1. Introduction
Ionizing radiation (IR) is used as a treatment for many cancers and is an important diagnostic tool, but it is also a genetic toxicant that can negatively affect various cellular processes [
1]. Fortunately, improvements in the ability to detect and treat cancer have resulted in a 60% survival rate of five years after cancer diagnosis, and about two-thirds of these survivors are receiving radiation therapy. Therefore, the late effect of radiation is an area of clinical interest [
2]. IR also induces factors that are important in modulating mitochondrial biogenesis, which is related to cell survival: mitochondria have also been reported to be the primary target for radiation-induced apoptosis [
3]. In addition, this organelle may play a role in radiation-induced intra- and intercellular signaling [
4,
5]. A subcellular proteomic analysis further revealed that proteins involved in energy metabolism processes are regulated by IR exposure in vivo [
6]. However, the effects of IR on mitochondria have been much less intensively investigated than those on the cell nucleus.
Proper control of mitochondrial DNA (mtDNA) copy numbers is believed to be important for normal cell function [
7]. An increase in the mtDNA copy number after radiation stimulation, termed “mitochondrial polyploidization” [
8], is believed to be a compensatory mechanism or an adaptive response of mitochondria to maintain function in post-irradiated cells and malignantly transformed progeny that survive after radiation exposure [
6,
9]. The benefit of such an increase in the mtDNA copy number after irradiation is currently a matter of debate. In addition, mitochondria play an important role in the regulation of several cellular functions, including stress response, apoptosis, and metabolic processes such as gluconeogenesis and β-oxidation, and are sensitive to radiation and may have radiation damage within hours after exposure [
10].
The increase in mtDNA content after irradiation may thus lead to the overproduction of mitochondrially encoded subunits. Notably, peroxisome proliferator-activated receptor coactivator 1 (PGC-1) is also associated with mitochondrial proliferation/numbers [
11]. PGC-1, a transcriptional coactivator that is essential for mitochondrial biosynthesis, activates genes that regulate energy homeostasis and metabolism. In this context, PGC-1 modifies the metabolic rate and expression of genes involved in gluconeogenesis, fat oxidation, and mitochondrial biosynthesis [
12]. PGC-1 also plays a key role in the oxidative metabolism of brown fat and muscle by increasing mitochondrial biogenesis and augmenting the expression of uncoupling proteins (UCPs) and enzymes of the electron transport system [
13].
Adenosine monophosphate-activated kinase (AMPK) was first identified as a kinase that functions to inhibit fatty acid synthesis through phosphorylation and inactivation of acetyl coenzyme A carboxylase (ACC). Phosphorylation of ACC activates carnitine palmitoyl transferase 1 (CPT-1), thereby promoting the shuttling of fatty acids into the mitochondrial matrix for β-oxidation. AMPK is also known to enhance oxidative phosphorylation (OXPHOS) by promoting expression of the mitochondrial enzymes malate and succinate dehydrogenase [
14]. AMPK, one of the most potent regulators of PGC-1 activity, can act through the phosphorylation of PGC-1 to increase both the charge and transcription of PGC-1. AMPK requires PGC-1 for many of its most important effects on mitochondrial gene expression in skeletal muscle, both in culture and in vivo [
13,
15].
Cell survival and growth require metabolic pathways that produce energy, precursors for macromolecular synthesis, and substrates for other essential functions [
16]. The major nutrient substrates glucose, glutamine, and fatty acids can be completely oxidized to CO
2 and H
2O via the tricarboxylic acid (TCA) cycle. This requires operation of the mitochondrial electron transport chain (ETC), which couples a reaction using oxygen as a terminal electron acceptor to the production of adenosine triphosphate (ATP) through OXPHOS. Since living cells do not store ATP, they must produce it continuously and on demand, and therefore they constantly consume oxygen and fuel substrates [
17]. Accordingly, understanding the molecular and bioenergetics events that promote early and late oxidative stress in irradiated cells/tissues will be informative for counteracting the adverse health effects of IR [
18].
In mammals, muscles make up almost half of body weight, but it is common that they are generally resistant to radiation and are not an important part of body changes following irradiation [
19]. However, transient weakness (a type of injury) and metabolic disorders have been observed in animals after irradiation [
20], and childhood radiation therapy can cause muscle atrophy, weakness, impaired mobility, and fibrosis [
2,
21]. There is currently no information about the effects of IR on mitochondrial energy metabolism in skeletal muscle [
22]. Thus, we hypothesized that IR-induced changes in mitochondrial energy metabolism include the activation of AMPK, PGC-1, CPT-1, and UCP-2 and play a role in maintaining mitochondrial activity and biogenesis in skeletal muscle. To this end, we examined IR regulation of potential roles in maintaining mitochondrial energy metabolism and mitochondrial homeostasis. Using animal and cell culture models, we observed that mitochondrial substrate utilization and intracellular metabolic activity were reprogrammed upon radiation exposure. In particular, we found that the production of acetyl-coenzyme A (CoA) from pyruvate was decreased by IR, which was a compensatory effect of using glutamine and fatty acids to supply α-ketoglutarate and palmitate in the TCA cycle. Taken together, these data highlight the capacity of IR to modulate mitochondrial activity in skeletal muscle.
2. Materials and Methods
2.1. Animals
Imprinting control region (ICR)-strain male mice (6–8 weeks old; average weight 22 ± 2 g) purchased from Koatech Co. (Pyeongtaek, South Korea) were maintained under specific pathogen-free conditions in a 12:12-h light–dark cycle (lights on from 8:00 a.m. to 8:00 p.m.) at a controlled temperature (22 ± 3 °C) and humidity (50% ± 20%). Mice were fed standard animal food pellets and tap water ad libitum and were acclimatized to laboratory conditions for 1 wk before use. All animal experiments were conducted according to our institution’s guidelines for the ethical use of animals, with the approval of the Institutional Animal Care and Use Committee of the Korea Institute of Radiological and Medical Sciences (KIRAMS; approved protocol KIRAMS 2014–17). Animals were euthanized in a CO2 chamber at a low flow rate (20–30% of the volume of the cage per minute), and blood and muscle tissues were collected. Muscle tissue samples, collected according to their muscle type, were fixed overnight in 10% buffered formalin, embedded in paraffin, sectioned for immunohistochemistry (IHC), snap-frozen in liquid nitrogen, and stored at -80 °C. Samples were subsequently analyzed for mRNA and protein expression.
2.2. Cell Culture
Mouse C2C12 skeletal myoblast-derived cells (ATCC; Manassas, VA, USA) were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% antibiotic mixture in a humidified atmosphere of 95% air and 5% CO2 at 37 °C. Differentiation of C2C12 myoblasts was induced by transferring confluent cells to low-serum (1% FBS) DMEM and culturing for 4 to 5 days to allow the formation of myotubes. The differentiation medium was changed every 48 h.
2.3. Irradiation
Total body irradiation (TBI) of animals was accomplished at room temperature using an X-RAD 320 X-ray source (Precision X-Ray; North Branford, CT, USA) operated at 260 kVp and 10 mA using an F1 filter (2 mm aluminum) at a dose rate of 2 Gy/min. The reference dose rate was set to realistic radiation conditions in air on the animal plate with a UNIDOSE® universal dosimeter (PTW; Freiburg, Germany). Each mouse was kept in a perforated plastic container. Mice undergoing radiation exposure were placed on a rotating platform to ensure even dose delivery to all tissues. Cells were irradiated at a dose rate of 3.81 Gy/min using a 137Cs gamma radiation source (Atomic Energy of Canada; Mississauga, ON, Canada) and then assessed at time points indicated in the text.
2.4. ELISA
Mouse blood samples were obtained and serum was immediately separated by centrifugation at 3000 rpm for 15 min at 4 °C. Muscle tissue samples (snap-frozen in liquid nitrogen) and C2C12 myotubes were disrupted using a TissueLyser II (Qiagen; Hilden, Germany) and homogenized in the extraction solution provided in the assay kit. Samples were deproteinized using 10-kDa molecular weight cut-off spin columns (Biovision; Milpitas, CA, USA). Assay kits were used to measure concentrations of lactate, pyruvate, acetyl-CoA, malate, glutamine, α-ketoglutarate (Biovision), and NAD+/NADH (Bioassay System; Hayward, CA, USA) according to the manufacturer’s instructions.
2.5. Reverse Transcription Polymerase Chain Reaction (PCR) and Real-Time PCR
Total RNA was isolated using the QIAzol reagent (Qiagen) and quantified by formaldehyde–agarose gel electrophoresis. Single-strand cDNA was synthesized from total RNA (2 μg) using 0.27 μg of oligo dT and amfiRivert reverse transcriptase (GenDEPOT; Katy, TX, USA). The desired cDNA fragments were amplified by real-time quantitative PCR (qPCR) using the primers listed in
Table 1. Reactions were carried out in 20-μL reaction volumes containing 1 μL cDNA, 5× Hot FIREPol evaGreen qPCR Supermix (Solis BioDyne; Tartu, Estonia), and the relevant primers.
2.6. Measurement of Relative mtDNA Content
Total DNA was extracted from mouse muscle tissue and C2C12 myotubes using a QIAamp DNA Mini Kit (Qiagen), and mtDNA was measured by assessing the relative levels of ND2 (
Table 1) and genomic DNA (gDNA) in extracts of total DNA using PCR analyses. Reactions were carried out in 20-μL reaction volumes containing 10 ng DNA, 5× Hot FIREPol Blend Master Mix (Solis BioDyne), and the relevant primers. Amplified products were resolved on ethidium bromide (0.1 μg/mL)-stained 2% agarose gels, visualized using UV spectrophotometry, and quantified with ImageJ software.
2.7. Immunohistochemistry
Paraffin-embedded sections were deparaffinized with xylene and dehydrated with an increasing ethanol series. Endogenous peroxidases were quenched by brief exposure to 3% H2O2, and antigen retrieval in citrate buffer was used to enhance the signal. For immunohistochemistry, specimens were incubated overnight at 4 °C with primary antibodies against COXIV (1:100 dilution; Abcam; Cambridge, United Kingdom) and then with a fluorescein isothiocyanate (FITC)-conjugated secondary antibody (1:200 dilution; BD Transduction Laboratories; Lexington, KY, USA) for 1 h at room temperature in the dark. Specimens were treated with 2 μg/mL propidium iodide (PI) in phosphate-buffered saline (PBS) for 5 min, mounted on glass slides, and observed under a Zeiss LSM 510 META confocal microscope (Carl Zeiss, Oberkochen, Germany).
2.8. Immunocytochemistry
C2C12 myoblasts were seeded on cell culture slides (SPL Life Sciences; Pochen-si, Gyeonggi-do, Korea) and allowed to form myotubes under the differentiation-inducing conditions described above. After treatment, myotubes were rinsed with PBS and immersed in 70% ethanol overnight. Cell culture slides were rinsed with PBS and fixed in 4% paraformaldehyde for 15 min at room temperature. This was followed by permeabilization with 0.1% Triton X-100 for 10 min and rinsing with PBS. Cells were blocked through incubation with sterile PBS containing 2% bovine serum albumin (BSA) for 1 h. Thereafter, cells were rinsed with PBS/0.1% BSA and incubated overnight at 4 °C with primary antibodies against COXIV (1:100 dilution; Abcam) and then with a FITC-conjugated secondary antibody (1:200 dilution; Invitrogen; Carlsbad, CA, USA) for 1 h at room temperature in the dark. Specimens were treated with 2 μg/mL PI in PBS for 5 min, mounted on glass slides, and observed under a Zeiss LSM 510 META confocal microscope (Carl Zeiss).
2.9. Western Blot Analysis
Total cell lysates were prepared in lysis buffer (50 mM Tris pH 7.4, 150 mM NaCl, 5 mM EDTA, 1% Triton X-100, 1% sodium deoxycholic acid, 30 mM Na2HPO4, 50 mM NaF, 1 mM Na3VO4) containing freshly added protease inhibitor cocktail (GenDEPOT). Nuclear, mitochondrial, and plasma membrane extracts were prepared using an NE-PER Nuclear and Cytoplasmic Extraction Kit (Pierce), a Mitochondria Isolation Kit (Abcam), and a Plasma Membrane Protein Extraction Kit (Abcam), respectively. Extracts (30 mg) were mixed with sodium dodecyl sulfate (SDS) sample buffer, boiled for 5 min, separated by SDS-PAGE (polyacrylamide gel electrophoresis) on 8% or 10% gels, and transferred to nitrocellulose membranes (MilliporeSigma, Burlington, MA, USA). Blots were incubated with primary antibodies against COXIV (ab14744), CPT-1 (ab128568) (Abcam), phospho-ACC (Ser79) (#3661), ACC (#3662), phospho-AMPK (Thr172) (#2531), AMPK (#2532), EGFR (#2232) (Cell Signaling Technology; Danvers, MA, USA), Glut-1 (sc-7903), Glut-4 (sc-7938), UCP-2 (sc-6525), Bcl-2 (sc-7382), U1 SnRNP 70 (sc-390899), and/or α-actin (sc-32251) (Santa Cruz Biotechnology; Santa Cruz, CA, USA), as described in the text.
2.10. Fluorometric and Fluorescence Microscopic Measurement of ROS Generation
C2C12 myoblasts were seeded on cell culture slides (SPL Life Sciences) and were allowed to form myotubes under differentiation-induced conditions, as described above. After treatment, the medium was removed, and prewarmed medium containing 200 nM MitoTracker Green (Invitrogen) was added. Myotubes were then incubated for 45 min in a humidified 5% CO2 atmosphere at 37 °C and fixed in 3.7% formaldehyde in prewarmed media. Myotubes were coverslip-mounted, cured, and imaged using a fluorescence microscope (Olympus, Tokyo, Japan). Medium containing MitoTracker Green was removed, and cells were suspended in prewarmed media. Group mean fluorescence was measured using a SpectraMax M2 Microplate reader (Molecular Devices; San Jose, CA, USA). For measurement of ROS generation, cells were incubated with 10 μM 2′,7′-dichlorodihydrofluorescein diacetate (DCF-DA; Sigma-Aldrich; St. Louis, MO, USA) for 30 min, and the intensity of DCF-DA fluorescence was determined using a fluorescence microplate reader.
2.11. Seahorse XF24 Metabolic Flux Analysis
Post-IR, the culture medium was removed and replaced with Seahorse XF Medium containing 25 mM glucose and 4 mM glutamine (CO2-free) and was incubated at 37 °C. Cellular oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were measured as described by the manufacturer. Optimization of reagents was performed using the Seahorse XF Mito Stress Test kit (Agilent; Santa Clara, CA, USA), applying the protocol and algorithm in the XF24 analyzer. Glycolysis was measured using a Seahorse XF Glycolysis Stress Test kit (Agilent), as described by the manufacturer, and is presented as ECAR. Three major fuel sources (glucose, glutamine, and fatty acids) were measured using a Seahorse XF Mito Fuel Flex Test kit (Agilent), as described by the manufacturer, and are presented as OCR. The Seahorse XF24 Extracellular Analyzer was run using 8-min cyclic protocol commands (mix for 3 min, let stand 2 min, and measure for 3 min) in triplicate.
2.12. Measurement of [18F]- Fluorodeoxyglucose (FDG) Uptake
On the day of the experiment, 2 mL of uptake medium (DMEM with 1 mg/mL glucose) containing 74 kBq (2 μCi) of [18F]-FDG was added to each well after rinsing the wells once in cold uptake medium. Myoblasts were incubated at 37 °C for 1 h to allow FDG to accumulate in cells. After washing twice in cold PBS, cells were lysed with 0.2% SDS, and radioactivity was immediately measured using a 1480 WIZARD gamma counter (PerkinElmer; Waltham, MA, USA).
2.13. Measurement of ATP
ATP was measured using an ATP Determination kit (Invitrogen) as described by the manufacturer. Briefly, myotubes were homogenized in lysis buffer (50 mM Tris-HCl pH 7.5, 1% Triton X-100, 0.1% SDS, 150 mM NaCl) freshly supplemented with the protease cocktail inhibitor Complete (GenDEPOT). ATP levels were assessed using a Micro-Lumat Plus LB96V fluorescence plate reader (Berthold Technologies; Bad Wildbad, Germany) and normalized to total protein, determined using a Bradford-based assay (GenDEPOT).
2.14. Statistical Analysis
Data are expressed as means ± standard deviations (SDs) of at least three independent experiments. Comparisons between two groups were analyzed by unpaired two-tailed Student’s t-tests, performed using Excel (Microsoft). A P-value <0.05 was regarded as significant (individual P-values are indicated in figure legends).
4. Discussion
The mitochondrion has traditionally been viewed as an organelle that functions as the “powerhouse” of mammalian cells, generating cellular energy in the form of ATP through the process of OXPHOS. It was first proposed by Harman [
25] that defects in mitochondria are involved in severe clinical sequelae that affect the whole organism, or at least the central organs—the brain, heart, liver, and skeletal muscle—that have the highest energy consumption. Furthermore, a subcellular proteomic analysis has revealed that irradiation alters proteins involved in various energy metabolism-related processes. Therefore, more specific protection of mitochondria and mitochondrial energy metabolism would be expected to reduce damage to normal cells exposed to IR [
6,
26].
In this study, we tested radiation effects on mitochondrial energy metabolism in skeletal muscle, an organ with a high energetic demand that requires efficient mitochondrial function, and identified a number of important consequences of IR exposure. First, we provide strong evidence that mitochondrial metabolism is reprogrammed in response to radiation. Metabolic assays further indicated how metabolism was changed, showing that glutamine was metabolized to produce α-ketoglutarate, thereby enabling the TCA cycle to continue to produce energy. Surprisingly, radiation exposure also affected pyruvate, acetyl-CoA, malate, α-ketoglutamate, and glutamate/glutamine and significantly increased lactate secretion (
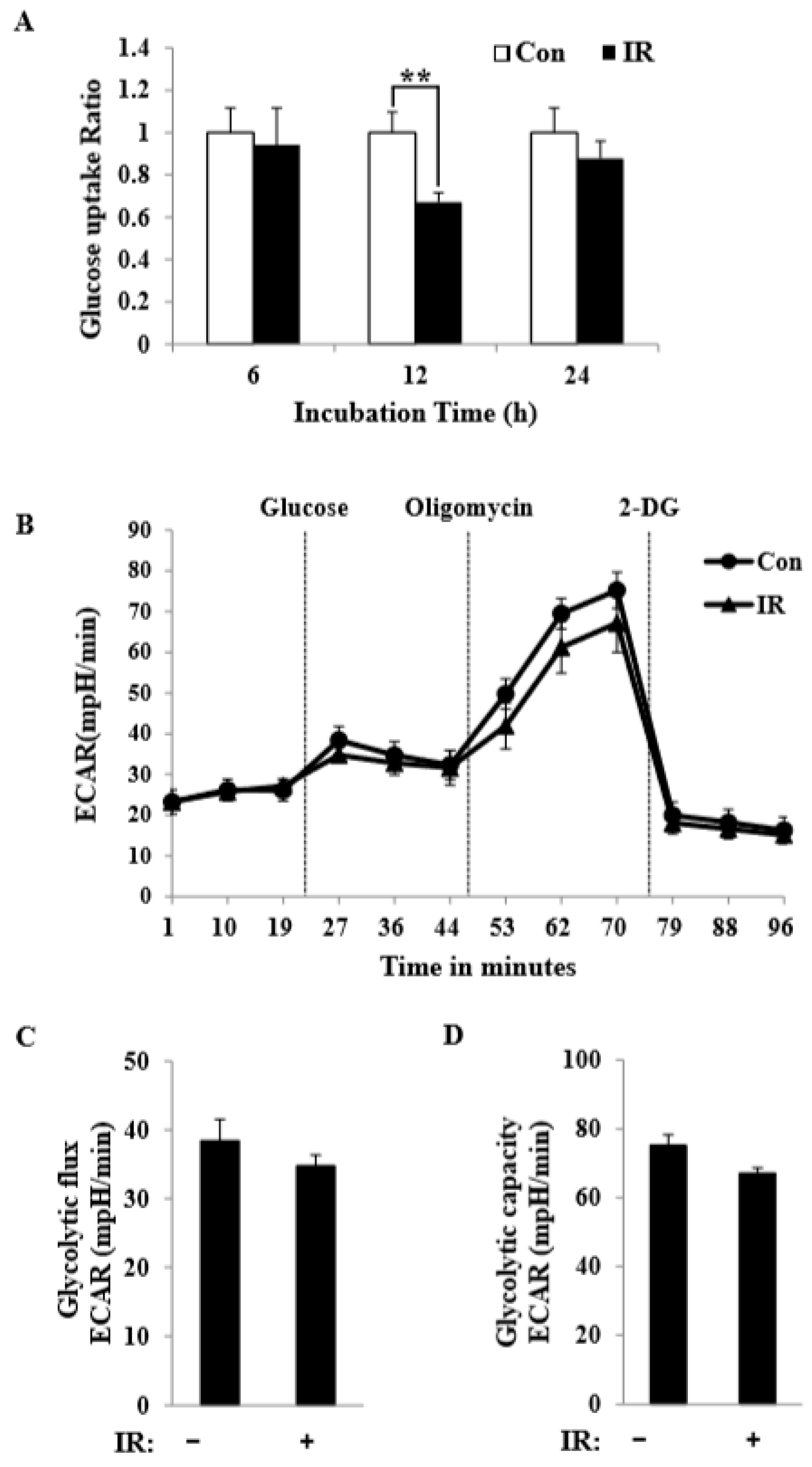
Figure 1). We further observed a decrease in glucose uptake and glycolytic metabolism in skeletal muscle (
Figure 5), suggesting that cells increase their reliance on glutamine anaplerosis when mitochondrial pyruvate transport is limited by reprogramming TCA metabolism. Second, based on previous work, we expected that radiation exposure would increase mitochondrial biogenesis, and we hypothesized that these effects would be dependent upon mitochondrial content. Further analyses revealed that radiation exposure increased mitochondrial mass in skeletal muscle (
Figure 2 and
Figure 3), suggesting that increased mitochondrial biogenesis may underlie the ability of IR to alter mitochondrial function. Finally, as might be expected, we observed an increase in AMPK activity following irradiation that correlated with increased mitochondrial metabolism, as assessed by both oxygen consumption and substrate utilization (
Figure 4 and
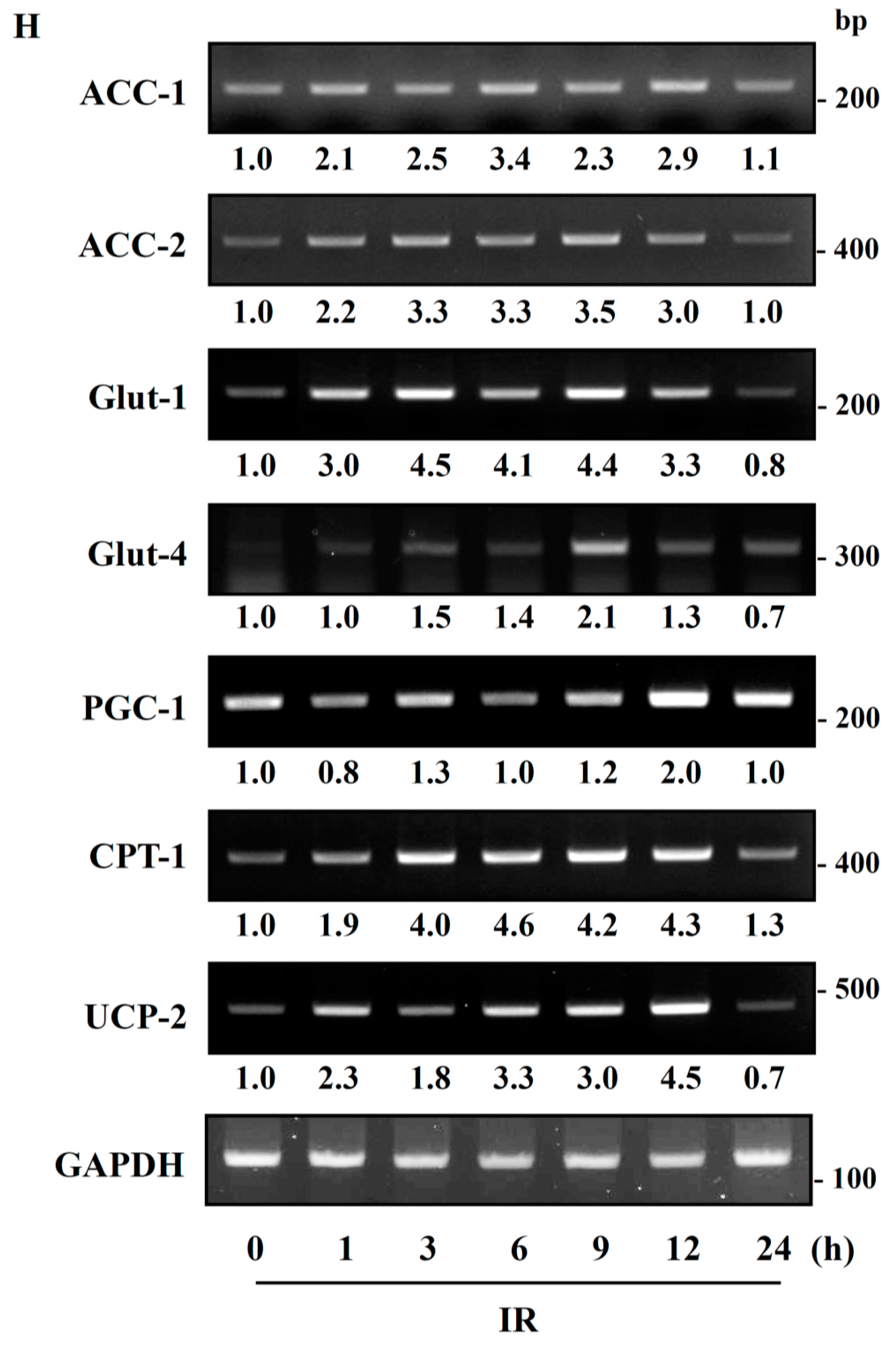
Figure 6). Taken together, our data suggest that IR exposure induced the phosphorylation of AMPK, which upregulated the mitochondria biogenesis-associated proteins PGC-1, UCP-2, and CPT-1 through the phosphorylation of ACC (
Figure 7).
IR alters mitochondrial functions, increases mitochondrial oxidative stress, and causes specific changes in the expression of mitochondrial genes related to cell survival [
3,
27,
28]. In sum, both genetic and metabolic susceptibility to IR greatly affect the outcome of radiation exposure. At the organelle level, mitochondria, as the major sites of oxidative metabolism, are almost certain to be affected. Although many studies have addressed the effects of IR on biochemical and morphological changes in mitochondria, studies of radiation-induced changes in the bioenergetics of this organelle are lacking. This is particularly important since mitochondrial structure and bioenergetics may be tightly linked [
18]. In this study, we not only determined basal respiration, but also identified the type of exogenous nutrient substrates that are capable of being oxidized and the rates at which they can be oxidized under the experimental conditions used. Our results show that IR-exposed C2C12 myotubes possessed the ability to oxidize fatty acid and glutamine at higher rates than control C2C12 myotubes (
Figure 6C). The synergistic effect of inhibiting these three mitochondrial substrate oxidation pathways highlights the plasticity of mitochondrial metabolism in respiring cells and independently demonstrates that radiation-exposed C2C12 myotubes employed fatty acid and amino acid oxidation to meet their bioenergetics demands. This suggests that C2C12 myotubes exposed to radiation shift away from glycolytic metabolism and adopt a more oxidative metabolism, perhaps through reprogramming of their metabolic network via certain epigenetic events.
Recent studies have shown that macroautophagy and chaperone-mediated autophagy are required to maintain the energy metabolism necessary to survive a variety of stimuli, including radiation [
17]. Linking the observed changes in respiration and endogenous substrates, we speculate that autophagy may contribute, at least in part, to the provision of endogenous substrates in the form of fatty acids and amino acids. AMPK-dependent autophagy and/or mitophagy may represent important components of such metabolic control. Under energy stress conditions, mitochondrial homeostasis is ensured by AMPK-dependent mitophagy [
29], which may serve to remove damaged, inefficient mitochondria following irradiation and thereby limit oxygen consumption [
14]. Therefore, pharmacological activators of AMPK might act to protect against radiation damage. It further seems likely that unidentified substrates, substrate signaling networks, and pharmacological activators of AMPK in metabolic cells/tissues remain to be discovered and might serve as the basis for the development of strategies to defend against radiation-induced damage responses [
30]. Mitochondrial dysfunction-associated senescence (MiDAS) [
30], characterized by an imbalance in energy levels caused by a decreased NAD
+/NADH ratio, is associated with cell cycle arrest resulting from stimulation of the cellular energy sensor, AMPK. Whether changes in AMPK and NAD
+/NADH metabolism are causative factors or occur in parallel with IR-induced remodeling of the energy metabolism of skeletal muscle remains to be clarified [
31].











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}