Cellular Stress Responses in Radiotherapy

 ,
,
Abstract
1. Introduction
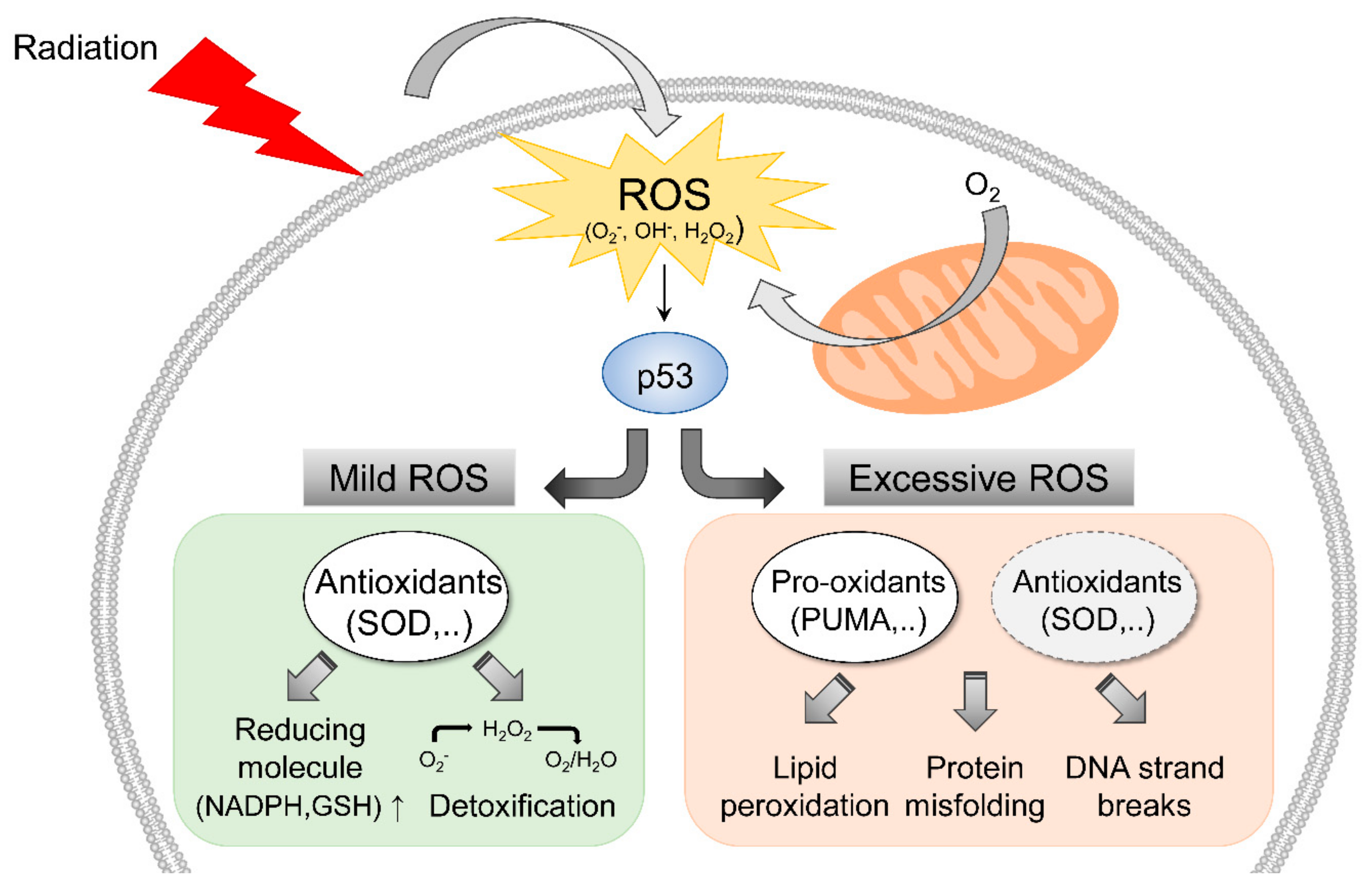
2. Radiation-Induced ROS Response
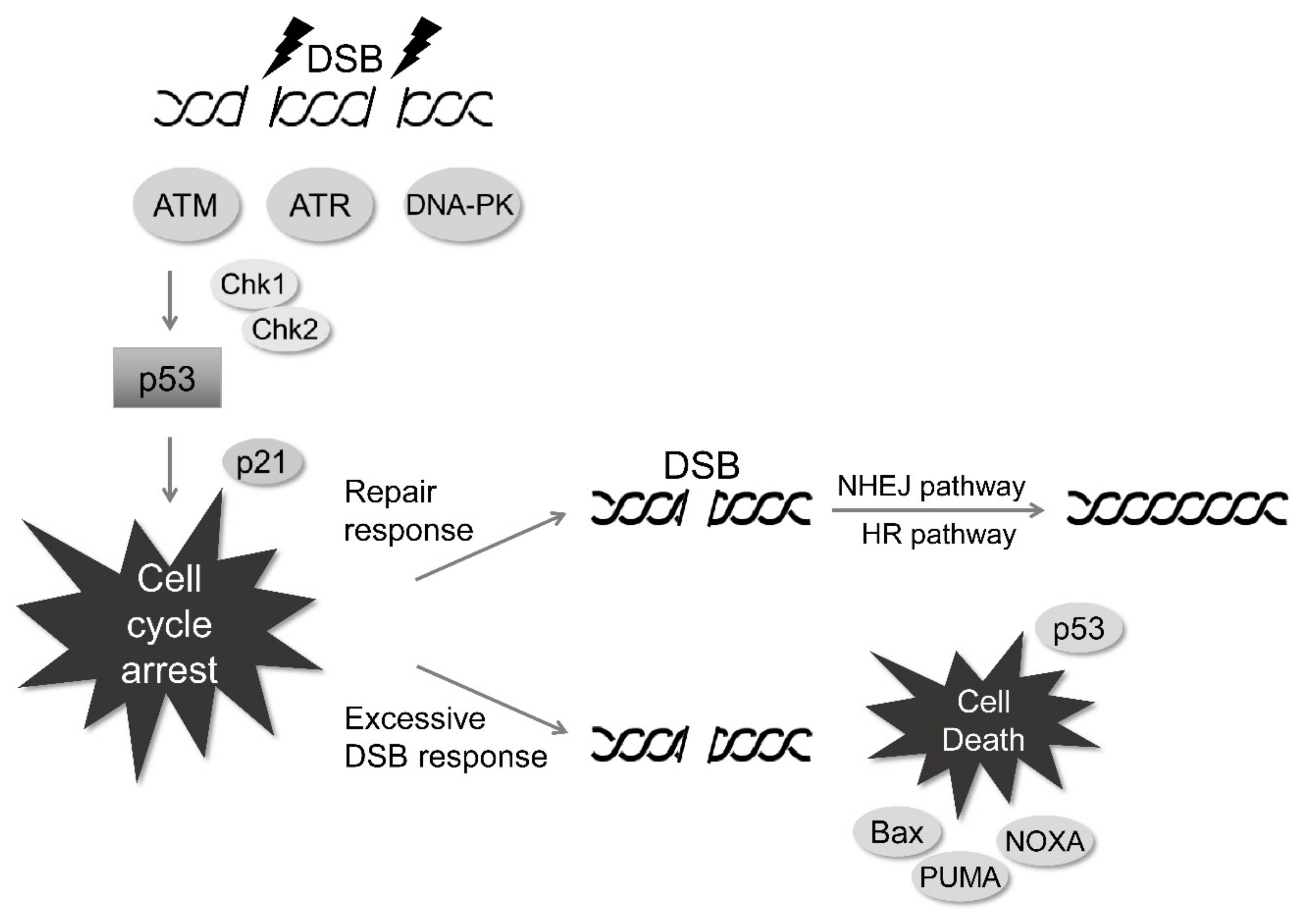
3. Radiation-Induced DNA Damage Response
4. Radiation-Induced Subcellular Organelle Response
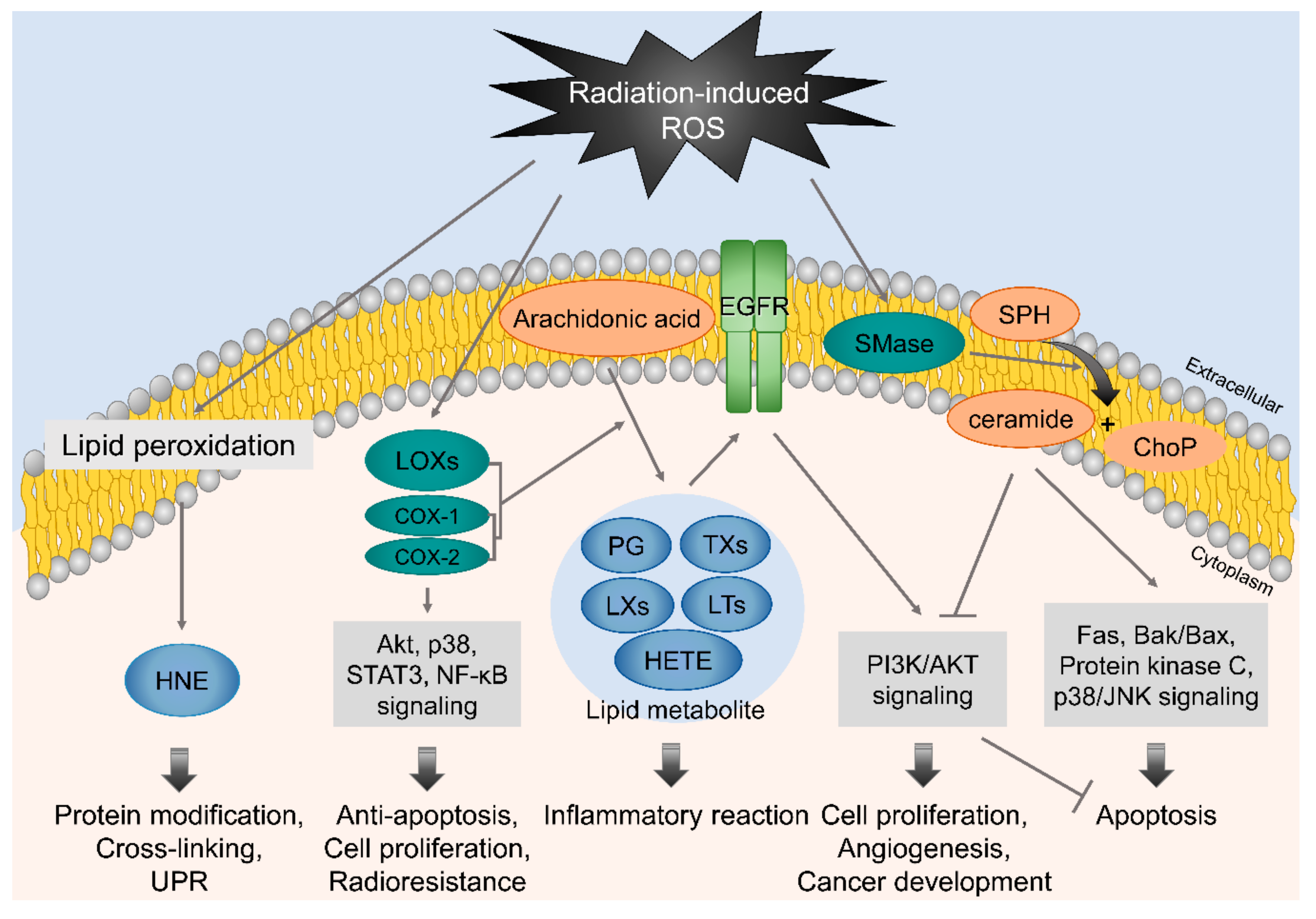
4.1. Membrane-Associated Signaling in Response to Irradiation
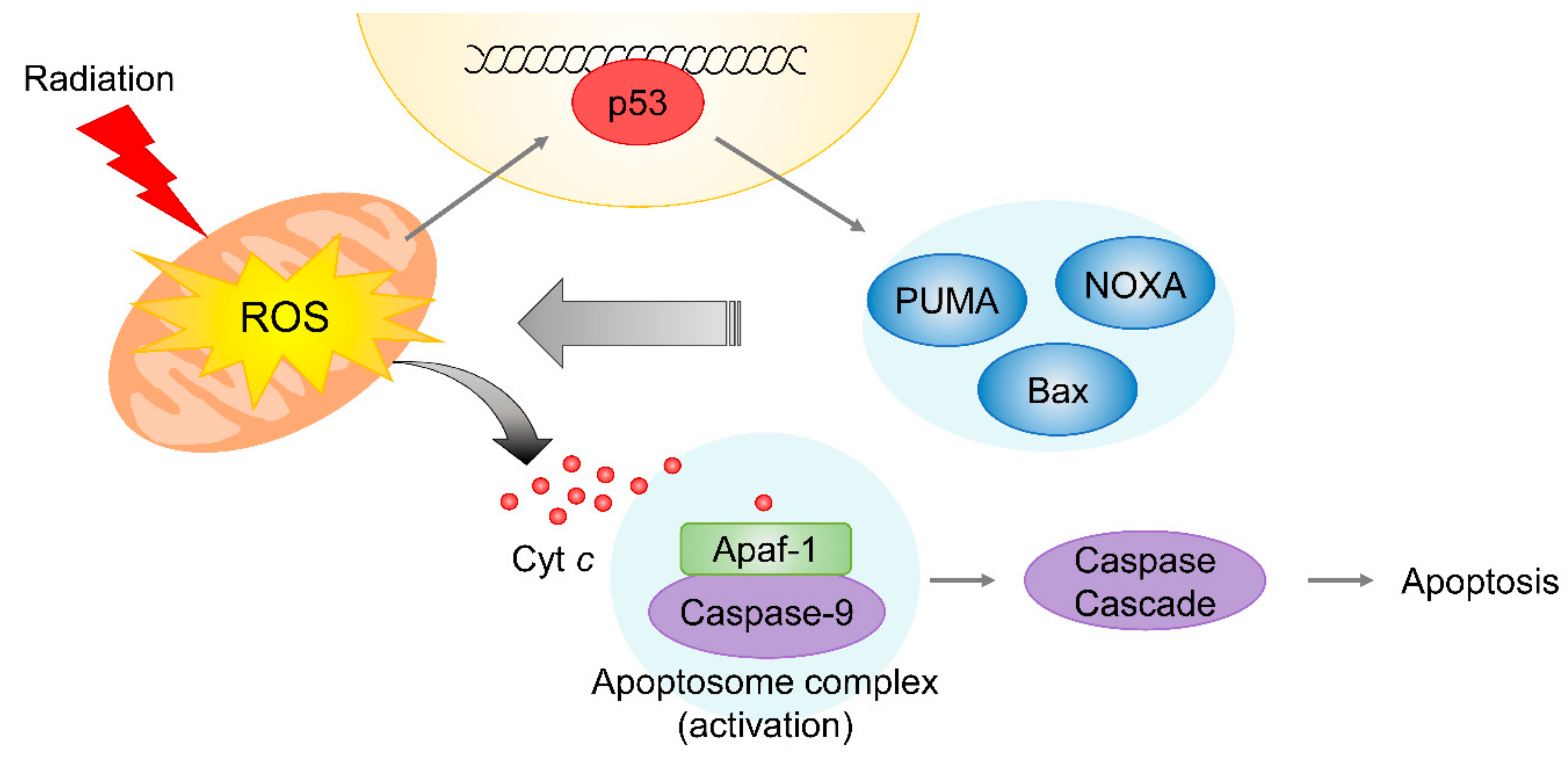
4.2. Mitochondrial Damage Induced by Radiation
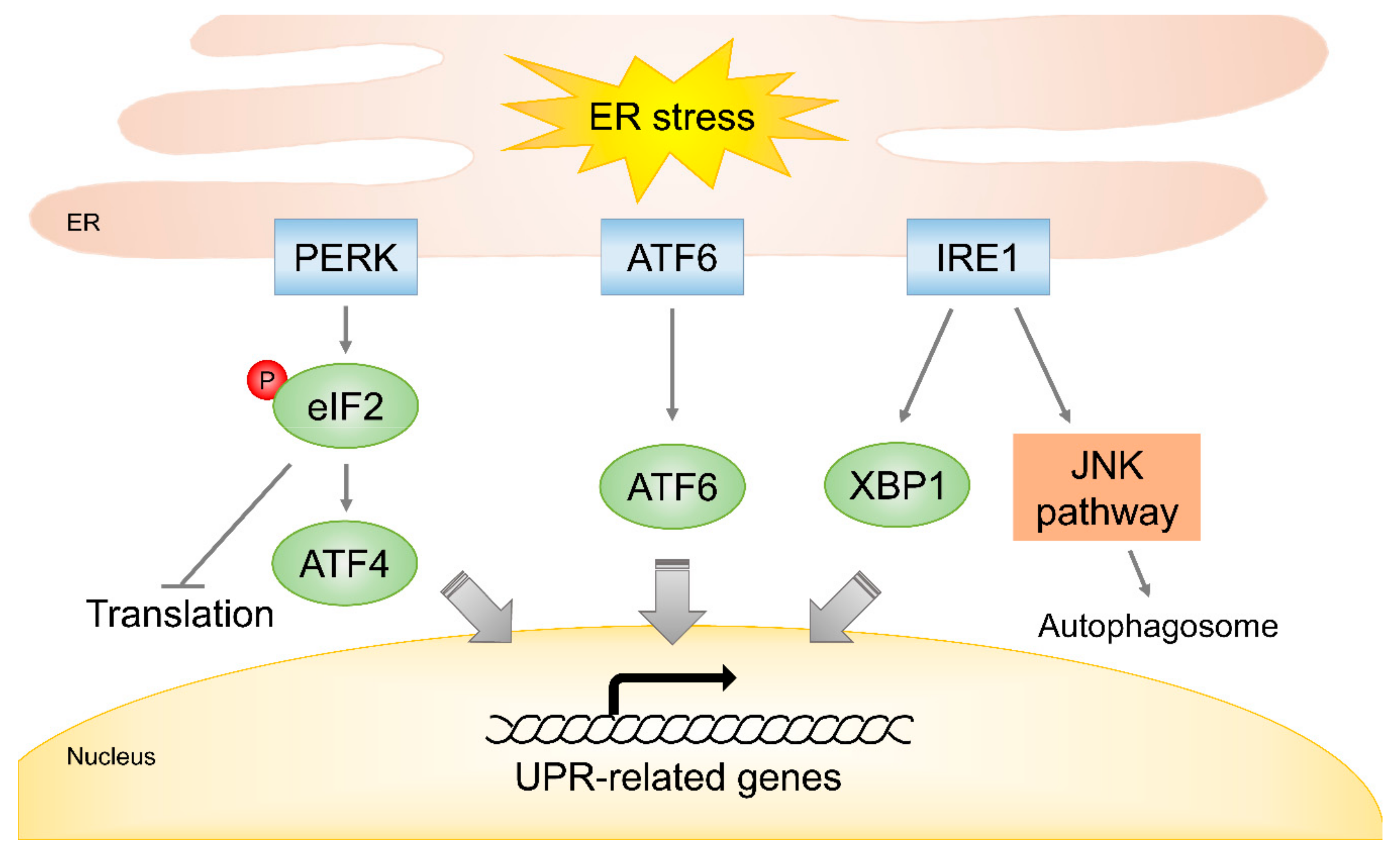
4.3. Endoplasmic Reticulum Stress in Response to Radiation
5. Radiation-Induced Autophagy
6. Clinical Approaches to Radiosensitization Based on the Regulation of Cell Stress Responses
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Allen, C.; Her, S.; Jaffray, D.A. Radiotherapy for Cancer: Present and Future. Adv. Drug Deliv. Rev. 2017, 109, 1–2. [Google Scholar] [CrossRef] [PubMed]
- De Ruysscher, D.; Niedermann, G.; Burnet, N.G.; Siva, S.; Lee, A.W.M.; Hegi-Johnson, F. Radiotherapy toxicity. Nat. Rev. Dis. Primers 2019, 5, 13. [Google Scholar] [CrossRef] [PubMed]
- Son, B.; Kwon, T.; Lee, S.; Han, I.; Kim, W.; Youn, H.; Youn, B. CYP2E1 regulates the development of radiation-induced pulmonary fibrosis via ER stress- and ROS-dependent mechanisms. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 313, L916–L929. [Google Scholar] [CrossRef]
- Wang, J.S.; Wang, H.J.; Qian, H.L. Biological effects of radiation on cancer cells. Mil. Med. Res. 2018, 5. [Google Scholar] [CrossRef] [PubMed]
- Lomax, M.; Folkes, L.; O’Neill, P. Biological Consequences of Radiation-induced DNA Damage: Relevance to Radiotherapy. Clin. Oncol. 2013, 25, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, U.S.; Tan, B.W.Q.; Vellayappan, B.A.; Jeyasekharan, A.D. ROS and the DNA damage response in cancer. Redox. Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, J.; Nairy, R.K.; Langhnoja, J.; Tripathi, A.; Patil, R.K.; Pillai, P.P.; Mustak, M.S. ER stress and genomic instability induced by gamma radiation in mice primary cultured glial cells. Metab. Brain Dis. 2018, 33, 855–868. [Google Scholar] [CrossRef] [PubMed]
- Diehn, M.; Cho, R.W.; Lobo, N.A.; Kalisky, T.; Dorie, M.J.; Kulp, A.N.; Qian, D.; Lam, J.S.; Ailles, L.E.; Wong, M.; et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009, 458, 780–783. [Google Scholar] [CrossRef]
- Nagelkerke, A.; Bussink, J.; van der Kogel, A.J.; Sweep, F.C.; Span, P.N. The PERK/ATF4/LAMP3-arm of the unfolded protein response affects radioresistance by interfering with the DNA damage response. Radiother. Oncol. 2013, 108, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.A.; Das, B.C.; Ray, S.K. Targeting autophagy for combating chemoresistance and radioresistance in glioblastoma. Apoptosis 2018, 23, 563–575. [Google Scholar] [CrossRef]
- Kwon, T.; Youn, H.; Son, B.; Kim, D.; Seong, K.M.; Park, S.; Kim, W.; Youn, B. DANGER is involved in high glucose-induced radioresistance through inhibiting DAPK-mediated anoikis in non-small cell lung cancer. Oncotarget 2016, 7, 7193–7206. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Kim, W.; Kwon, T.; Youn, H.; Kim, J.S.; Youn, B. Plasminogen activator inhibitor-1 enhances radioresistance and aggressiveness of non-small cell lung cancer cells. Oncotarget 2016, 7, 23961–23974. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Youn, H.; Lee, S.; Kim, E.; Kim, D.; Sub Lee, J.; Lee, J.M.; Youn, B. RNF138-mediated ubiquitination of rpS3 is required for resistance of glioblastoma cells to radiation-induced apoptosis. Exp. Mol. Med. 2018, 50. [Google Scholar] [CrossRef] [PubMed]
- Son, B.; Jun, S.Y.; Seo, H.; Youn, H.; Yang, H.J.; Kim, W.; Kim, H.K.; Kang, C.; Youn, B. Inhibitory effect of traditional oriental medicine-derived monoamine oxidase B inhibitor on radioresistance of non-small cell lung cancer. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.; Chang, H.; Li, H.; Wang, S. Induction of reactive oxygen species: An emerging approach for cancer therapy. Apoptosis 2017, 22, 1321–1335. [Google Scholar] [CrossRef]
- Tulard, A.; Hoffschir, F.; de Boisferon, F.H.; Luccioni, C.; Bravard, A. Persistent oxidative stress after ionizing radiation is involved in inherited radiosensitivity. Free Radic. Biol. Med. 2003, 35, 68–77. [Google Scholar] [CrossRef]
- Kim, W.; Youn, H.; Kang, C.; Youn, B. Inflammation-induced radioresistance is mediated by ROS-dependent inactivation of protein phosphatase 1 in non-small cell lung cancer cells. Apoptosis 2015, 20, 1242–1252. [Google Scholar] [CrossRef]
- Leach, J.K.; van Tuyle, G.; Lin, P.S.; Schmidt-Ullrich, R.; Mikkelsen, R.B. Ionizing radiation-induced, mitochondria-dependent generation of reactive oxygen/nitrogen. Cancer Res. 2001, 61, 3894–3901. [Google Scholar]
- Kam, W.W.; Banati, R.B. Effects of ionizing radiation on mitochondria. Free Radic. Biol. Med. 2013, 65, 607–619. [Google Scholar] [CrossRef]
- Bhuyan, K.C.; Bhuyan, D.K. Superoxide dismutase of the eye: Relative functions of superoxide dismutase and catalase in protecting the ocular lens from oxidative damage. Biochim. Biophys. Acta 1978, 542, 28–38. [Google Scholar] [CrossRef]
- Glorieux, C.; Calderon, P.B. Catalase, a remarkable enzyme: Targeting the oldest antioxidant enzyme to find a new cancer treatment approach. Biol. Chem. 2017, 398, 1095–1108. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.P.; Amstad, P.; He, P.; Robles, A.; Lupold, S.; Kaneko, I.; Ichimiya, M.; Sengupta, S.; Mechanic, L.; Okamura, S.; et al. p53-induced up-regulation of MnSOD and GPx but not catalase increases oxidative stress and apoptosis. Cancer Res. 2004, 64, 2350–2356. [Google Scholar] [CrossRef]
- Budanov, A.V. Stress-responsive sestrins link p53 with redox regulation and mammalian target of rapamycin signaling. Antioxid. Redox Signal. 2011, 15, 1679–1690. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Zhang, C.; Wu, R.; Sun, Y.; Levine, A.; Feng, Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc. Natl. Acad. Sci. USA 2010, 107, 7455–7460. [Google Scholar] [CrossRef]
- Liu, B.; Bhatt, D.; Oltvai, Z.N.; Greenberger, J.S.; Bahar, I. Significance of p53 dynamics in regulating apoptosis in response to ionizing radiation, and polypharmacological strategies. Sci. Rep. 2014, 4. [Google Scholar] [CrossRef]
- Shi, Y.; Nikulenkov, F.; Zawacka-Pankau, J.; Li, H.; Gabdoulline, R.; Xu, J.; Eriksson, S.; Hedstrom, E.; Issaeva, N.; Kel, A.; et al. ROS-dependent activation of JNK converts p53 into an efficient inhibitor of oncogenes leading to robust apoptosis. Cell Death Differ. 2014, 21, 612–623. [Google Scholar] [CrossRef]
- Italiano, D.; Lena, A.M.; Melino, G.; Candi, E. Identification of NCF2/p67phox as a novel p53 target gene. Cell Cycle 2012, 11, 4589–4596. [Google Scholar] [CrossRef]
- Bernerd, F.; Sarasin, A.; Magnaldo, T. Galectin-7 overexpression is associated with the apoptotic process in UVB-induced sunburn keratinocytes. Proc. Natl. Acad. Sci. USA 1999, 96, 11329–11334. [Google Scholar] [CrossRef]
- Faraonio, R.; Vergara, P.; di Marzo, D.; Pierantoni, M.G.; Napolitano, M.; Russo, T.; Cimino, F. p53 suppresses the Nrf2-dependent transcription of antioxidant response genes. J. Biol. Chem. 2006, 281, 39776–39784. [Google Scholar] [CrossRef] [PubMed]
- Rey, S.; Schito, L.; Koritzinsky, M.; Wouters, B.G. Molecular targeting of hypoxia in radiotherapy. Adv. Drug Deliv. Rev. 2017, 109, 45–62. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.S.; Tsai, H.Y.; Drake, P.; Wang, F.N.; Chiang, C.S. Gadolinium-doped iron oxide nanoparticles induced magnetic field hyperthermia combined with radiotherapy increases tumour response by vascular disruption and improved oxygenation. Int. J. Hyperth. 2017, 33, 770–778. [Google Scholar] [CrossRef] [PubMed]
- Movafagh, S.; Crook, S.; Vo, K. Regulation of hypoxia-inducible factor-1a by reactive oxygen species: New developments in an old debate. J. Cell. Biochem. 2015, 116, 696–703. [Google Scholar] [CrossRef] [PubMed]
- Viallard, C.; Larrivee, B. Tumor angiogenesis and vascular normalization: Alternative therapeutic targets. Angiogenesis 2017, 20, 409–426. [Google Scholar] [CrossRef] [PubMed]
- Cairns, R.A.; Papandreou, I.; Sutphin, P.D.; Denko, N.C. Metabolic targeting of hypoxia and HIF1 in solid tumors can enhance cytotoxic chemotherapy. Proc. Natl. Acad. Sci. USA 2007, 104, 9445–9450. [Google Scholar] [CrossRef] [PubMed]
- Harada, H. Hypoxia-inducible factor 1-mediated characteristic features of cancer cells for tumor radioresistance. J. Radiat. Res. 2016, 57, i99–i105. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.F. DNA damage produced by ionizing radiation in mammalian cells: Identities, mechanisms of formation, and reparability. Prog. Nucleic Acid Res. Mol. Biol. 1988, 35, 95–125. [Google Scholar]
- Almeida, K.H.; Sobol, R.W. A unified view of base excision repair: Lesion-dependent protein complexes regulated by post-translational modification. DNA Repair 2007, 6, 695–711. [Google Scholar] [CrossRef]
- Akbari, M.; Pena-Diaz, J.; Andersen, S.; Liabakk, N.B.; Otterlei, M.; Krokan, H.E. Extracts of proliferating and non-proliferating human cells display different base excision pathways and repair fidelity. DNA Repair 2009, 8, 834–843. [Google Scholar] [CrossRef]
- Svilar, D.; Goellner, E.M.; Almeida, K.H.; Sobol, R.W. Base excision repair and lesion-dependent subpathways for repair of oxidative DNA damage. Antioxid. Redox Signal. 2011, 14, 2491–2507. [Google Scholar] [CrossRef] [PubMed]
- Lan, L.; Nakajima, S.; Oohata, Y.; Takao, M.; Okano, S.; Masutani, M.; Wilson, S.H.; Yasui, A. In situ analysis of repair processes for oxidative DNA damage in mammalian cells. Proc. Natl. Acad. Sci. USA 2004, 101, 13738–13743. [Google Scholar] [CrossRef] [PubMed]
- Mortusewicz, O.; Rothbauer, U.; Cardoso, M.C.; Leonhardt, H. Differential recruitment of DNA Ligase I and III to DNA repair sites. Nucleic Acids Res. 2006, 34, 3523–3532. [Google Scholar] [CrossRef] [PubMed]
- McKinnon, P.J.; Caldecott, K.W. DNA strand break repair and human genetic disease. Annu. Rev. Genom. Hum. Genet. 2007, 8, 37–55. [Google Scholar] [CrossRef] [PubMed]
- Mavragani, I.V.; Nikitaki, Z.; Souli, M.P.; Aziz, A.; Nowsheen, S.; Aziz, K.; Rogakou, E.; Georgakilas, A.G. Complex DNA Damage: A Route to Radiation-Induced Genomic Instability and Carcinogenesis. Cancers 2017, 9. [Google Scholar] [CrossRef]
- Georgakilas, A.G.; O’Neill, P.; Stewart, R.D. Induction and repair of clustered DNA lesions: What do we know so far? Radiat. Res. 2013, 180, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Shikazono, N.; Noguchi, M.; Fujii, K.; Urushibara, A.; Yokoya, A. The yield, processing, and biological consequences of clustered DNA damage induced by ionizing radiation. J. Radiat. Res. 2009, 50, 27–36. [Google Scholar] [CrossRef]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef]
- Mailand, N.; Bekker-Jensen, S.; Faustrup, H.; Melander, F.; Bartek, J.; Lukas, C.; Lukas, J. RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell 2007, 131, 887–900. [Google Scholar] [CrossRef]
- Thompson, L.H. Recognition, signaling, and repair of DNA double-strand breaks produced by ionizing radiation in mammalian cells: The molecular choreography. Mutat. Res. 2012, 751, 158–246. [Google Scholar] [CrossRef]
- Doil, C.; Mailand, N.; Bekker-Jensen, S.; Menard, P.; Larsen, D.H.; Pepperkok, R.; Ellenberg, J.; Panier, S.; Durocher, D.; Bartek, J.; et al. RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell 2009, 136, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, W.C.; Denning, M.F. P21Waf1 control of epithelial cell cycle and cell fate. Crit. Rev. Oral Biol. Med. 2002, 13, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.; Asaithamby, A. Repurposing DNA repair factors to eradicate tumor cells upon radiotherapy. Transl. Cancer Res. 2017, 6, S822–S839. [Google Scholar] [CrossRef] [PubMed]
- Bernhard, E.J.; Maity, A.; Muschel, R.J.; McKenna, W.G. Effects of ionizing radiation on cell cycle progression. A review. Radiat. Environ. Biophys. 1995, 34, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Dogu, Y.; Diaz, J. Mathematical model of a network of interaction between p53 and Bcl-2 during genotoxic-induced apoptosis. Biophys. Chem. 2009, 143, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Kuribayashi, K.; Finnberg, N.; Jeffers, J.R.; Zambetti, G.P.; El-Deiry, W.S. The relative contribution of pro-apoptotic p53-target genes in the triggering of apoptosis following DNA damage in vitro and in vivo. Cell Cycle 2011, 10, 2380–2389. [Google Scholar] [CrossRef] [PubMed]
- Oda, E.; Ohki, R.; Murasawa, H.; Nemoto, J.; Shibue, T.; Yamashita, T.; Tokino, T.; Taniguchi, T.; Tanaka, N. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 2000, 288, 1053–1058. [Google Scholar] [CrossRef] [PubMed]
- Lou, Z.; Minter-Dykhouse, K.; Franco, S.; Gostissa, M.; Rivera, M.A.; Celeste, A.; Manis, J.P.; van Deursen, J.; Nussenzweig, A.; Paull, T.T.; et al. MDC1 maintains genomic stability by participating in the amplification of ATM-dependent DNA damage signals. Mol. Cell 2006, 21, 187–200. [Google Scholar] [CrossRef]
- Minter-Dykhouse, K.; Ward, I.; Huen, M.S.; Chen, J.; Lou, Z. Distinct versus overlapping functions of MDC1 and 53BP1 in DNA damage response and tumorigenesis. J. Cell Biol. 2008, 181, 727–735. [Google Scholar] [CrossRef]
- Boulianne, B.; Feldhahn, N. Transcribing malignancy: Transcription-associated genomic instability in cancer. Oncogene 2018, 37, 971–981. [Google Scholar] [CrossRef]
- Corre, I.; Niaudet, C.; Paris, F. Plasma membrane signaling induced by ionizing radiation. Mutat. Res. 2010, 704, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Wong-Ekkabut, J.; Xu, Z.; Triampo, W.; Tang, I.M.; Tieleman, D.P.; Monticelli, L. Effect of lipid peroxidation on the properties of lipid bilayers: A molecular dynamics study. Biophys. J. 2007, 93, 4225–4236. [Google Scholar] [CrossRef] [PubMed]
- Poli, G.; Schaur, R.J.; Siems, W.G.; Leonarduzzi, G. 4-hydroxynonenal: A membrane lipid oxidation product of medicinal interest. Med. Res. Rev. 2008, 28, 569–631. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.H.; Yen, J.H.; Weng, C.Y.; Wang, L.; Ha, C.L.; Wu, M.J. Lipid peroxidation end product 4-hydroxy-trans-2-nonenal triggers unfolded protein response and heme oxygenase-1 expression in PC12 cells: Roles of ROS and MAPK pathways. Toxicology 2014, 315, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Kolesnick, R. The therapeutic potential of modulating the ceramide/sphingomyelin pathway. J. Clin. Invest. 2002, 110, 3–8. [Google Scholar] [CrossRef]
- Liao, W.C.; Haimovitz-Friedman, A.; Persaud, R.S.; McLoughlin, M.; Ehleiter, D.; Zhang, N.; Gatei, M.; Lavin, M.; Kolesnick, R.; Fuks, Z. Ataxia telangiectasia-mutated gene product inhibits DNA damage-induced apoptosis via ceramide synthase. J. Biol. Chem. 1999, 274, 17908–17917. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Son, B.; Lee, S.; Do, H.; Youn, B. Targeting the enzymes involved in arachidonic acid metabolism to improve radiotherapy. Cancer Metastasis Rev. 2018, 37, 213–225. [Google Scholar] [CrossRef]
- Wang, D.; Dubois, R.N. Eicosanoids and cancer. Nat. Rev. Cancer 2010, 10, 181–193. [Google Scholar] [CrossRef]
- Multhoff, G.; Radons, J. Radiation, inflammation, and immune responses in cancer. Front. Oncol. 2012, 2. [Google Scholar] [CrossRef]
- Meng, Z.; Gan, Y.H. Activating PTEN by COX-2 inhibitors antagonizes radiation-induced AKT activation contributing to radiosensitization. Biochem. Biophys. Res. Commun. 2015, 460, 198–204. [Google Scholar] [CrossRef]
- Lin, F.; Luo, J.; Gao, W.; Wu, J.; Shao, Z.; Wang, Z.; Meng, J.; Ou, Z.; Yang, G. COX-2 promotes breast cancer cell radioresistance via p38/MAPK-mediated cellular anti-apoptosis and invasiveness. Tumour Biol. 2013, 34, 2817–2826. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.Y.; Lee, H.T.; Chen, C.M.; Shen, C.C.; Ma, H.I. Celecoxib suppresses the phosphorylation of STAT3 protein and can enhance the radiosensitivity of medulloblastoma-derived cancer stem-like cells. Int. J. Mol. Sci. 2014, 15, 11013–11029. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Wang, Z.; Chong, T.; Yang, J.; Li, H.; Chen, H. Curcumin enhances the radiosensitivity of renal cancer cells by suppressing NF-kappaB signaling pathway. Biomed. Pharmacother. 2017, 94, 974–981. [Google Scholar] [CrossRef]
- Yamamori, T.; Yasui, H.; Yamazumi, M.; Wada, Y.; Nakamura, Y.; Nakamura, H.; Inanami, O. Ionizing radiation induces mitochondrial reactive oxygen species production accompanied by upregulation of mitochondrial electron transport chain function and mitochondrial content under control of the cell cycle checkpoint. Free Radic. Biol. Med. 2012, 53, 260–270. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Li, N.; Wang, Y.; Wang, Y.; Zhang, X.; Zhang, H. Effects of X-irradiation on mitochondrial DNA damage and its supercoiling formation change. Mitochondrion 2011, 11, 886–892. [Google Scholar] [CrossRef] [PubMed]
- Cain, K.; Bratton, S.B.; Langlais, C.; Walker, G.; Brown, D.G.; Sun, X.M.; Cohen, G.M. Apaf-1 oligomerizes into biologically active approximately 700-kDa and inactive approximately 1.4-MDa apoptosome complexes. J. Biol. Chem. 2000, 275, 6067–6070. [Google Scholar] [CrossRef] [PubMed]
- Gatenby, R.A.; Gillies, R.J. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 2004, 4, 891–899. [Google Scholar] [CrossRef]
- Simonnet, H.; Alazard, N.; Pfeiffer, K.; Gallou, C.; Beroud, C.; Demont, J.; Bouvier, R.; Schagger, H.; Godinot, C. Low mitochondrial respiratory chain content correlates with tumor aggressiveness in renal cell carcinoma. Carcinogenesis 2002, 23, 759–768. [Google Scholar] [CrossRef]
- Ganapathy-Kanniappan, S.; Geschwind, J.F. Tumor glycolysis as a target for cancer therapy: Progress and prospects. Mol. Cancer 2013, 12, 152–162. [Google Scholar] [CrossRef]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef]
- Meijer, T.W.; Kaanders, J.H.; Span, P.N.; Bussink, J. Targeting hypoxia, HIF-1, and tumor glucose metabolism to improve radiotherapy efficacy. Clin. Cancer Res. 2012, 18, 5585–5594. [Google Scholar] [CrossRef] [PubMed]
- Wanka, C.; Steinbach, J.P.; Rieger, J. Tp53-induced glycolysis and apoptosis regulator (TIGAR) protects glioma cells from starvation-induced cell death by up-regulating respiration and improving cellular redox homeostasis. J. Biol. Chem. 2012, 287, 33436–33446. [Google Scholar] [CrossRef] [PubMed]
- Pena-Rico, M.A.; Calvo-Vidal, M.N.; Villalonga-Planells, R.; Martinez-Soler, F.; Gimenez-Bonafe, P.; Navarro-Sabate, A.; Tortosa, A.; Bartrons, R.; Manzano, A. TP53 induced glycolysis and apoptosis regulator (TIGAR) knockdown results in radiosensitization of glioma cells. Radiother. Oncol. 2011, 101, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Bosch-Marce, M.; Shimoda, L.A.; Tan, Y.S.; Baek, J.H.; Wesley, J.B.; Gonzalez, F.J.; Semenza, G.L. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J. Biol. Chem. 2008, 283, 10892–10903. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H. ER stress and diseases. Febs J. 2007, 274, 630–658. [Google Scholar] [CrossRef]
- Brush, M.H.; Weiser, D.C.; Shenolikar, S. Growth arrest and DNA damage-inducible protein GADD34 targets protein phosphatase 1 alpha to the endoplasmic reticulum and promotes dephosphorylation of the alpha subunit of eukaryotic translation initiation factor 2. Mol. Cell. Biol. 2003, 23, 1292–1303. [Google Scholar] [CrossRef] [PubMed]
- Oyadomari, S.; Mori, M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004, 11, 381–389. [Google Scholar] [CrossRef]
- Maytin, E.V.; Ubeda, M.; Lin, J.C.; Habener, J.F. Stress-inducible transcription factor CHOP/gadd153 induces apoptosis in mammalian cells via p38 kinase-dependent and -independent mechanisms. Exp. Cell Res. 2001, 267, 193–204. [Google Scholar] [CrossRef]
- Yoshida, H.; Okada, T.; Haze, K.; Yanagi, H.; Yura, T.; Negishi, M.; Mori, K. ATF6 activated by proteolysis binds in the presence of NF-Y (CBF) directly to the cis-acting element responsible for the mammalian unfolded protein response. Mol. Cell. Biol. 2000, 20, 6755–6767. [Google Scholar] [CrossRef]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef]
- Ogata, M.; Hino, S.; Saito, A.; Morikawa, K.; Kondo, S.; Kanemoto, S.; Murakami, T.; Taniguchi, M.; Tanii, I.; Yoshinaga, K.; et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol. Cell. Biol. 2006, 26, 9220–9231. [Google Scholar] [CrossRef] [PubMed]
- McCullough, K.D.; Martindale, J.L.; Klotz, L.O.; Aw, T.Y.; Holbrook, N.J. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol. Cell. Biol. 2001, 21, 1249–1259. [Google Scholar] [CrossRef] [PubMed]
- Dadey, D.Y.; Kapoor, V.; Khudanyan, A.; Urano, F.; Kim, A.H.; Thotala, D.; Hallahan, D.E. The ATF6 pathway of the ER stress response contributes to enhanced viability in glioblastoma. Oncotarget 2016, 7, 2080–2092. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Kim, E.; Kim, W.; Seong, K.M.; Youn, H.; Kim, J.W.; Kim, J.; Youn, B. Rhamnetin and cirsiliol induce radiosensitization and inhibition of epithelial-mesenchymal transition (EMT) by miR-34a-mediated suppression of Notch-1 expression in non-small cell lung cancer cell lines. J. Biol. Chem. 2013, 288, 27343–27357. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Lee, Y.J.; Kang, S.; Lim, Y.B. Ionizing radiation activates PERK/eIF2alpha/ATF4 signaling via ER stress-independent pathway in human vascular endothelial cells. Int. J. Radiat. Biol. 2014, 90, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wang, Y.; Pang, X.; Su, Y.; Ai, G.; Wang, T. ER stress induced by ionising radiation in IEC-6 cells. Int. J. Radiat. Biol. 2010, 86, 429–435. [Google Scholar] [CrossRef]
- Wang, Y.; Alam, G.N.; Ning, Y.; Visioli, F.; Dong, Z.; Nor, J.E.; Polverini, P.J. The unfolded protein response induces the angiogenic switch in human tumor cells through the PERK/ATF4 pathway. Cancer Res. 2012, 72, 5396–5406. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, R.; Lipson, K.L.; Sargent, K.E.; Mercurio, A.M.; Hunt, J.S.; Ron, D.; Urano, F. Transcriptional regulation of VEGF-A by the unfolded protein response pathway. PLoS ONE 2010, 5. [Google Scholar] [CrossRef]
- Honscheid, P.; Datta, K.; Muders, M.H. Autophagy: Detection, regulation and its role in cancer and therapy response. Int. J. Radiat. Biol. 2014, 90, 628–635. [Google Scholar] [CrossRef]
- Yang, Y.; Yang, Y.; Yang, X.; Zhu, H.; Guo, Q.; Chen, X.; Zhang, H.; Cheng, H.; Sun, X. Autophagy and its function in radiosensitivity. Tumour Biol. 2015, 36, 4079–4087. [Google Scholar] [CrossRef]
- Ko, A.; Kanehisa, A.; Martins, I.; Senovilla, L.; Chargari, C.; Dugue, D.; Marino, G.; Kepp, O.; Michaud, M.; Perfettini, J.L.; et al. Autophagy inhibition radiosensitizes in vitro, yet reduces radioresponses in vivo due to deficient immunogenic signalling. Cell Death Differ. 2014, 21, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Noman, M.Z.; Janji, B.; Berchem, G.; Mami-Chouaib, F.; Chouaib, S. Hypoxia-induced autophagy: A new player in cancer immunotherapy? Autophagy 2012, 8, 704–706. [Google Scholar] [CrossRef] [PubMed]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouyssegur, J.; Mazure, N.M. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell. Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef] [PubMed]
- Maier, P.; Hartmann, L.; Wenz, F.; Herskind, C. Cellular Pathways in Response to Ionizing Radiation and Their Targetability for Tumor Radiosensitization. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef]
- Li, H.; Jin, X.; Chen, B.; Li, P.; Li, Q. Autophagy-regulating microRNAs: Potential targets for improving radiotherapy. J. Cancer Res. Clin. Oncol. 2018, 144, 1623–1634. [Google Scholar] [CrossRef]
- Palumbo, S.; Pirtoli, L.; Tini, P.; Cevenini, G.; Calderaro, F.; Toscano, M.; Miracco, C.; Comincini, S. Different involvement of autophagy in human malignant glioma cell lines undergoing irradiation and temozolomide combined treatments. J. Cell. Biochem. 2012, 113, 2308–2318. [Google Scholar] [CrossRef]
- Levine, B.; Yuan, J. Autophagy in cell death: An innocent convict? J. Clin. Invest. 2005, 115, 2679–2688. [Google Scholar] [CrossRef]
- Zhuang, W.; Li, B.; Long, L.; Chen, L.; Huang, Q.; Liang, Z. Induction of autophagy promotes differentiation of glioma-initiating cells and their radiosensitivity. Int. J. Cancer 2011, 129, 2720–2731. [Google Scholar] [CrossRef]
- Cui, L.; Song, Z.; Liang, B.; Jia, L.; Ma, S.; Liu, X. Radiation induces autophagic cell death via the p53/DRAM signaling pathway in breast cancer cells. Oncol. Rep. 2016, 35, 3639–3647. [Google Scholar] [CrossRef]
- Kim, K.W.; Mutter, R.W.; Cao, C.; Albert, J.M.; Freeman, M.; Hallahan, D.E.; Lu, B. Autophagy for cancer therapy through inhibition of pro-apoptotic proteins and mammalian target of rapamycin signaling. J. Biol. Chem. 2006, 281, 36883–36890. [Google Scholar] [CrossRef]
- Jing, Q.; Li, G.; Chen, X.; Liu, C.; Lu, S.; Zheng, H.; Ma, H.; Qin, Y.; Zhang, D.; Zhang, S.; et al. Wnt3a promotes radioresistance via autophagy in squamous cell carcinoma of the head and neck. J. Cell. Mol. Med. 2019, 23, 4711–4722. [Google Scholar] [CrossRef]
- Hu, J.L.; He, G.Y.; Lan, X.L.; Zeng, Z.C.; Guan, J.; Ding, Y.; Qian, X.L.; Liao, W.T.; Ding, Y.Q.; Liang, L. Inhibition of ATG12-mediated autophagy by miR-214 enhances radiosensitivity in colorectal cancer. Oncogenesis 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Pelicano, H.; Carney, D.; Huang, P. ROS stress in cancer cells and therapeutic implications. Drug Resist. Updat. 2004, 7, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Goodman, J.; Hochstein, P. Generation of free radicals and lipid peroxidation by redox cycling of adriamycin and daunomycin. Biochem. Biophys. Res. Commun. 1977, 77, 797–803. [Google Scholar] [CrossRef]
- Alexander, M.S.; Wilkes, J.G.; Schroeder, S.R.; Buettner, G.R.; Wagner, B.A.; Du, J.; Gibson-Corley, K.; O’Leary, B.R.; Spitz, D.R.; Buatti, J.M.; et al. Pharmacologic Ascorbate Reduces Radiation-Induced Normal Tissue Toxicity and Enhances Tumor Radiosensitization in Pancreatic Cancer. Cancer Res. 2018, 78, 6838–6851. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Cieslak, J.A.; Welsh, J.L.; Sibenaller, Z.A.; Allen, B.G.; Wagner, B.A.; Kalen, A.L.; Doskey, C.M.; Strother, R.K.; Button, A.M.; et al. Pharmacological Ascorbate Radiosensitizes Pancreatic Cancer. Cancer Res. 2015, 75, 3314–3326. [Google Scholar] [CrossRef] [PubMed]
- Mehta, M.P.; Shapiro, W.R.; Phan, S.C.; Gervais, R.; Carrie, C.; Chabot, P.; Patchell, R.A.; Glantz, M.J.; Recht, L.; Langer, C.; et al. Motexafin gadolinium combined with prompt whole brain radiotherapy prolongs time to neurologic progression in non-small-cell lung cancer patients with brain metastases: Results of a phase III trial. Int. J. Radiat. Oncol. Biol. Phys. 2009, 73, 1069–1076. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Zhao, Y.; Yu, S.; Zhang, M. Activated PI3K/Akt/COX-2 pathway induces resistance to radiation in human cervical cancer HeLa cells. Cancer Biother. Radiopharm. 2010, 25, 317–323. [Google Scholar] [CrossRef]
- Chatterjee, M.; Das, S.; Roy, K.; Chatterjee, M. Overexpression of 5-lipoxygenase and its relation with cell proliferation and angiogenesis in 7,12-dimethylbenz(alpha)anthracene-induced rat mammary carcinogenesis. Mol. Carcinog. 2013, 52, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Dadey, D.Y.A.; Kapoor, V.; Khudanyan, A.; Thotala, D.; Hallahan, D.E. PERK Regulates Glioblastoma Sensitivity to ER Stress Although Promoting Radiation Resistance. Mol. Cancer Res. 2018, 16, 1447–1453. [Google Scholar] [CrossRef]
- Rosenfeld, M.R.; Ye, X.; Supko, J.G.; Desideri, S.; Grossman, S.A.; Brem, S.; Mikkelson, T.; Wang, D.; Chang, Y.C.; Hu, J.; et al. A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy 2014, 10, 1359–1368. [Google Scholar] [CrossRef] [PubMed]
- Rojas-Puentes, L.L.; Gonzalez-Pinedo, M.; Crismatt, A.; Ortega-Gomez, A.; Gamboa-Vignolle, C.; Nunez-Gomez, R.; Dorantes-Gallareta, Y.; Arce-Salinas, C.; Arrieta, O. Phase II randomized, double-blind, placebo-controlled study of whole-brain irradiation with concomitant chloroquine for brain metastases. Radiat. Oncol. 2013, 8, 209. [Google Scholar] [CrossRef] [PubMed]
- Hennessey, D.; Martin, L.M.; Atzberger, A.; Lynch, T.H.; Hollywood, D.; Marignol, L. Exposure to hypoxia following irradiation increases radioresistance in prostate cancer cells. Urol. Oncol. 2013, 31, 1106–1116. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Suo, C.; Zheng, C.; Zhang, H. Hypoxia and Metabolism in Metastasis. Adv. Exp. Med. Biol. 2019, 1136, 87–95. [Google Scholar] [PubMed]
- Manoochehri Khoshinani, H.; Afshar, S.; Najafi, R. Hypoxia: A Double-Edged Sword in Cancer Therapy. Cancer Invest. 2016, 34, 536–545. [Google Scholar] [CrossRef] [PubMed]
- Dewhirst, M.W.; Vujaskovic, Z.; Jones, E.; Thrall, D. Re-setting the biologic rationale for thermal therapy. Int. J. Hyperthermia 2005, 21, 779–790. [Google Scholar] [CrossRef]
- Elming, P.B.; Sorensen, B.S.; Oei, A.L.; Franken, N.A.P.; Crezee, J.; Overgaard, J.; Horsman, M.R. Hyperthermia: The Optimal Treatment to Overcome Radiation Resistant Hypoxia. Cancers 2019, 11. [Google Scholar] [CrossRef]
- Peeken, J.C.; Vaupel, P.; Combs, S.E. Integrating Hyperthermia into Modern Radiation Oncology: What Evidence Is Necessary? Front. Oncol. 2017, 7. [Google Scholar] [CrossRef]
- Lee, S.; Son, B.; Park, G.; Kim, H.; Kang, H.; Jeon, J.; Youn, H.; Youn, B. Immunogenic Effect of Hyperthermia on Enhancing Radiotherapeutic Efficacy. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef]
- Lauber, K.; Brix, N.; Ernst, A.; Hennel, R.; Krombach, J.; Anders, H.; Belka, C. Targeting the heat shock response in combination with radiotherapy: Sensitizing cancer cells to irradiation-induced cell death and heating up their immunogenicity. Cancer Lett. 2015, 368, 209–229. [Google Scholar] [CrossRef]
- Mahmood, J.; Shukla, H.D.; Soman, S.; Samanta, S.; Singh, P.; Kamlapurkar, S.; Saeed, A.; Amin, N.P.; Vujaskovic, Z. Immunotherapy, Radiotherapy, and Hyperthermia: A Combined Therapeutic Approach in Pancreatic Cancer Treatment. Cancers 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Laszlo, A.; Fleischer, I. Heat-induced perturbations of DNA damage signaling pathways are modulated by molecular chaperones. Cancer Res. 2009, 69, 2042–2049. [Google Scholar] [CrossRef] [PubMed]
- Van den Tempel, N.; Laffeber, C.; Odijk, H.; van Cappellen, W.A.; van Rhoon, G.C.; Franckena, M.; Kanaar, R. The effect of thermal dose on hyperthermia-mediated inhibition of DNA repair through homologous recombination. Oncotarget 2017, 8, 44593–44604. [Google Scholar] [PubMed]
- Van Oorschot, B.; Granata, G.; di Franco, S.; Ten Cate, R.; Rodermond, H.M.; Todaro, M.; Medema, J.P.; Franken, N.A. Targeting DNA double strand break repair with hyperthermia and DNA-PKcs inhibition to enhance the effect of radiation treatment. Oncotarget 2016, 7, 65504–65513. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Stress-Responsive Signaling for Radioresistance | Associated Molecules | Refs | |
|---|---|---|---|
| ROS stress response | Upregulation of antioxidants | p53, SODs, glutathione peroxidase 1, sestrin | [24,25,26] |
| Adaptation to hypoxia and inhibition of ROS production | HIF1, VEGF, PDK1 | [33,34,35,36] | |
| DNA damage response | Upregulation of DNA damage-sensing and repair proteins | ATM, γH2AX, DNA-PK, ATR, MDC1, BRCA1, BRCA2 | [58,59,60] |
| Subcellular organelle response | Production of bioactive lipid metabolites | HNE (non-protein), COXs, LOXs | [63,70,71,72,73] |
| Glycolytic reprogramming and mitochondrial malfunction | HIF1, PDK1 | [81,82,83,84] | |
| Activation of UPR signaling | PERK, ATF4, ATF6, IRE1 | [93,94,95,96,97,98] | |
| Autophagy | Activation of cytoprotective autophagy | ATGs, ULK1, Beclin-1 | [100,101,102,103,104,105] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, W.; Lee, S.; Seo, D.; Kim, D.; Kim, K.; Kim, E.; Kang, J.; Seong, K.M.; Youn, H.; Youn, B. Cellular Stress Responses in Radiotherapy. Cells 2019, 8, 1105. https://doi.org/10.3390/cells8091105
Kim W, Lee S, Seo D, Kim D, Kim K, Kim E, Kang J, Seong KM, Youn H, Youn B. Cellular Stress Responses in Radiotherapy. Cells. 2019; 8(9):1105. https://doi.org/10.3390/cells8091105
Chicago/Turabian StyleKim, Wanyeon, Sungmin Lee, Danbi Seo, Dain Kim, Kyeongmin Kim, EunGi Kim, JiHoon Kang, Ki Moon Seong, HyeSook Youn, and BuHyun Youn. 2019. "Cellular Stress Responses in Radiotherapy" Cells 8, no. 9: 1105. https://doi.org/10.3390/cells8091105
APA StyleKim, W., Lee, S., Seo, D., Kim, D., Kim, K., Kim, E., Kang, J., Seong, K. M., Youn, H., & Youn, B. (2019). Cellular Stress Responses in Radiotherapy. Cells, 8(9), 1105. https://doi.org/10.3390/cells8091105