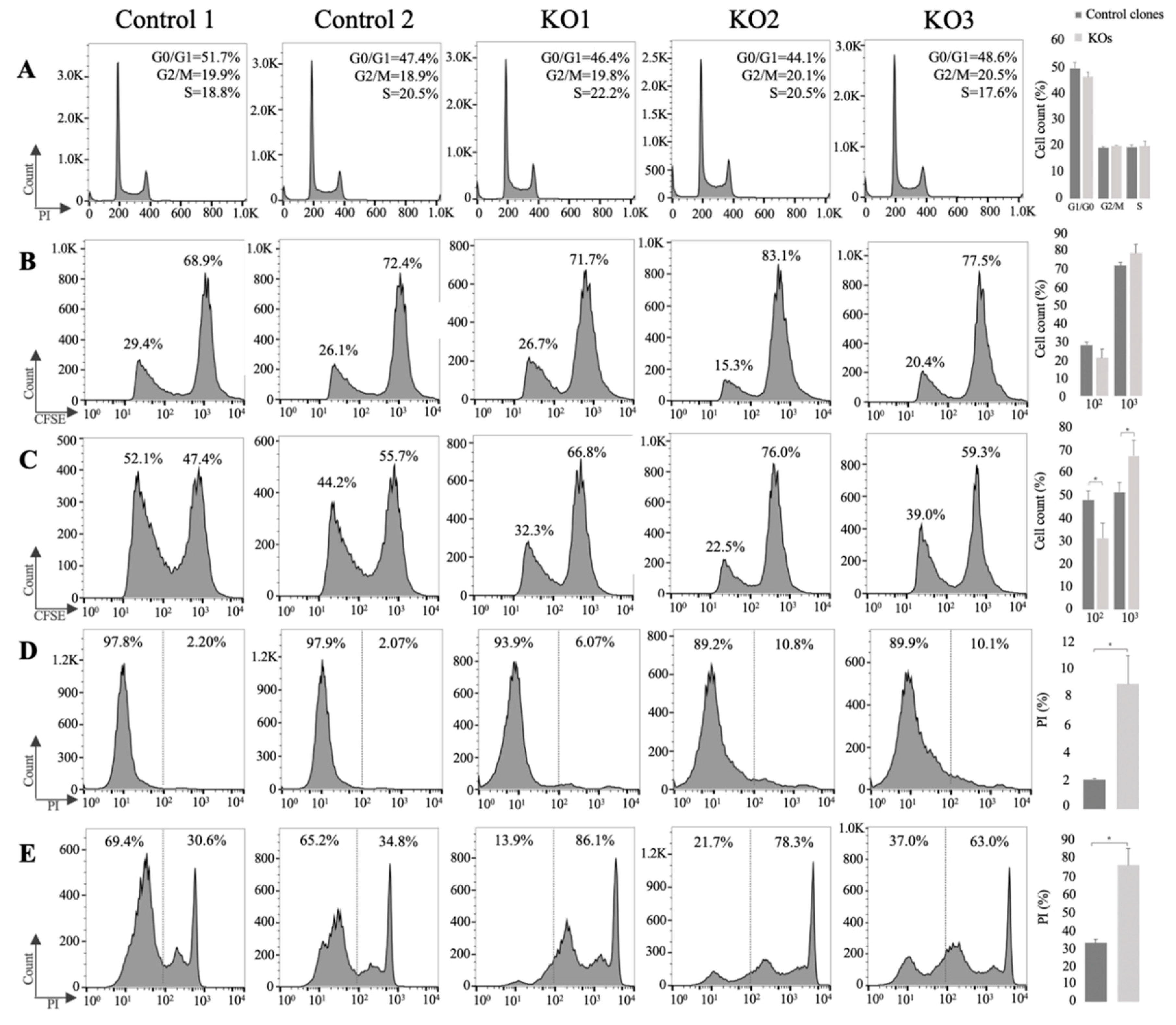
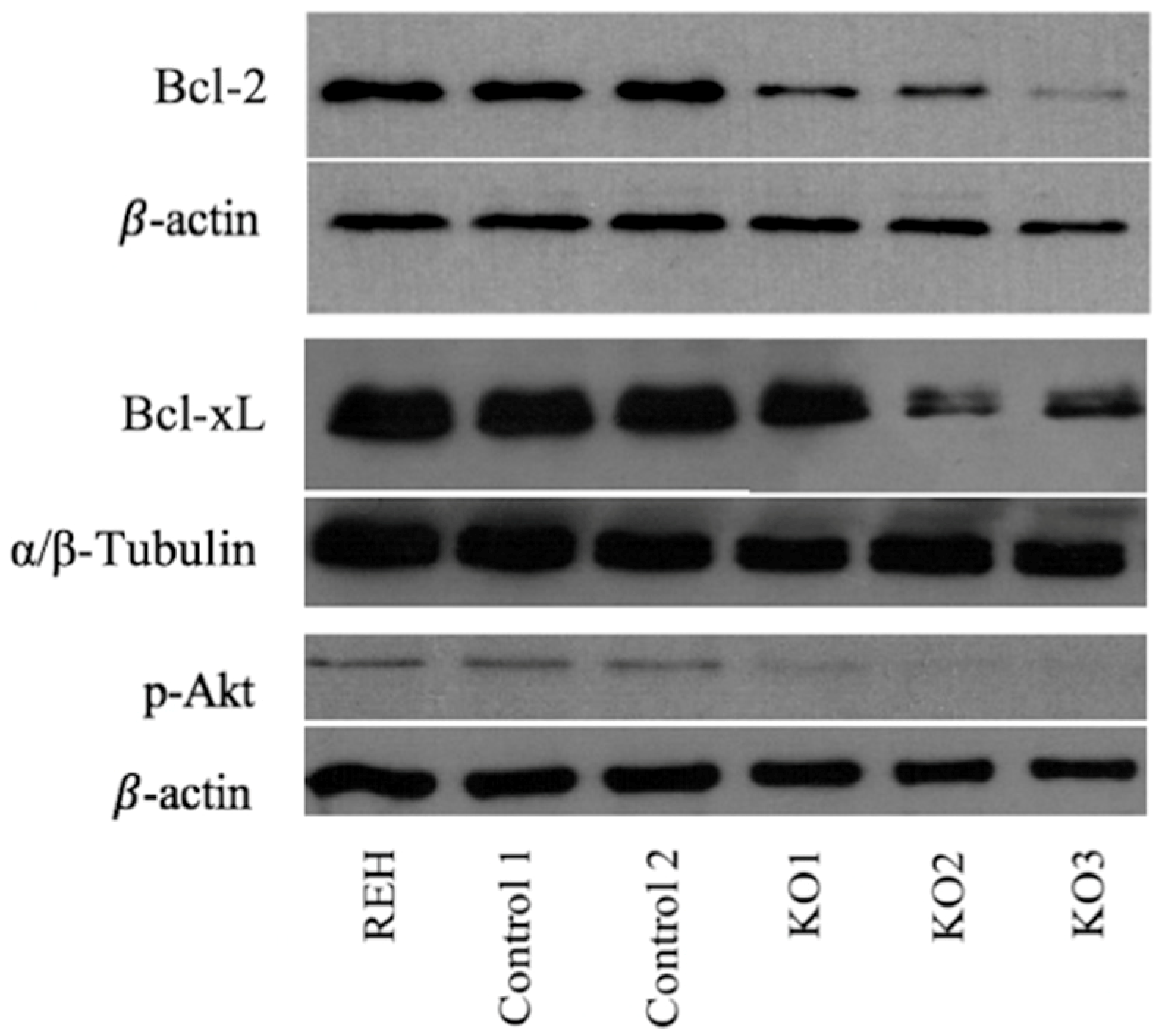
3.3. E/R Abrogation Reduces Proliferative Capacity and Resistance to Apoptosis In vitro
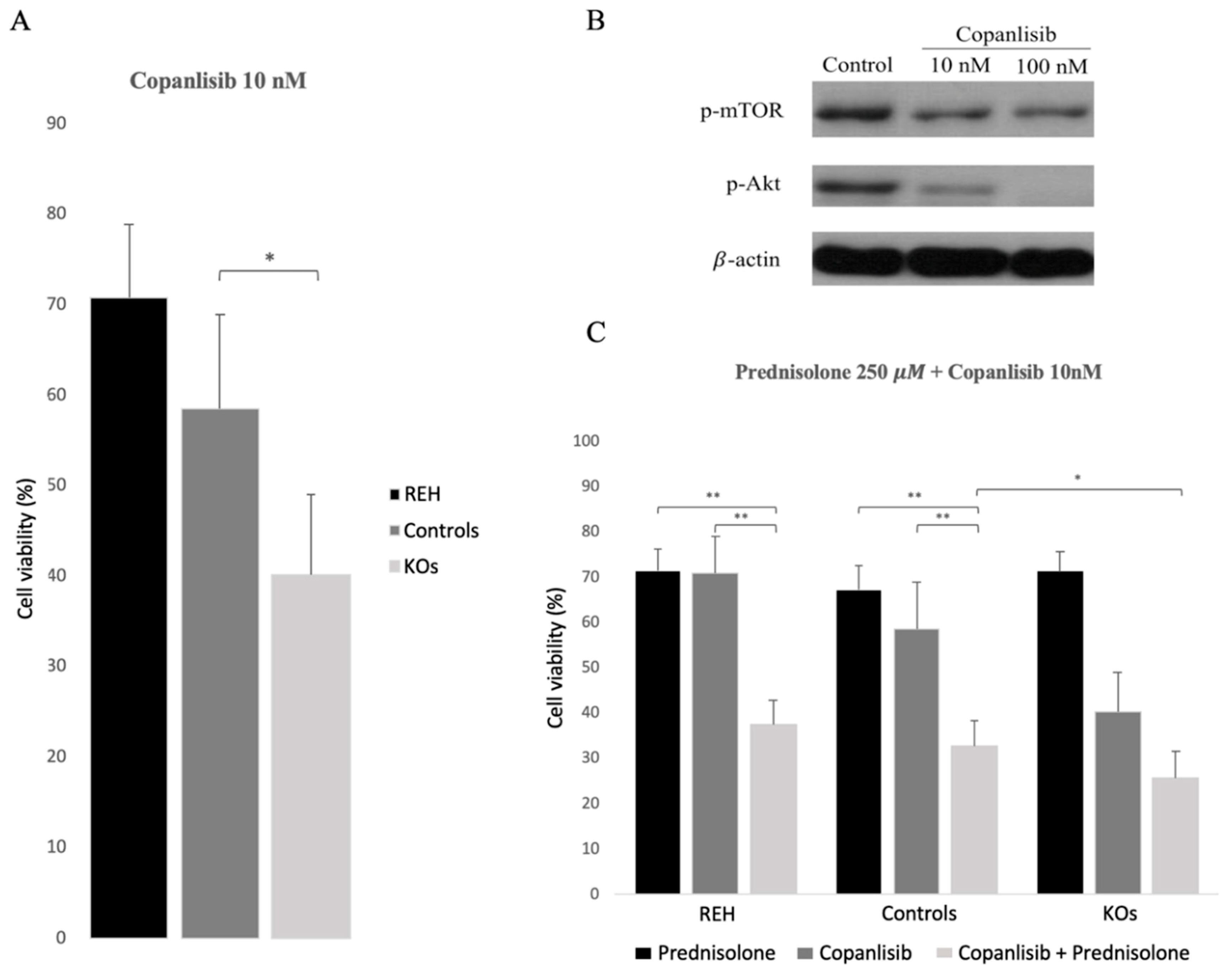
3.4. Abrogation of ETV6/RUNX1 Expression Enhances Sensitivity to the PI3K Inhibitor Copanlisib
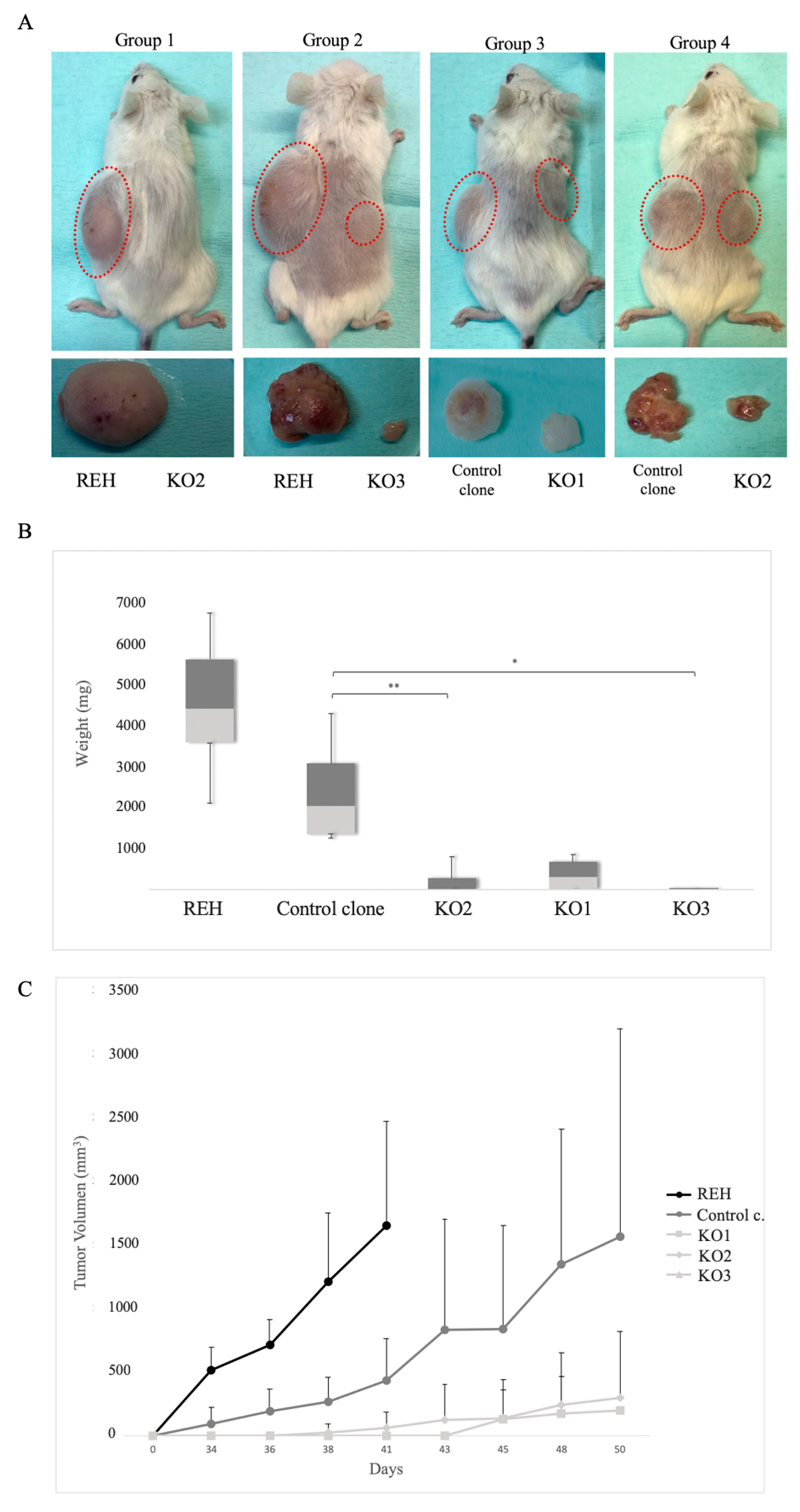
3.5. E/R Repression Impairs the Tumourigenicity In vivo
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pui, C.H.; Relling, M.V.; Downing, J.R. Acute lymphoblastic leukemia. N. Engl. J. Med. 2004, 350, 1535–1548. [Google Scholar]
- Shurtleff, S.A.; Buijs, A.; Behm, F.G.; Rubnitz, J.E.; Raimondi, S.C.; Hancock, M.L.; Chan, G.C.; Pui, C.H.; Grosveld, G.; Downing, J.R. TEL/AML1 fusion resulting from a cryptic t(12;21) is the most common genetic lesion in pediatric ALL and defines a subgroup of patients with an excellent prognosis. Leukemia 1995, 9, 1985–1989. [Google Scholar] [PubMed]
- Conter, V.; Bartram, C.R.; Valsecchi, M.G.; Schrauder, A.; Panzer-Grumayer, R.; Moricke, A.; Arico, M.; Zimmermann, M.; Mann, G.; De Rossi, G.; et al. Molecular response to treatment redefines all prognostic factors in children and adolescents with B-cell precursor acute lymphoblastic leukemia: Results in 3184 patients of the AIEOP-BFM ALL 2000 study. Blood 2010, 115, 3206–3214. [Google Scholar] [CrossRef] [PubMed]
- Moorman, A.V.; Ensor, H.M.; Richards, S.M.; Chilton, L.; Schwab, C.; Kinsey, S.E.; Vora, A.; Mitchell, C.D.; Harrison, C.J. Prognostic effect of chromosomal abnormalities in childhood B-cell precursor acute lymphoblastic leukaemia: Results from the UK Medical Research Council ALL97/99 randomised trial. Lancet Oncol. 2010, 11, 429–438. [Google Scholar] [CrossRef]
- Pui, C.-H.; Robison, L.L.; Look, A.T. Acute lymphoblastic leukaemia. Lancet 2008, 371, 1030–1043. [Google Scholar] [CrossRef]
- Rubnitz, J.E.; Downing, J.R.; Pui, C.H. Significance of the TEL-AML fusion gene in childhood AML. Leukemia 1999, 13, 1470–1471. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Uckun, F.M.; Pallisgaard, N.; Hokland, P.; Navara, C.; Narla, R.; Gaynon, P.S.; Sather, H.; Heerema, N. Expression of TEL-AML1 fusion transcripts and response to induction therapy in standard risk acute lymphoblastic leukemia. Leuk. Lymphoma 2001, 42, 41–56. [Google Scholar] [CrossRef]
- Bokemeyer, A.; Eckert, C.; Meyr, F.; Koerner, G.; von Stackelberg, A.; Ullmann, R.; Turkmen, S.; Henze, G.; Seeger, K. Copy number genome alterations are associated with treatment response and outcome in relapsed childhood ETV6/RUNX1-positive acute lymphoblastic leukemia. Haematologica 2014, 99, 706–714. [Google Scholar] [CrossRef]
- Kuster, L.; Grausenburger, R.; Fuka, G.; Kaindl, U.; Krapf, G.; Inthal, A.; Mann, G.; Kauer, M.; Rainer, J.; Kofler, R.; et al. ETV6/RUNX1-positive relapses evolve from an ancestral clone and frequently acquire deletions of genes implicated in glucocorticoid signaling. Blood 2011, 117, 2658–2667. [Google Scholar] [CrossRef]
- Raynaud, S.; Cave, H.; Baens, M.; Bastard, C.; Cacheux, V.; Grosgeorge, J.; Guidal-Giroux, C.; Guo, C.; Vilmer, E.; Marynen, P.; et al. The 12;21 translocation involving TEL and deletion of the other TEL allele: Two frequently associated alterations found in childhood acute lymphoblastic leukemia. Blood 1996, 87, 2891–2899. [Google Scholar] [CrossRef]
- Sun, C.; Chang, L.; Zhu, X. Pathogenesis of ETV6/RUNX1-positive childhood acute lymphoblastic leukemia and mechanisms underlying its relapse. Oncotarget 2017, 8, 35445–35459. [Google Scholar] [CrossRef] [PubMed]
- Zaliova, M.; Madzo, J.; Cario, G.; Trka, J. Revealing the role of TEL/AML1 for leukemic cell survival by RNAi-mediated silencing. Leukemia 2011, 25, 313–320. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Diakos, C.; Krapf, G.; Gerner, C.; Inthal, A.; Lemberger, C.; Ban, J.; Dohnal, A.M.; Panzer-Grumayer, E.R. RNAi-mediated silencing of TEL/AML1 reveals a heat-shock protein and survivin-dependent mechanism for survival. Blood 2007, 109, 2607–2610. [Google Scholar] [CrossRef] [PubMed]
- Fuka, G.; Kantner, H.P.; Grausenburger, R.; Inthal, A.; Bauer, E.; Krapf, G.; Kaindl, U.; Kauer, M.; Dworzak, M.N.; Stoiber, D.; et al. Silencing of ETV6/RUNX1 abrogates PI3K/AKT/mTOR signaling and impairs reconstitution of leukemia in xenografts. Leukemia 2012, 26, 927–933. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Tunon, I.; Hernandez-Sanchez, M.; Ordonez, J.L.; Alonso-Perez, V.; Alamo-Quijada, M.; Benito, R.; Guerrero, C.; Hernandez-Rivas, J.M.; Sanchez-Martin, M. The CRISPR/Cas9 system efficiently reverts the tumorigenic ability of BCR/ABL in vitro and in a xenograft model of chronic myeloid leukemia. Oncotarget 2017, 8, 26027–26040. [Google Scholar] [CrossRef]
- Martinez, N.; Drescher, B.; Riehle, H.; Cullmann, C.; Vornlocher, H.P.; Ganser, A.; Heil, G.; Nordheim, A.; Krauter, J.; Heidenreich, O. The oncogenic fusion protein RUNX1–CBFA2T1 supports proliferation and inhibits senescence in t(8;21)-positive leukaemic cells. BMC Cancer 2004, 4, 44. [Google Scholar] [CrossRef]
- Supek, F.; Bosnjak, M.; Skunca, N.; Smuc, T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef]
- Abramoff, M.D.; Magalhães, P.J.; Ram, S.J. Image Processing with ImageJ. Biophotonics Int. 2004, 11, 36–42. [Google Scholar]
- Collins, T.J. ImageJ for microscopy. Biotechniques 2007, 43, 25–30. [Google Scholar] [CrossRef]
- Ordonez, J.L.; Amaral, A.T.; Carcaboso, A.M.; Herrero-Martin, D.; del Carmen Garcia-Macias, M.; Sevillano, V.; Alonso, D.; Pascual-Pasto, G.; San-Segundo, L.; Vila-Ubach, M.; et al. The PARP inhibitor olaparib enhances the sensitivity of Ewing sarcoma to trabectedin. Oncotarget 2015, 6, 18875–18890. [Google Scholar] [CrossRef]
- Brownlie, R.J.; Zamoyska, R. T cell receptor signalling networks: Branched, diversified and bounded. Nat. Rev. Immunol. 2013, 13, 257–269. [Google Scholar] [CrossRef]
- Corthals, S.L.; Wynne, K.; She, K.; Shimizu, H.; Curman, D.; Garbutt, K.; Reid, G.S. Differential immune effects mediated by Toll-like receptors stimulation in precursor B-cell acute lymphoblastic leukaemia. Br. J. Haematol. 2006, 132, 452–458. [Google Scholar] [CrossRef]
- Melo, R.C.C.; Longhini, A.L.; Bigarella, C.L.; Baratti, M.O.; Traina, F.; Favaro, P.; de Melo Campos, P.; Saad, S.T. CXCR7 is highly expressed in acute lymphoblastic leukemia and potentiates CXCR4 response to CXCL12. PLoS ONE 2014, 9, e85926. [Google Scholar] [CrossRef]
- Polak, R.; Bierings, M.B.; van der Leije, C.S.; Sanders, M.A.; Roovers, O.; Marchante, J.R.M.; Boer, J.M.; Cornelissen, J.J.; Pieters, R.; den Boer, M.L.; et al. Autophagy inhibition as a potential future targeted therapy for ETV6-RUNX1-driven B-cell precursor acute lymphoblastic leukemia. Haematologica 2019, 104, 738–748. [Google Scholar] [CrossRef]
- Ross, M.E.; Zhou, X.; Song, G.; Shurtleff, S.A.; Girtman, K.; Williams, W.K.; Liu, H.C.; Mahfouz, R.; Raimondi, S.C.; Lenny, N.; et al. Classification of pediatric acute lymphoblastic leukemia by gene expression profiling. Blood 2003, 102, 2951–2959. [Google Scholar] [CrossRef]
- Yeoh, E.J.; Ross, M.E.; Shurtleff, S.A.; Williams, W.K.; Patel, D.; Mahfouz, R.; Behm, F.G.; Raimondi, S.C.; Relling, M.V.; Patel, A.; et al. Classification, subtype discovery, and prediction of outcome in pediatric acute lymphoblastic leukemia by gene expression profiling. Cancer Cell 2002, 1, 133–143. [Google Scholar] [CrossRef]
- Bauer, E.; Schlederer, M.; Scheicher, R.; Horvath, J.; Aigner, P.; Schiefer, A.I.; Kain, R.; Regele, H.; Hoermann, G.; Steiner, G.; et al. Cooperation of ETV6/RUNX1 and BCL2 enhances immunoglobulin production and accelerates glomerulonephritis in transgenic mice. Oncotarget 2016, 7, 12191–12205. [Google Scholar] [CrossRef][Green Version]
- Torrano, V.; Procter, J.; Cardus, P.; Greaves, M.; Ford, A.M. ETV6-RUNX1 promotes survival of early B lineage progenitor cells via a dysregulated erythropoietin receptor. Blood 2011, 118, 4910–4918. [Google Scholar] [CrossRef]
- Krause, G.; Hassenruck, F.; Hallek, M. Copanlisib for treatment of B-cell malignancies: The development of a PI3K inhibitor with considerable differences to idelalisib. Drug Des. Dev. Therapy 2018, 12, 2577–2590. [Google Scholar] [CrossRef]
- Liu, N.; Rowley, B.R.; Bull, C.O.; Schneider, C.; Haegebarth, A.; Schatz, C.A.; Fracasso, P.R.; Wilkie, D.P.; Hentemann, M.; Wilhelm, S.M.; et al. BAY 80–6946 is a highly selective intravenous PI3K inhibitor with potent p110alpha and p110delta activities in tumor cell lines and xenograft models. Mol. Cancer Ther. 2013, 12, 2319–2330. [Google Scholar] [CrossRef]
- Montano, A.; Forero-Castro, M.; Hernandez-Rivas, J.M.; Garcia-Tunon, I.; Benito, R. Targeted genome editing in acute lymphoblastic leukemia: A review. BMC Biotechnol. 2018, 18, 45. [Google Scholar] [CrossRef] [PubMed]
- Culbertson, M.R.; Leeds, P.F. Looking at mRNA decay pathways through the window of molecular evolution. Curr. Opin. Genet. Dev. 2003, 13, 207–214. [Google Scholar] [CrossRef]
- Fuka, G.; Kauer, M.; Kofler, R.; Haas, O.A.; Panzer-Grumayer, R. The leukemia-specific fusion gene ETV6/RUNX1 perturbs distinct key biological functions primarily by gene repression. PLoS ONE 2011, 6, e26348. [Google Scholar] [CrossRef] [PubMed]
- Hiebert, S.W.; Sun, W.; Davis, J.N.; Golub, T.; Shurtleff, S.; Buijs, A.; Downing, J.R.; Grosveld, G.; Roussell, M.F.; Gilliland, D.G.; et al. The t(12;21) translocation converts AML-1B from an activator to a repressor of transcription. Mol. Cell. Biol. 1996, 16, 1349–1355. [Google Scholar] [CrossRef]
- Tijssen, M.R.; Cvejic, A.; Joshi, A.; Hannah, R.L.; Ferreira, R.; Forrai, A.; Bellissimo, D.C.; Oram, S.H.; Smethurst, P.A.; Wilson, N.K.; et al. Genome-wide analysis of simultaneous GATA1/2, RUNX1, FLI1, and SCL binding in megakaryocytes identifies hematopoietic regulators. Dev. Cell 2011, 20, 597–609. [Google Scholar] [CrossRef]
- Inthal, A.; Krapf, G.; Beck, D.; Joas, R.; Kauer, M.O.; Orel, L.; Fuka, G.; Mann, G.; Panzer-Grumayer, E.R. Role of the erythropoietin receptor in ETV6/RUNX1-positive acute lymphoblastic leukemia. Clin. Cancer Res. 2008, 14, 7196–7204. [Google Scholar] [CrossRef]
- Liang, G.; Bansal, G.; Xie, Z.; Druey, K.M. RGS16 inhibits breast cancer cell growth by mitigating phosphatidylinositol 3-kinase signaling. J. Biol. Chem. 2009, 284, 21719–21727. [Google Scholar] [CrossRef]
- Liu, T.; Bohlken, A.; Kuljaca, S.; Lee, M.; Nguyen, T.; Smith, S.; Cheung, B.; Norris, M.D.; Haber, M.; Holloway, A.J.; et al. The retinoid anticancer signal: Mechanisms of target gene regulation. Br. J. Cancer 2005, 93, 310–318. [Google Scholar] [CrossRef]
- Vasilatos, S.N.; Katz, T.A.; Oesterreich, S.; Wan, Y.; Davidson, N.E.; Huang, Y. Crosstalk between lysine-specific demethylase 1 (LSD1) and histone deacetylases mediates antineoplastic efficacy of HDAC inhibitors in human breast cancer cells. Carcinogenesis 2013, 34, 1196–1207. [Google Scholar] [CrossRef]
- Berthebaud, M.; Riviere, C.; Jarrier, P.; Foudi, A.; Zhang, Y.; Compagno, D.; Galy, A.; Vainchenker, W.; Louache, F. RGS16 is a negative regulator of SDF-1-CXCR4 signaling in megakaryocytes. Blood 2005, 106, 2962–2968. [Google Scholar] [CrossRef]
- Genitsari, S.; Stiakaki, E.; Perdikogianni, C.; Martimianaki, G.; Pelagiadis, I.; Pesmatzoglou, M.; Kalmanti, M.; Dimitriou, H. Biological Features of Bone Marrow Mesenchymal Stromal Cells in Childhood Acute Lymphoblastic Leukemia. Turk. J. Haematol. Off. J. Turk. Soc. Haematol. 2018, 35, 19–26. [Google Scholar] [CrossRef]
- Nwabo Kamdje, A.H.; Kamga, P.T.; Simo, R.T.; Vecchio, L.; Seke Etet, P.F.; Muller, J.M.; Bassi, G.; Lukong, E.; Goel, R.K.; Amvene, J.M.; et al. Mesenchymal stromal cells’ role in tumor microenvironment: Involvement of signaling pathways. Cancer Biol. Med. 2017, 14, 129–141. [Google Scholar] [CrossRef]
- Bonilla, X.; Vanegas, N.P.; Vernot, J.P. Acute Leukemia Induces Senescence and Impaired Osteogenic Differentiation in Mesenchymal Stem Cells Endowing Leukemic Cells with Functional Advantages. Stem Cells Int. 2019, 2019, 3864948. [Google Scholar] [CrossRef]
- Yan, W.; Guo, H.; Suo, F.; Han, C.; Zheng, H.; Chen, T. The effect of miR-146a on STAT1 expression and apoptosis in acute lymphoblastic leukemia Jurkat cells. Oncol. Lett. 2017, 13, 151–154. [Google Scholar] [CrossRef]
- Binsky, I.; Lantner, F.; Grabovsky, V.; Harpaz, N.; Shvidel, L.; Berrebi, A.; Goldenberg, D.M.; Leng, L.; Bucala, R.; Alon, R.; et al. TAp63 regulates VLA-4 expression and chronic lymphocytic leukemia cell migration to the bone marrow in a CD74-dependent manner. J. Immunol. 2010, 184, 4761–4769. [Google Scholar] [CrossRef]
- Lantner, F.; Starlets, D.; Gore, Y.; Flaishon, L.; Yamit-Hezi, A.; Dikstein, R.; Leng, L.; Bucala, R.; Machluf, Y.; Oren, M.; et al. CD74 induces TAp63 expression leading to B-cell survival. Blood 2007, 110, 4303–4311. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Montaño, A.; Ordoñez, J.L.; Alonso-Pérez, V.; Hernández-Sánchez, J.; Santos, S.; González, T.; Benito, R.; García-Tuñón, I.; Hernández-Rivas, J.M. ETV6/RUNX1 Fusion Gene Abrogation Decreases the Oncogenicity of Tumour Cells in a Preclinical Model of Acute Lymphoblastic Leukaemia. Cells 2020, 9, 215. https://doi.org/10.3390/cells9010215
Montaño A, Ordoñez JL, Alonso-Pérez V, Hernández-Sánchez J, Santos S, González T, Benito R, García-Tuñón I, Hernández-Rivas JM. ETV6/RUNX1 Fusion Gene Abrogation Decreases the Oncogenicity of Tumour Cells in a Preclinical Model of Acute Lymphoblastic Leukaemia. Cells. 2020; 9(1):215. https://doi.org/10.3390/cells9010215
Chicago/Turabian StyleMontaño, Adrián, Jose Luis Ordoñez, Verónica Alonso-Pérez, Jesús Hernández-Sánchez, Sandra Santos, Teresa González, Rocío Benito, Ignacio García-Tuñón, and Jesús María Hernández-Rivas. 2020. "ETV6/RUNX1 Fusion Gene Abrogation Decreases the Oncogenicity of Tumour Cells in a Preclinical Model of Acute Lymphoblastic Leukaemia" Cells 9, no. 1: 215. https://doi.org/10.3390/cells9010215
APA StyleMontaño, A., Ordoñez, J. L., Alonso-Pérez, V., Hernández-Sánchez, J., Santos, S., González, T., Benito, R., García-Tuñón, I., & Hernández-Rivas, J. M. (2020). ETV6/RUNX1 Fusion Gene Abrogation Decreases the Oncogenicity of Tumour Cells in a Preclinical Model of Acute Lymphoblastic Leukaemia. Cells, 9(1), 215. https://doi.org/10.3390/cells9010215