Trehalose Alleviates Crystalline Silica-Induced Pulmonary Fibrosis via Activation of the TFEB-Mediated Autophagy-Lysosomal System in Alveolar Macrophages

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Antibodies and Other Reagents
2.2. Animals and Treatment
2.3. Isolation of Primary Alveolar Macrophages (AMs)
2.4. Cell Culture and Treatment
2.5. Lentiviral Short Hairpin RNA (shRNA) Transfection
2.6. Cell Viability Assay
2.7. mRFP-GFP-LC3 Transfection
2.8. Lysotracker Red and Acridine Orange Staining
2.9. Enzyme-Linked Immunosorbent Assay (ELISA)
2.10. Immunoblotting
2.11. Immunofluorescence
2.12. Quantitative PCR (qPCR)
2.13. Histology and Immunohistochemistry
2.14. TUNEL Assay
2.15. Statistical Analysis
3. Results
3.1. CS Induces TFEB Nuclear Localization and Increases TFEB Expression In Vivo
3.2. TFEB Modulates CS-Induced Macrophages Apoptosis and Secretion of Inflammatory Cytokines In Vitro
3.3. TFEB Overexpression Abrogates CS-Induced Macrophage Apoptosis and Secretion of Inflammatory Cytokines via Regulation of the Autophagy-Lysosome Pathway
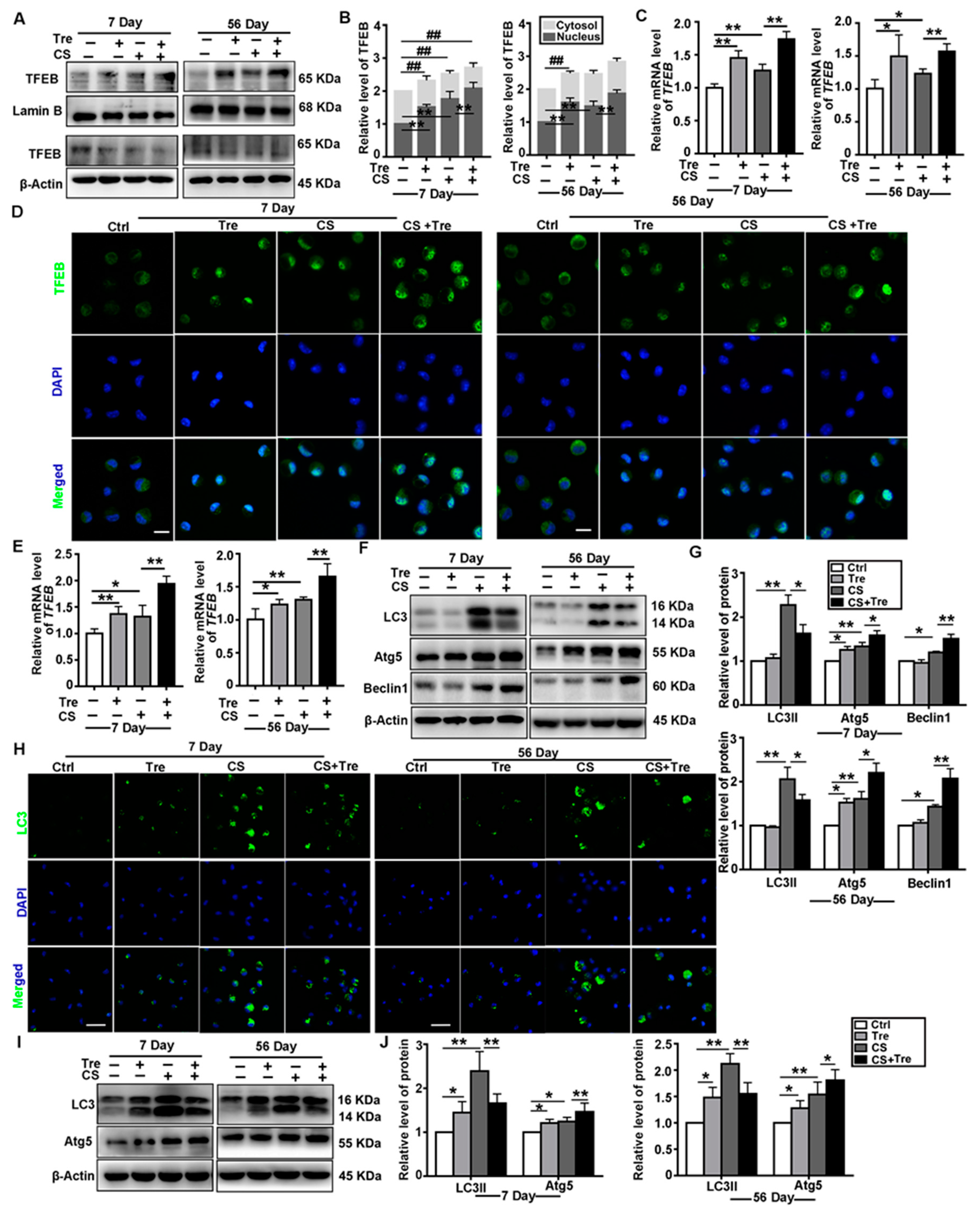
3.4. Tre Activates TFEB and Affects Autophagy-Associated Proteins in a Silicosis Mouse Model
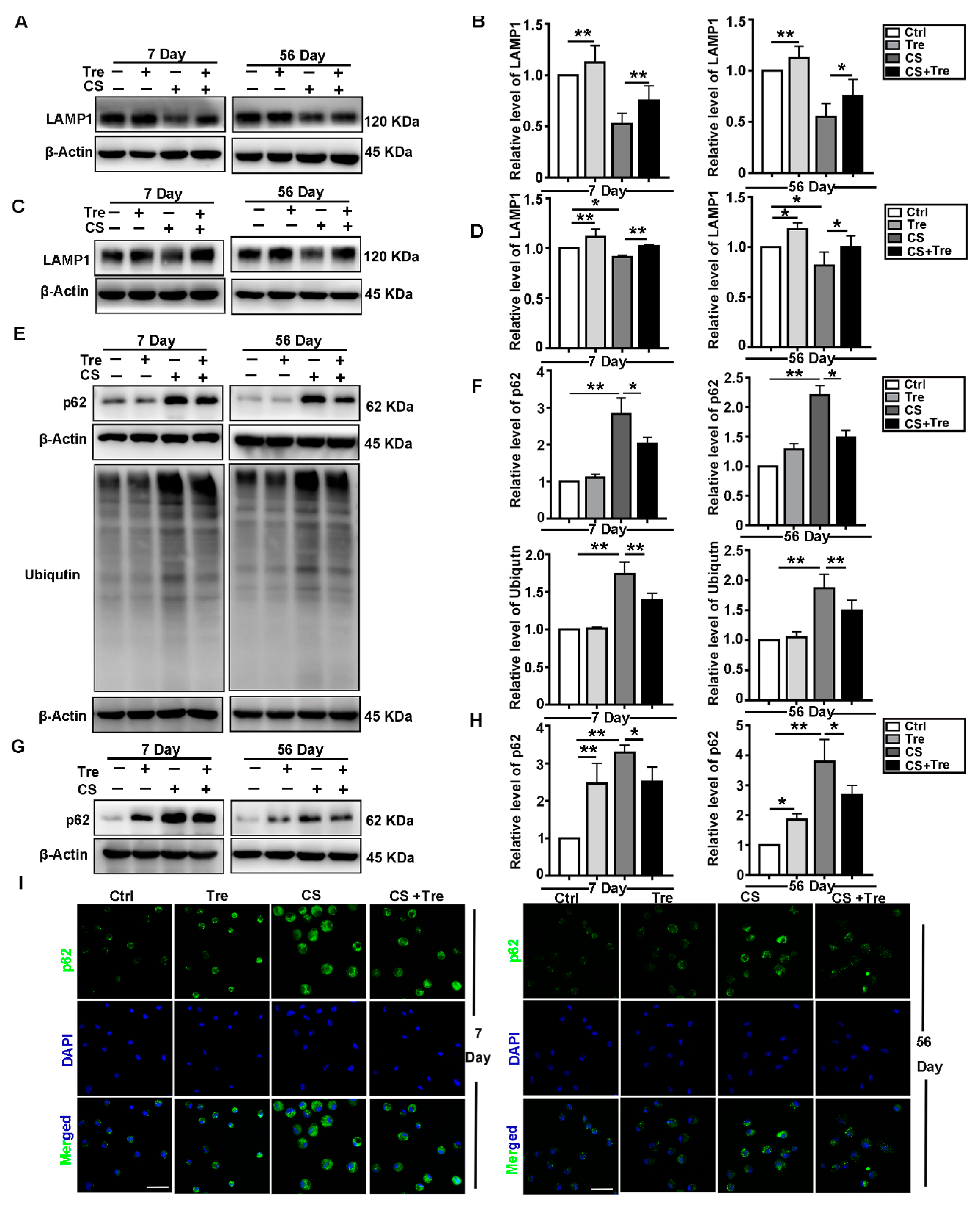
3.5. Tre Relieves CS-Induced Lysosome Damage and Disorder of Autophagic Substrates Degradation In Vivo
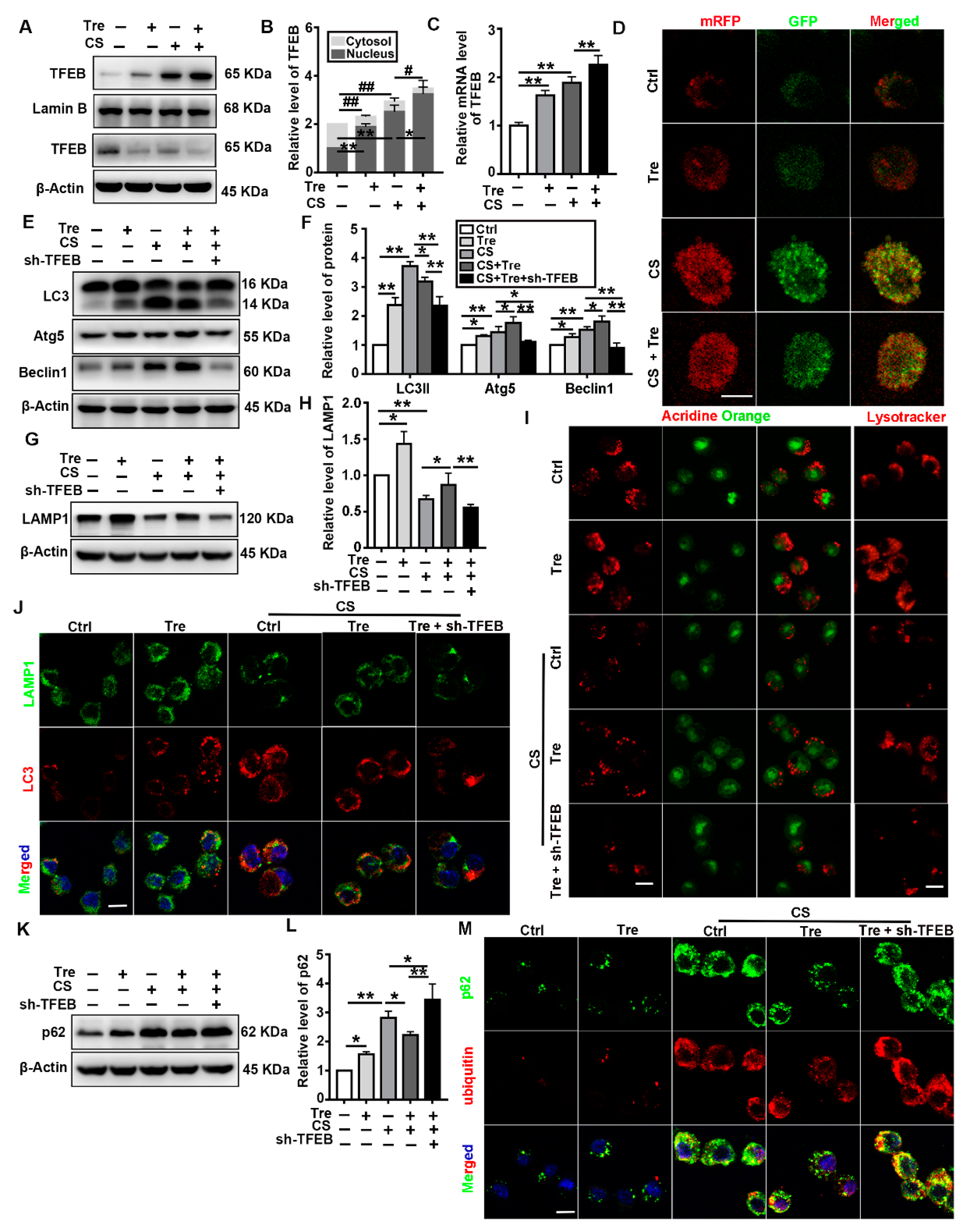
3.6. Tre Activates TFEB and Relieves CS-Induced Lysosomes Damage and Disorder of Autophagic Substrate Degradation through TFEB Activation In Vitro
3.7. Tre Relieves CS-Induced Apoptosis and Inflammation through TFEB Activation
3.8. Tre Relieves Pulmonary Fibrosis in Silicosis Model Mice
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Leung, C.C.; Yu, I.T.S.; Chen, W. Silicosis. Lancet 2012, 379, 2008–2018. [Google Scholar] [CrossRef]
- Li, C.; Du, S.; Lu, Y.; Lu, X.; Liu, F.; Chen, Y.; Weng, D.; Chen, J. Blocking the 4-1BB Pathway Ameliorates Crystalline Silica-induced Lung Inflammation and Fibrosis in Mice. Theranostics 2016, 6, 2052–2067. [Google Scholar] [CrossRef] [PubMed]
- The Lancet Respiratory Medicine. The world is failing on silicosis. Lancet Respir. Med. 2019, 7, 283. [Google Scholar] [CrossRef]
- Hamilton, R.F., Jr.; Thakur, S.A.; Holian, A. Silica binding and toxicity in alveolar macrophages. Free Radic. Biol. Med. 2008, 44, 1246–1258. [Google Scholar] [CrossRef] [PubMed]
- Hughes, C.S.; Colhoun, L.M.; Bains, B.K.; Kilgour, J.D.; Burden, R.E.; Burrows, J.F.; Lavelle, E.C.; Gilmore, B.F.; Scott, C.J. Extracellular cathepsin S and intracellular caspase 1 activation are surrogate biomarkers of particulate-induced lysosomal disruption in macrophages. Part. Fibre Toxicol. 2015, 13, 19. [Google Scholar] [CrossRef]
- Hornung, V.; Bauernfeind, F.; Halle, A.; Samstad, E.O.; Kono, H.; Rock, K.L.; Fitzgerald, K.A.; Latz, E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 2008, 9, 847–856. [Google Scholar] [CrossRef]
- Yao, S.Q.; Rojanasakul, L.W.; Chen, Z.Y.; Xu, Y.J.; Bai, Y.P.; Chen, G.; Zhang, X.Y.; Zhang, C.M.; Yu, Y.Q.; Shen, F.H.; et al. Fas/FasL pathway-mediated alveolar macrophage apoptosis involved in human silicosis. Apoptosis 2011, 16, 1195. [Google Scholar] [CrossRef]
- Huaux, F. New developments in the understanding of immunology in silicosis. Curr. Opin. Allergy Clin. Immunol. 2007, 7, 168–173. [Google Scholar] [CrossRef]
- Lopes-Pacheco, M.; Bandeira, E.; Morales, M.M. Cell-Based Therapy for Silicosis. Stem Cells Int. 2016, 2016, 5091838. [Google Scholar] [CrossRef]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef]
- Yang, Z.; Klionsky, D.J. Mammalian autophagy: Core molecular machinery and signaling regulation. Curr. Opin. Cell Biol. 2010, 22, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Biswas, R.; Trout, K.L.; Jessop, F.; Harkema, J.R.; Holian, A. Imipramine blocks acute silicosis in a mouse model. Part. Fibre Toxicol. 2017, 14, 36. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Yuan, J.; Yao, S.; Jin, Y.; Chen, G.; Tian, W.; Xi, J.; Xu, Z.; Weng, D.; Chen, J. Lipopolysaccharides may aggravate apoptosis through accumulation of autophagosomes in alveolar macrophages of human silicosis. Autophagy 2015, 11, 2346–2357. [Google Scholar] [CrossRef] [PubMed]
- Jessop, F.; Hamilton, R.F.; Rhoderick, J.F.; Shaw, P.K.; Holian, A. Autophagy deficiency in macrophages enhances NLRP3 inflammasome activity and chronic lung disease following silica exposure. Toxicol. Appl. Pharmacol. 2016, 309, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Du, S.; Li, C.; Lu, Y.; Lei, X.; Zhang, Y.; Li, S.; Liu, F.; Chen, Y.; Weng, D.; Chen, J. Dioscin Alleviates Crystalline Silica-Induced Pulmonary Inflammation and Fibrosis through Promoting Alveolar Macrophage Autophagy. Theranostics 2019, 9, 1878–1892. [Google Scholar] [CrossRef] [PubMed]
- Han, R.; Ji, X.; Rong, R.; Li, Y.; Yao, W.; Yuan, J.; Wu, Q.; Yang, J.; Yan, W.; Han, L.; et al. MiR-449a regulates autophagy to inhibit silica-induced pulmonary fibrosis through targeting Bcl2. J. Mol. Med. 2016, 94, 1267–1279. [Google Scholar] [CrossRef]
- Napolitano, G.; Ballabio, A. TFEB at a glance. J. Cell Sci. 2016, 129, 2475–2481. [Google Scholar] [CrossRef]
- Sardiello, M.; Palmieri, M.; di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A gene network regulating lysosomal biogenesis and function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Garcia Arencibia, M.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB links autophagy to lysosomal biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef]
- Jia, D.; Wang, Y.Y.; Wang, P.; Huang, Y.; Liang, D.Y.; Wang, D.; Cheng, C.; Zhang, C.; Guo, L.; Liang, P.; et al. SVIP alleviates CCl4-induced liver fibrosis via activating autophagy and protecting hepatocytes. Cell Death Dis. 2019, 10, 71. [Google Scholar] [CrossRef]
- Kim, S.H.; Kim, G.; Han, D.H.; Lee, M.; Kim, I.; Kim, B.; Kim, K.H.; Song, Y.M.; Yoo, J.E.; Wang, H.J.; et al. Ezetimibe ameliorates steatohepatitis via AMP activated protein kinase-TFEB-mediated activation of autophagy and NLRP3 inflammasome inhibition. Autophagy 2017, 13, 1767–1781. [Google Scholar] [CrossRef] [PubMed]
- Emanuele, E. Can trehalose prevent neurodegeneration? Insights from experimental studies. Curr. Drug Targets 2014, 15, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Evans, T.D.; Jeong, S.J.; Zhang, X.; Sergin, I.; Razani, B. TFEB and trehalose drive the macrophage autophagy-lysosome system to protect against atherosclerosis. Autophagy 2018, 14, 724–726. [Google Scholar] [CrossRef] [PubMed]
- Sergin, I.; Evans, T.D.; Zhang, X.; Bhattacharya, S.; Stokes, C.J.; Song, E.; Ali, S.; Dehestani, B.; Holloway, K.B.; Micevych, P.S.; et al. Exploiting macrophage autophagy-lysosomal biogenesis as a therapy for atherosclerosis. Nat. Commun. 2017, 8, 15750. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Li, C.; Zhang, Y.; He, X.; Chen, X.; Zeng, X.; Liu, F.; Chen, Y.; Chen, J. Targeting Mechanics-Induced Fibroblast Activation through CD44-RhoA-YAP Pathway Ameliorates Crystalline Silica-Induced Silicosis. Theranostics 2019, 9, 4993–5008. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Lu, Y.; Du, S.; Li, S.; Zhang, Y.; Liu, F.; Chen, Y.; Weng, D.; Chen, J. Dioscin Exerts Protective Effects Against Crystalline Silica-induced Pulmonary Fibrosis in Mice. Theranostics 2017, 7, 4255–4275. [Google Scholar] [CrossRef] [PubMed]
- Tai, H.; Wang, Z.; Gong, H.; Han, X.; Zhou, J.; Wang, X.; Wei, X.; Ding, Y.; Huang, N.; Qin, J.; et al. Autophagy impairment with lysosomal and mitochondrial dysfunction is an important characteristic of oxidative stress-induced senescence. Autophagy 2017, 13, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Y.; Yang, H.; Wang, M.G.; Yang, D.B.; Wang, Z.Y.; Wang, L. Trehalose protects against cadmium-induced cytotoxicity in primary rat proximal tubular cells via inhibiting apoptosis and restoring autophagic flux. Cell Death Dis. 2017, 8, e3099. [Google Scholar] [CrossRef]
- Lu, Y.; Li, C.; Du, S.; Chen, X.; Zeng, X.; Liu, F.; Chen, Y.; Chen, J. 4-1BB Signaling Promotes Alveolar Macrophages-Mediated Pro-Fibrotic Responses and Crystalline Silica-Induced Pulmonary Fibrosis in Mice. Front. Immunol. 2018, 9, 1848. [Google Scholar] [CrossRef]
- Aflaki, E.; Moaven, N.; Borger, D.K.; Lopez, G.; Westbroek, W.; Chae, J.J.; Marugan, J.; Patnaik, S.; Maniwang, E.; Gonzalez, A.N.; et al. Lysosomal storage and impaired autophagy lead to inflammasome activation in Gaucher macrophages. Aging Cell 2016, 15, 77–88. [Google Scholar] [CrossRef]
- Puertollano, R.; Ferguson, S.M.; Brugarolas, J.; Ballabio, A. The complex relationship between TFEB transcription factor phosphorylation and subcellular localization. EMBO J. 2018, 37, e98804. [Google Scholar] [CrossRef] [PubMed]
- Delbridge, A.R.; Grabow, S.; Strasser, A.; Vaux, D.L. Thirty years of BCL-2: Translating cell death discoveries into novel cancer therapies. Nat. Rev. Cancer 2016, 16, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Lie, P.P.; Nixon, R.A. Lysosome trafficking and signaling in health and neurodegenerative diseases. Neurobiol. Dis. 2019, 122, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Sahebkar, A.; Hatamipour, M.; Tabatabaei, S.A. Trehalose administration attenuates atherosclerosis in rabbits fed a high-fat diet. J. Cell. Biochem. 2019, 120, 9455–9459. [Google Scholar] [CrossRef] [PubMed]
- Khalifeh, M.; Barreto, G.E.; Sahebkar, A. Trehalose as a promising therapeutic candidate for the treatment of Parkinson’s disease. Br. J. Pharmacol. 2019, 176, 1173–1189. [Google Scholar] [CrossRef] [PubMed]
- Lotfi, P.; Tse, D.Y.; Di Ronza, A.; Seymour, M.L.; Martano, G.; Cooper, J.D.; Pereira, F.A.; Passafaro, M.; Wu, S.M.; Sardiello, M. Trehalose reduces retinal degeneration, neuroinflammation and storage burden caused by a lysosomal hydrolase deficiency. Autophagy 2018, 14, 1419–1434. [Google Scholar] [CrossRef] [PubMed]
- Cortes, C.J.; La Spada, A.R. The many faces of autophagy dysfunction in Huntington’s disease: From mechanism to therapy. Drug Discov. Today 2014, 19, 963–971. [Google Scholar] [CrossRef]
- Huynh, K. Atherosclerosis: Trehalose induces macrophage autophagy-lysosomal biogenesis. Nat. Rev. Cardiol. 2017, 14, 444. [Google Scholar] [CrossRef]
- Sciarretta, S.; Yee, D.; Nagarajan, N.; Bianchi, F.; Saito, T.; Valenti, V.; Tong, M.; Del Re, D.P.; Vecchione, C.; Schirone, L.; et al. Trehalose-Induced Activation of Autophagy Improves Cardiac Remodeling After Myocardial Infarction. J. Am. Coll. Cardiol. 2018, 71, 1999–2010. [Google Scholar] [CrossRef]
- Rusmini, P.; Cortese, K.; Crippa, V.; Cristofani, R.; Cicardi, M.E.; Ferrari, V.; Vezzoli, G.; Tedesco, B.; Meroni, M.; Messi, E.; et al. Trehalose induces autophagy via lysosomal-mediated TFEB activation in models of motoneuron degeneration. Autophagy 2019, 15, 631–651. [Google Scholar] [CrossRef]
- Wu, H.; Chen, H.; Zheng, Z.; Li, J.; Ding, J.; Huang, Z.; Jia, C.; Shen, Z.; Bao, G.; Wu, L.; et al. Trehalose promotes the survival of random-pattern skin flaps by TFEB mediated autophagy enhancement. Cell Death Dis. 2019, 10, 483. [Google Scholar] [CrossRef] [PubMed]
- Messer, J.S. The cellular autophagy/apoptosis checkpoint during inflammation. Cell. Mol. Life Sci. 2017, 74, 1281–1296. [Google Scholar] [CrossRef] [PubMed]
- Mejias-Pena, Y.; Estebanez, B.; Rodriguez-Miguelez, P.; Fernandez-Gonzalo, R.; Almar, M.; de Paz, J.A.; Gonzalez-Gallego, J.; Cuevas, M.J. Impact of resistance training on the autophagy-inflammation-apoptosis crosstalk in elderly subjects. Aging 2017, 9, 408. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; An, Y.; Zhang, X.; Wang, Z.; Duan, H. Experimental pulmonary fibrosis was suppressed by microRNA-506 through NF-kappa-mediated apoptosis and inflammation. Cell Tissue Res. 2019, 378, 255–265. [Google Scholar] [CrossRef]
- Di Malta, C.; Cinque, L.; Settembre, C. Transcriptional Regulation of Autophagy: Mechanisms and Diseases. Front. Cell Dev. Biol. 2019, 7, 114. [Google Scholar] [CrossRef]
- Raben, N.; Puertollano, R. TFEB and TFE3: Linking Lysosomes to Cellular Adaptation to Stress. Annu. Rev. Cell Dev. Biol. 2016, 32, 255–278. [Google Scholar] [CrossRef]
- Martina, J.A.; Diab, H.I.; Brady, O.A.; Puertollano, R. TFEB and TFE3 are novel components of the integrated stress response. EMBO J. 2016, 35, 479–495. [Google Scholar] [CrossRef]
- Stein, B.L.; Wexler, M.J. Preoperative parathyroid localization: A prospective evaluation of ultrasonography and thallium-technetium scintigraphy in hyperparathyroidism. J. Can. Chir. 1990, 33, 175–180. [Google Scholar]
- Thibodeau, M.S.; Giardina, C.; Knecht, D.A.; Helble, J.; Hubbard, A.K. Silica-induced apoptosis in mouse alveolar macrophages is initiated by lysosomal enzyme activity. Toxicol. Sci. 2004, 80, 34–48. [Google Scholar] [CrossRef]
- Settembre, C.; Zoncu, R.; Medina, D.L.; Vetrini, F.; Erdin, S.; Erdin, S.; Huynh, T.; Ferron, M.; Karsenty, G.; Vellard, M.C.; et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012, 31, 1095–1108. [Google Scholar] [CrossRef]
- Pastore, N.; Brady, O.A.; Diab, H.I.; Martina, J.A.; Sun, L.; Huynh, T.; Lim, J.A.; Zare, H.; Raben, N.; Ballabio, A.; et al. TFEB and TFE3 cooperate in the regulation of the innate immune response in activated macrophages. Autophagy 2016, 12, 1240–1258. [Google Scholar] [CrossRef] [PubMed]
- Emanuel, R.; Sergin, I.; Bhattacharya, S.; Turner, J.; Epelman, S.; Settembre, C.; Diwan, A.; Ballabio, A.; Razani, B. Induction of lysosomal biogenesis in atherosclerotic macrophages can rescue lipid-induced lysosomal dysfunction and downstream sequelae. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1942–1952. [Google Scholar] [CrossRef] [PubMed]
- Arguelles, J.C. Why can’t vertebrates synthesize trehalose? J. Mol. Evol. 2014, 79, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Ohtake, S.; Wang, Y.J. Trehalose: Current use and future applications. J. Pharm. Sci. 2011, 100, 2020–2053. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, M.; Pal, R.; Nelvagal, H.R.; Lotfi, P.; Stinnett, G.R.; Seymour, M.L.; Chaudhury, A.; Bajaj, L.; Bondar, V.V.; Bremner, L.; et al. mTORC1-independent TFEB activation via Akt inhibition promotes cellular clearance in neurodegenerative storage diseases. Nat. Commun. 2017, 8, 14338. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef]
- Levine, B.; Klionsky, D.J. Development by self-digestion: Molecular mechanisms and biological functions of autophagy. Dev. Cell 2004, 6, 463–477. [Google Scholar] [CrossRef]
- Liu, H.; Cheng, Y.; Yang, J.; Wang, W.; Fang, S.; Zhang, W.; Han, B.; Zhou, Z.; Yao, H.; Chao, J.; et al. BBC3 in macrophages promoted pulmonary fibrosis development through inducing autophagy during silicosis. Cell Death Dis. 2017, 8, e2657. [Google Scholar] [CrossRef]
- Liu, H.; Fang, S.; Wang, W.; Cheng, Y.; Zhang, Y.; Liao, H.; Yao, H.; Chao, J. Macrophage-derived MCPIP1 mediates silica-induced pulmonary fibrosis via autophagy. Part. Fibre Toxicol. 2016, 13, 55. [Google Scholar] [CrossRef]
- Hoffmann, A.C.; Minakaki, G.; Menges, S.; Salvi, R.; Savitskiy, S.; Kazman, A.; Vicente Miranda, H.; Mielenz, D.; Klucken, J.; Winkler, J.; et al. Extracellular aggregated alpha synuclein primarily triggers lysosomal dysfunction in neural cells prevented by trehalose. Sci. Rep. 2019, 9, 544. [Google Scholar] [CrossRef]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Levine, B.; Green, D.R.; Kroemer, G. Pharmacological modulation of autophagy: Therapeutic potential and persisting obstacles. Nat. Rev. Drug Discov. 2017, 16, 487–511. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Chen, Y.; Tooze, S.A. Autophagy pathway: Cellular and molecular mechanisms. Autophagy 2018, 14, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.A.; Gollan, B.; Helaine, S. Persistent bacterial infections and persister cells. Nat. Rev. Microbiol. 2017, 15, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C.; Cuervo, A.M.; Ravikumar, B.; Sarkar, S.; Korolchuk, V.; Kaushik, S.; Klionsky, D.J. In search of an “autophagomometer”. Autophagy 2009, 5, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Piguet, P.F.; Collart, M.A.; Grau, G.E.; Sappino, A.P.; Vassalli, P. Requirement of tumour necrosis factor for development of silica-induced pulmonary fibrosis. Nature 1990, 344, 245–247. [Google Scholar] [CrossRef] [PubMed]
- Minutoli, L.; Altavilla, D.; Bitto, A.; Polito, F.; Bellocco, E.; Lagana, G.; Fiumara, T.; Magazu, S.; Migliardo, F.; Venuti, F.S.; et al. Trehalose: A biophysics approach to modulate the inflammatory response during endotoxic shock. Eur. J. Pharmacol. 2008, 589, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Jurkuvenaite, A.; Benavides, G.A.; Komarova, S.; Doran, S.F.; Johnson, M.; Aggarwal, S.; Zhang, J.; Darley-Usmar, V.M.; Matalon, S. Upregulation of autophagy decreases chlorine-induced mitochondrial injury and lung inflammation. Free Radic. Biol. Med. 2015, 85, 83–94. [Google Scholar] [CrossRef]
- Anding, A.L.; Baehrecke, E.H. Cleaning House: Selective Autophagy of Organelles. Dev. Cell 2017, 41, 10–22. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, X.; Chen, S.; Li, C.; Ban, J.; Wei, Y.; He, Y.; Liu, F.; Chen, Y.; Chen, J. Trehalose Alleviates Crystalline Silica-Induced Pulmonary Fibrosis via Activation of the TFEB-Mediated Autophagy-Lysosomal System in Alveolar Macrophages. Cells 2020, 9, 122. https://doi.org/10.3390/cells9010122
He X, Chen S, Li C, Ban J, Wei Y, He Y, Liu F, Chen Y, Chen J. Trehalose Alleviates Crystalline Silica-Induced Pulmonary Fibrosis via Activation of the TFEB-Mediated Autophagy-Lysosomal System in Alveolar Macrophages. Cells. 2020; 9(1):122. https://doi.org/10.3390/cells9010122
Chicago/Turabian StyleHe, Xiu, Shi Chen, Chao Li, Jiaqi Ban, Yungeng Wei, Yangyang He, Fangwei Liu, Ying Chen, and Jie Chen. 2020. "Trehalose Alleviates Crystalline Silica-Induced Pulmonary Fibrosis via Activation of the TFEB-Mediated Autophagy-Lysosomal System in Alveolar Macrophages" Cells 9, no. 1: 122. https://doi.org/10.3390/cells9010122
APA StyleHe, X., Chen, S., Li, C., Ban, J., Wei, Y., He, Y., Liu, F., Chen, Y., & Chen, J. (2020). Trehalose Alleviates Crystalline Silica-Induced Pulmonary Fibrosis via Activation of the TFEB-Mediated Autophagy-Lysosomal System in Alveolar Macrophages. Cells, 9(1), 122. https://doi.org/10.3390/cells9010122