Increased Glycolysis and Higher Lactate Production in Hyperglycemic Myotubes

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Ethics Statement
2.3. Cell Culturing
2.4. Substrate Oxidation Assay
2.5. Mitochondrial Stress Assay
2.6. Glycolytic Rate Analysis
2.7. Measurement of ROS Concentration
2.8. Measurement of Lactate Concentration
2.9. Immunoblotting
2.10. Microarray Analysis
2.11. Statistics
3. Results
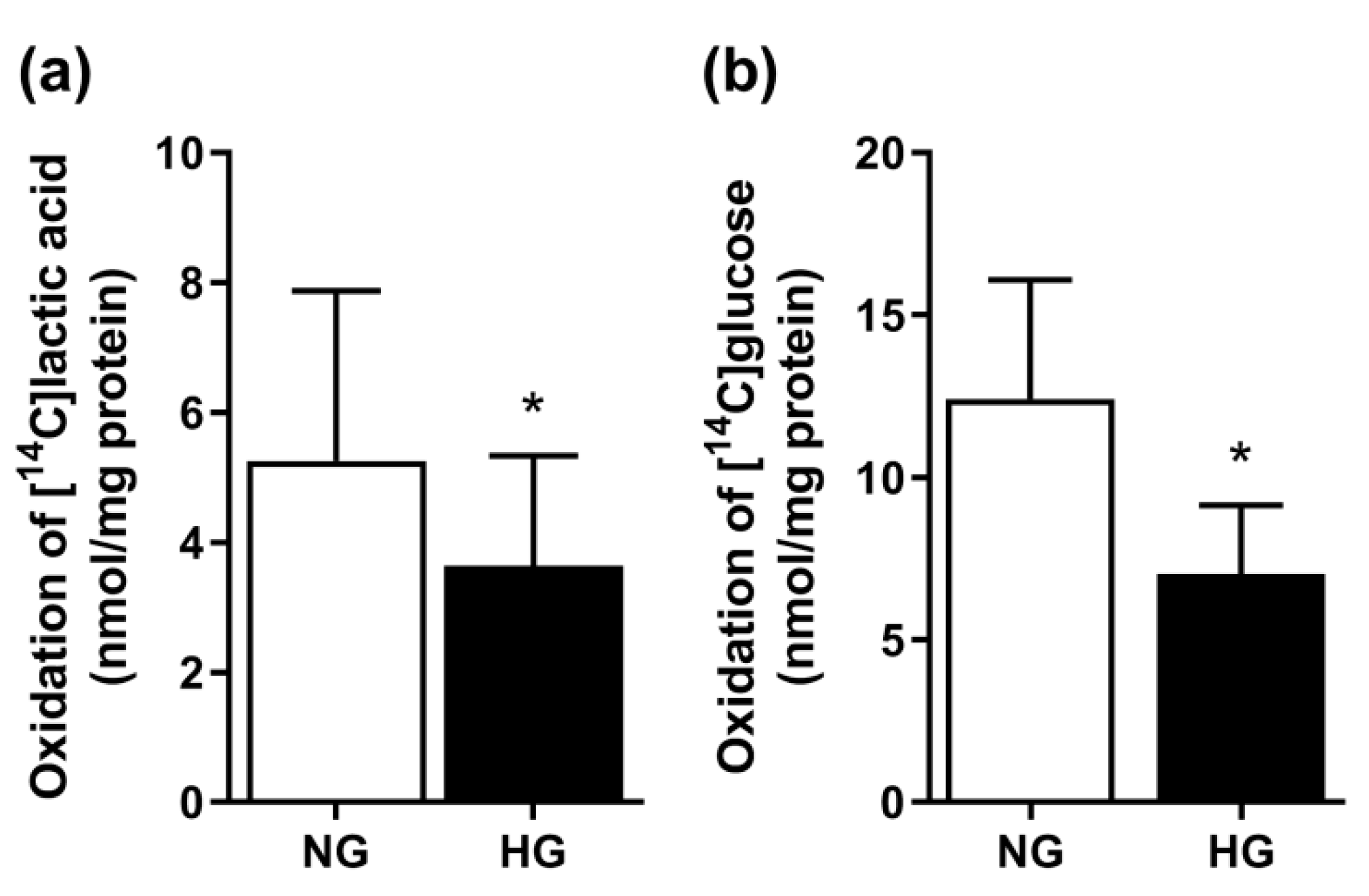
3.1. Effect of Chronic Hyperglycemia on Glucose and Lactic Acid Oxidation
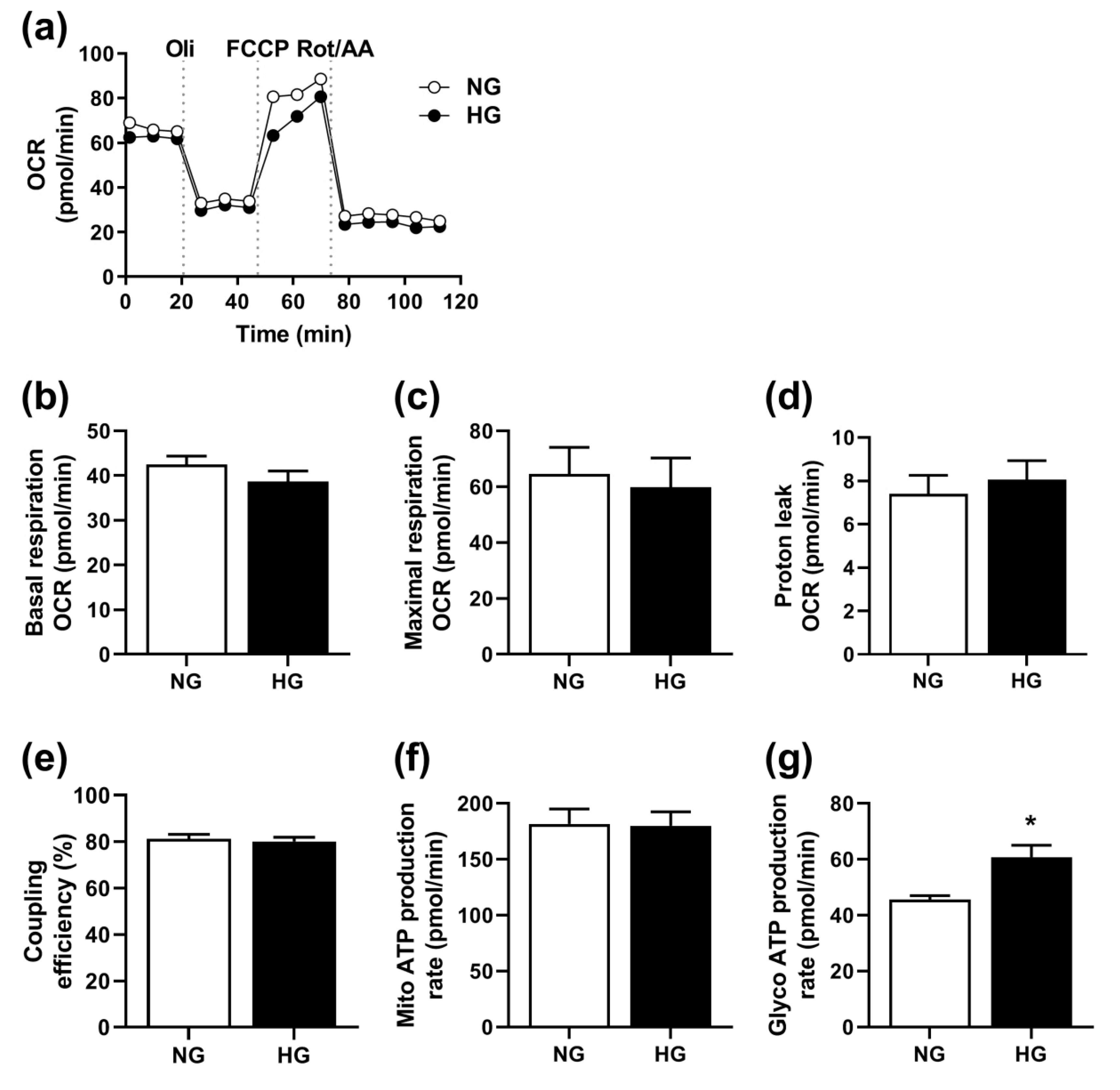
3.2. Effect of Hyperglycemia on Mitochondrial Function
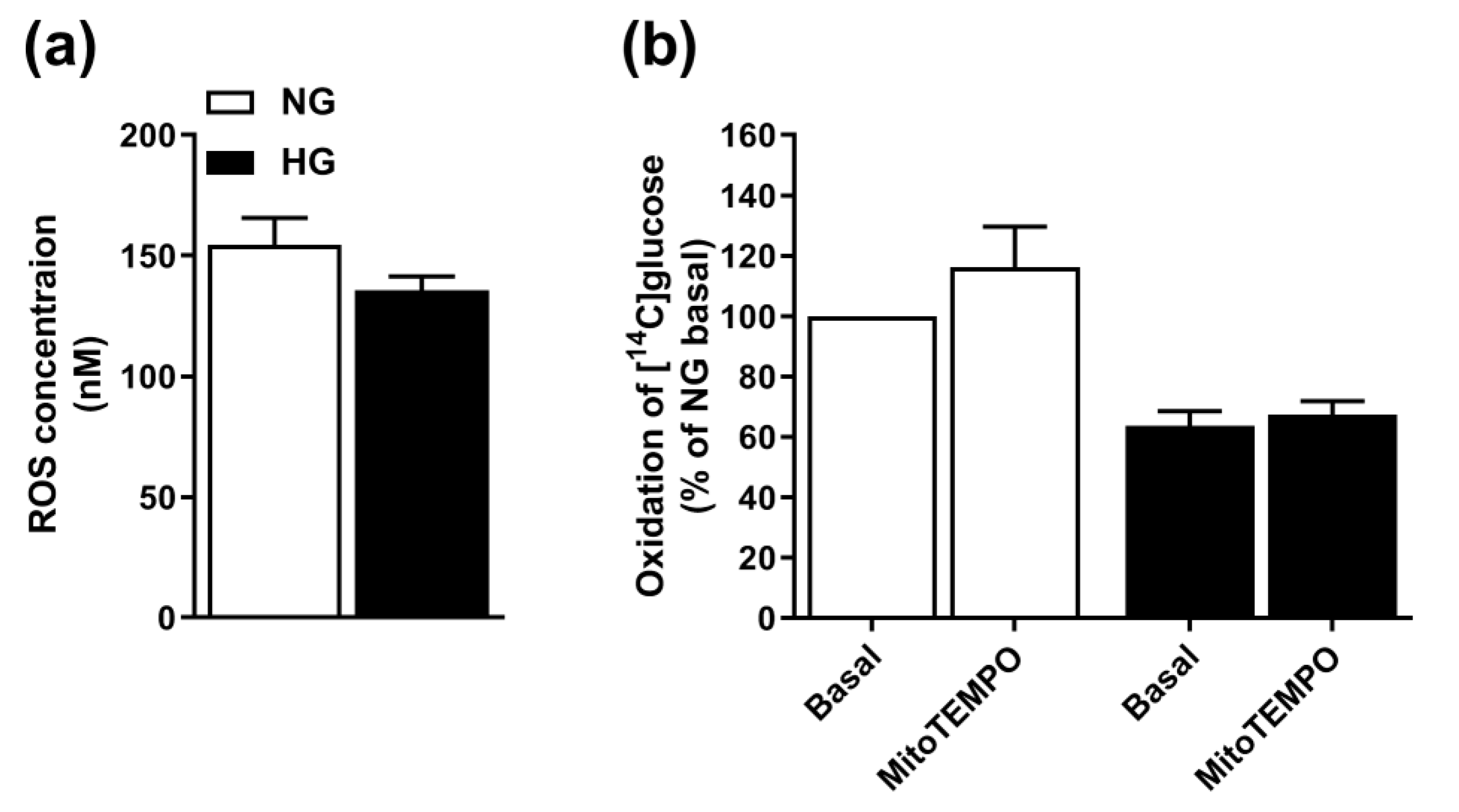
3.3. Involvement of ROS in Hyperglycemia
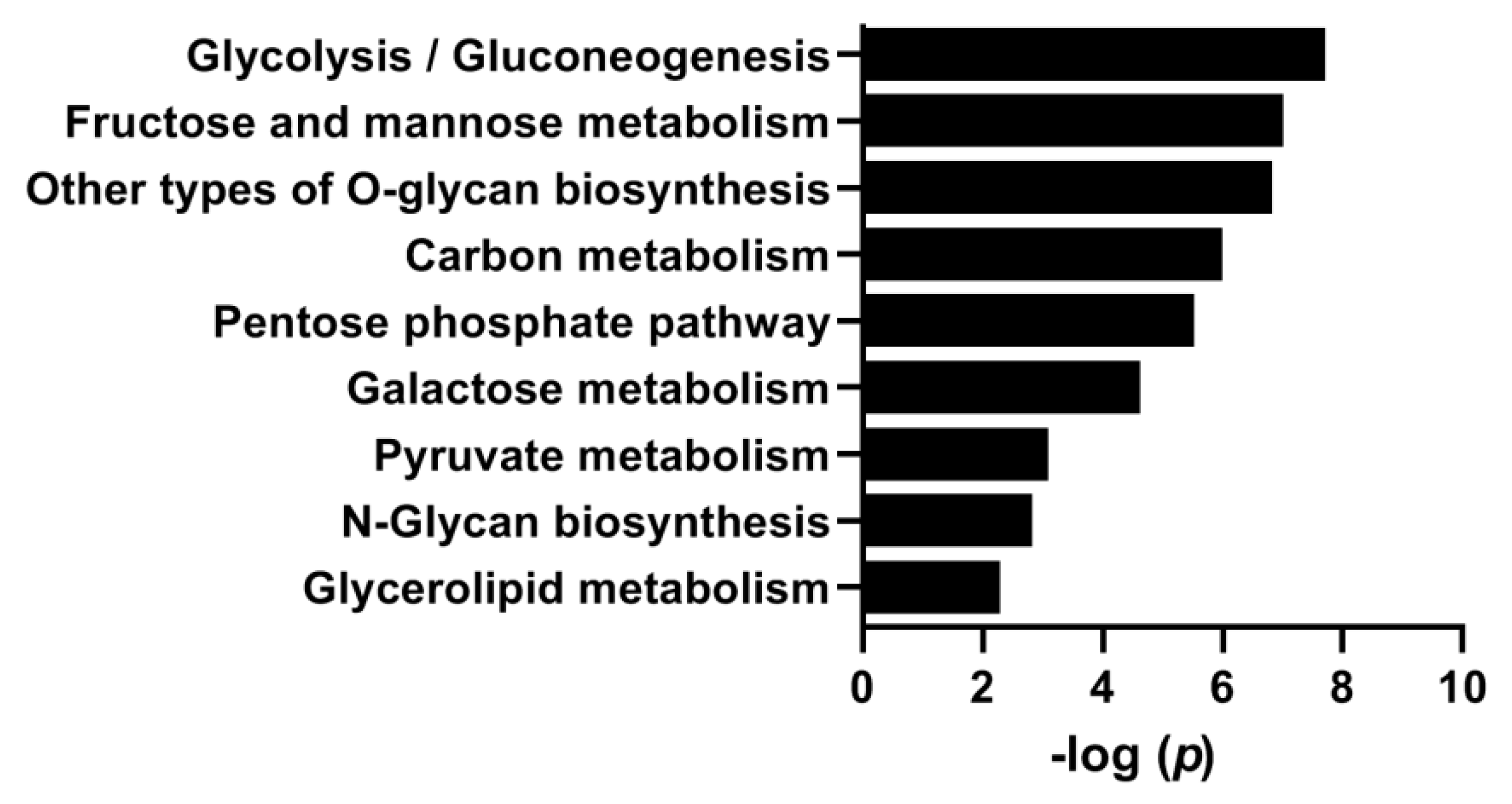
3.4. Effects of Hyperglycemia on Gene Expression
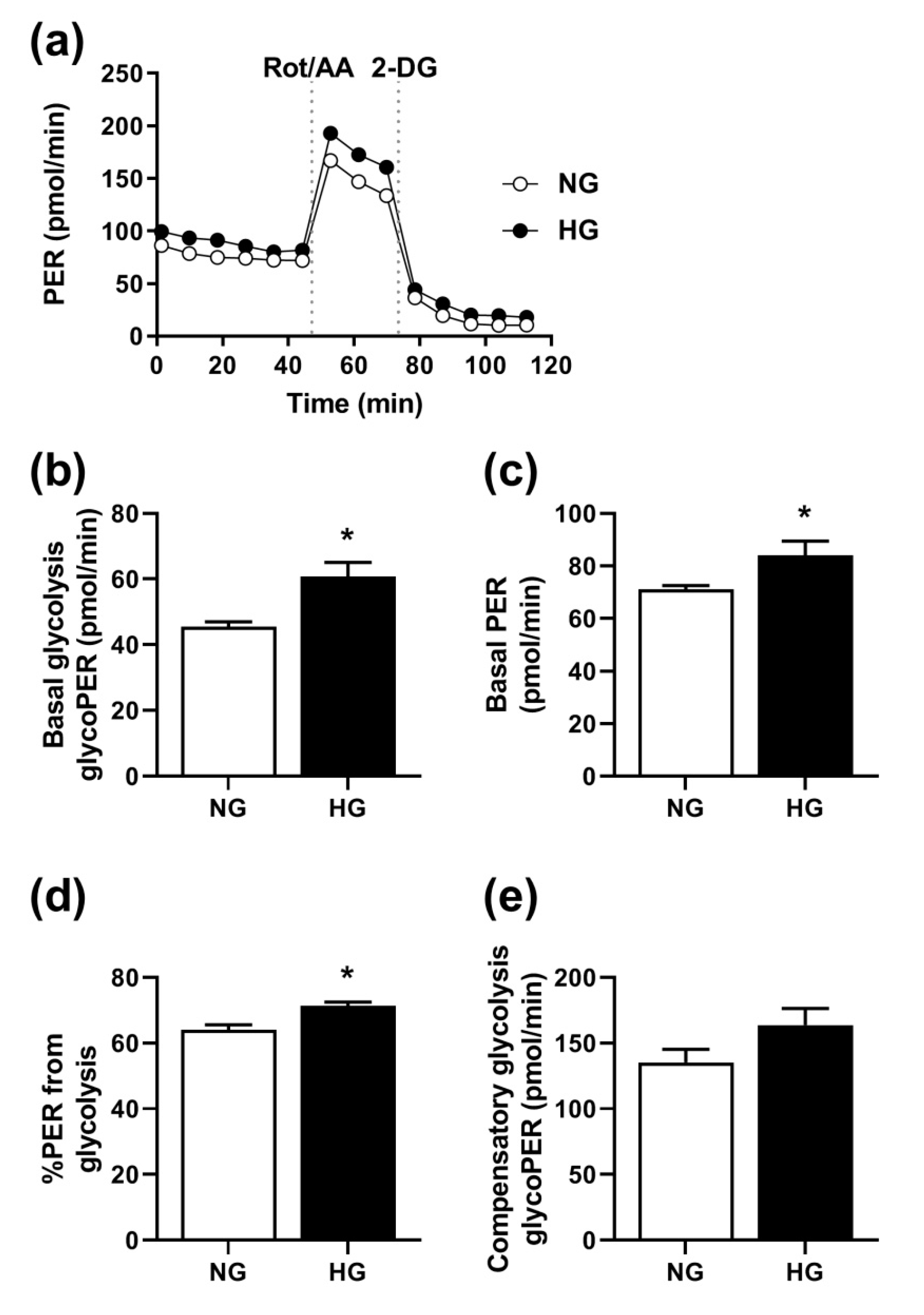
3.5. Effect of Hyperglycemia on Glycolysis
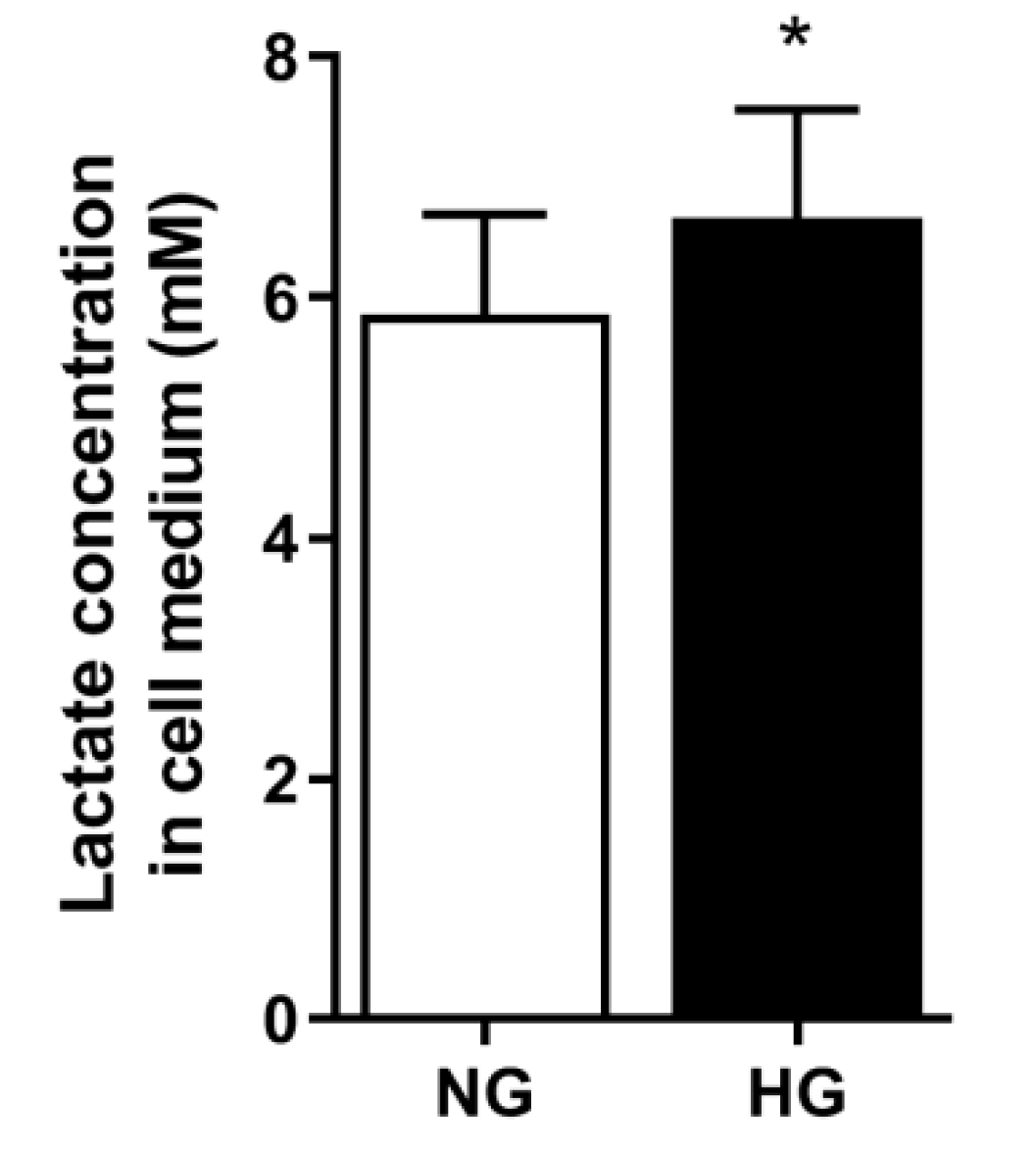
3.6. Lactate Concentration in Media
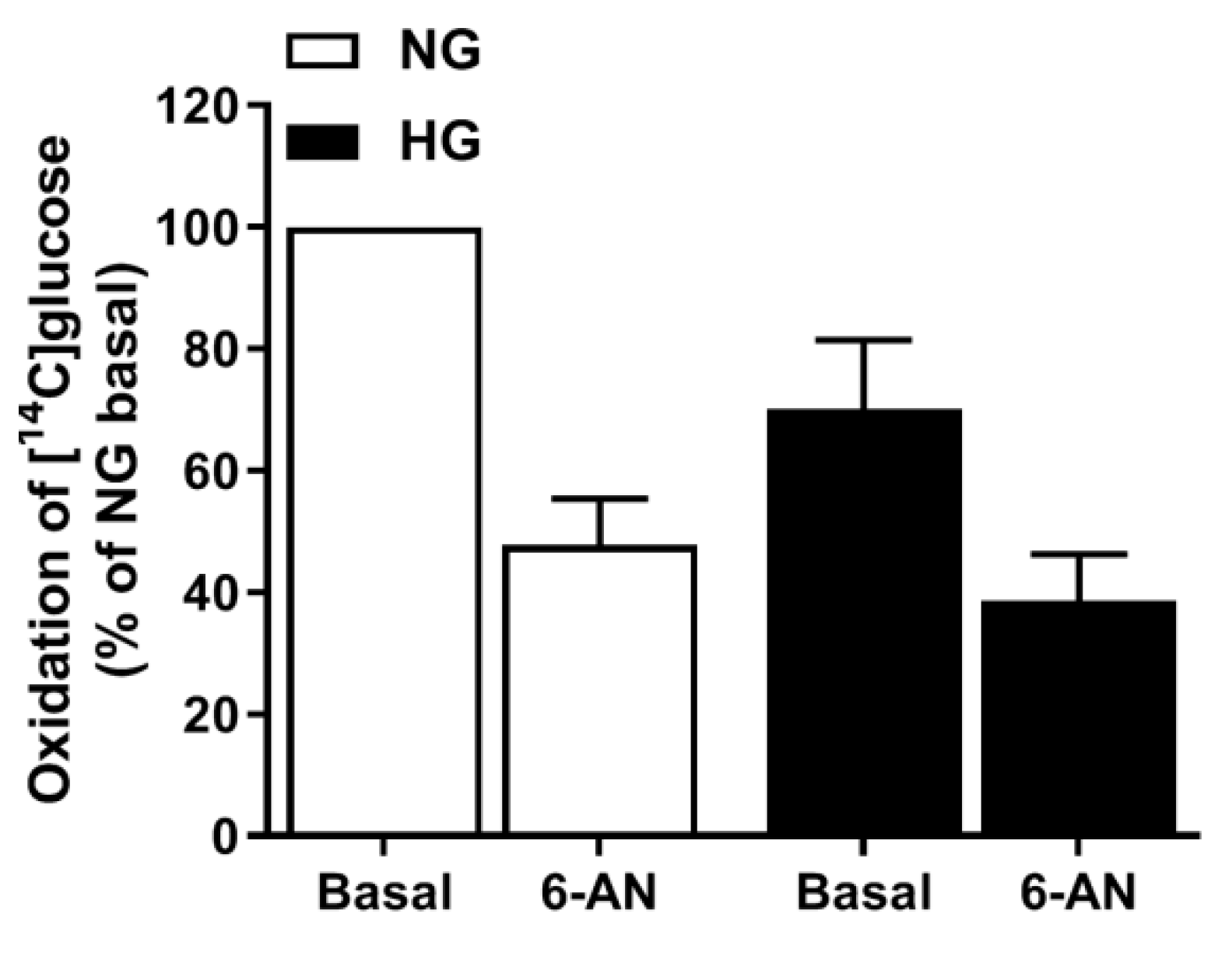
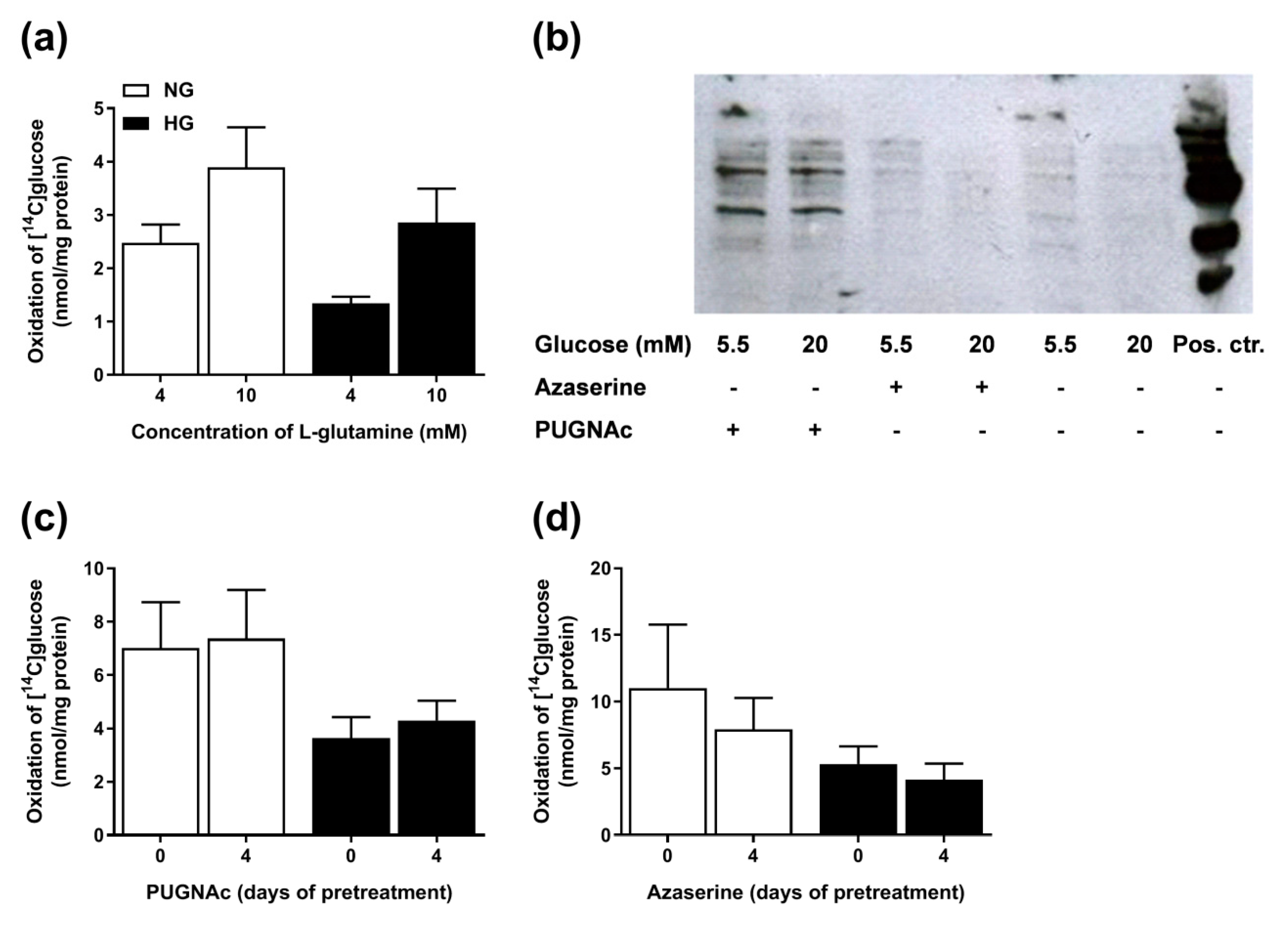
3.7. Involvement of the PPP in Hyperglycemia
3.8. Involvement of the Hexosamine Pathway
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rossetti, L. Glucose toxicity: The implications of hyperglycemia in the pathophysiology of diabetes mellitus. Clin. Investig. Med. 1995, 18, 255–260. [Google Scholar]
- Laakso, M. Hyperglycemia and cardiovascular disease in type 2 diabetes. Diabetes 1999, 48, 937–942. [Google Scholar] [CrossRef] [PubMed]
- Ceriello, A. Postprandial hyperglycemia and diabetes complications: Is it time to treat? Diabetes 2005, 54, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Testa, R.; Bonfigli, A.R.; Prattichizzo, F.; La Sala, L.; De Nigris, V.; Ceriello, A. The “Metabolic Memory” Theory and the Early Treatment of Hyperglycemia in Prevention of Diabetic Complications. Nutrients 2017, 9, 437. [Google Scholar] [CrossRef] [PubMed]
- Robertson, R.P. Chronic Oxidative Stress as a Central Mechanism for Glucose Toxicity in Pancreatic Islet Beta Cells in Diabetes. J. Boil. Chem. 2004, 279, 42351–42354. [Google Scholar] [CrossRef] [PubMed]
- Aas, V.; Hessvik, N.P.; Wettergreen, M.; Hvammen, A.W.; Hallen, S.; Thoresen, G.H.; Rustan, A.C. Chronic hyperglycemia reduces substrate oxidation and impairs metabolic switching of human myotubes. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2011, 1812, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Aas, V.; Kase, E.T.; Solberg, R.; Jensen, J.; Rustan, A.C. Chronic hyperglycaemia promotes lipogenesis and triacylglycerol accumulation in human skeletal muscle cells. Diabetologia 2004, 47, 1452–1461. [Google Scholar] [CrossRef]
- Yan, L.J. Pathogenesis of chronic hyperglycemia: From reductive stress to oxidative stress. J. Diabetes. Res. 2014, 2014. [Google Scholar] [CrossRef]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef]
- Chen, Y.D.; Varasteh, B.B.; Reaven, G.M. Plasma lactate concentration in obesity and type 2 diabetes. Diabete Metab. 1993, 19, 348–354. [Google Scholar]
- Leite, T.C.; Coelho, R.G.; Da Silva, D.; Coelho, W.S.; Marinho-Carvalho, M.M.; Sola-Penna, M. Lactate downregulates the glycolytic enzymes hexokinase and phosphofructokinase in diverse tissues from mice. FEBS Lett. 2011, 585, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, A.M.; Fabris, R.; Bassetto, F.; Serra, R.; Leturque, A.; Federspil, G.; Girard, J.; Vettor, R. Hyperlactatemia reduces muscle glucose uptake and GLUT-4 mRNA while increasing (E1alpha)PDH gene expression in rat. Am. J. Physiol. Content 1999, 276, 922–929. [Google Scholar]
- Lund, J.; Tangen, D.S.; Wiig, H.; Stadheim, H.K.; Helle, S.A.; Birk, J.B.; Ingemann-Hansen, T.; Rustan, A.C.; Thoresen, G.H.; Wojtaszewski, J.F. Glucose metabolism and metabolic flexibility in cultured skeletal muscle cells is related to exercise status in young male subjects. Arch. Physiol. Biochem. 2018, 124, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Wensaas, A.J.; Rustan, A.C.; Lövstedt, K.; Kull, B.; Wikström, S.; Drevon, C.A.; Hallén, S. Cell-based multiwell assays for the detection of substrate accumulation and oxidation. J. Lipid Res. 2007, 48, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Romero, N.; Swain, P.; Neilson, A. Improving Quantification of Cellular Glycolytic Rate Using Agilent Seahorse XF Technology (White Paper); Agilent Technologies, Inc.: Lexington, MA, USA, 2017; pp. 1–10. [Google Scholar]
- Selig, J.I.; Ouwens, M.D.; Fischer, J.W.; Lichtenberg, A.; Akhyari, P.; Barth, M. Impact of Hyperinsulinemia and Hyperglycemia on Valvular Interstitial Cells—A Link between Calcific Aortic Valve Disease and Type 2 Diabetes. Thorac. Cardiovasc. Surg. 2018, 66, S1–S110. [Google Scholar] [CrossRef]
- Selig, J.I.; Ouwens, D.M.; Raschke, S.; Thoresen, G.H.; Fischer, J.W.; Lichtenberg, A.; Akhyari, P.; Barth, M. Impact of hyperinsulinemia and hyperglycemia on valvular interstitial cells–A link between aortic heart valve degeneration and type 2 diabetes. Biochim. Biophys. Acta. Mol. Basis Dis. 2019, 1865, 2526–2537. [Google Scholar] [CrossRef] [PubMed]
- Hörbelt, T.; Knebel, B.; Fahlbusch, P.; Barbosa, D.; de Wiza, D.H.; Van de Velde, F.; Van Nieuwenhove, Y.; Lapauw, B.; Thoresen, G.H.; Al-Hasani, H. The adipokine sFRP4 induces insulin resistance and lipogenesis in the liver. Biochim. Biophys. Acta. Mol. Basis Dis. 2019, 1865, 2671–2684. [Google Scholar] [CrossRef]
- Lund, J.; Aas, V.; Tingstad, R.H.; Van Hees, A.; Nikolic, N. Utilization of lactic acid in human myotubes and interplay with glucose and fatty acid metabolism. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef]
- Richter, E.A.; Hansen, B.F.; Hansen, S.A. Glucose-induced insulin resistance of skeletal-muscle glucose transport and uptake. Biochem. J. 1988, 252, 733–737. [Google Scholar] [CrossRef]
- Hansen, B.F.; Hansen, S.A.; Ploug, T.; Bak, J.F.; Richter, E.A. Effects of glucose and insulin on development of impaired insulin action in muscle. Am. J. Physiol. Metab. 1992, 262, E440–E446. [Google Scholar] [CrossRef]
- Boden, G.; Ruiz, J.; Kim, C.J.; Chen, X. Effects of prolonged glucose infusion on insulin secretion, clearance, and action in normal subjects. Am. J. Physiol. Metab. 1996, 270, E251–E258. [Google Scholar] [CrossRef] [PubMed]
- Kawanaka, K.; Han, D.-H.; Gao, J.; Nolte, L.A.; Holloszy, J.O. Development of Glucose-induced Insulin Resistance in Muscle Requires Protein Synthesis. J. Boil. Chem. 2001, 276, 20101–20107. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, M. The Pathobiology of Diabetic Complications: A Unifying Mechanism. Diabetes 2005, 54, 1615–1625. [Google Scholar] [CrossRef] [PubMed]
- Palmeira, C.M.; Rolo, A.P.; Berthiaume, J.; Bjork, J.A.; Wallace, K.B. Hyperglycemia decreases mitochondrial function: The regulatory role of mitochondrial biogenesis. Toxicol. Appl. Pharmacol. 2007, 225, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Robotham, J.L.; Yoon, Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc. Natl. Acad. Sci. USA 2006, 103, 2653–2658. [Google Scholar] [CrossRef] [PubMed]
- Sedlic, F.; Muravyeva, M.Y.; Sepac, A.; Sedlic, M.; Williams, A.M.; Yang, M.; Bai, X.; Bosnjak, Z.J. Targeted Modification of Mitochondrial ROS Production Converts High Glucose-Induced Cytotoxicity to Cytoprotection: Effects on Anesthetic Preconditioning. J. Cell. Physiol. 2017, 232, 216–224. [Google Scholar] [CrossRef]
- Wautier, M.-P.; Chappey, O.; Corda, S.; Stern, D.M.; Schmidt, A.M.; Wautier, J.-L. Activation of NADPH oxidase by AGE links oxidant stress to altered gene expression via RAGE. Am. J. Physiol. Metab. 2001, 280, E685–E694. [Google Scholar] [CrossRef]
- Nishikawa, T.; Edelstein, D.; Brownlee, M. The missing link: A single unifying mechanism for diabetic complications. Kidney Int. 2000, 58, S26–S30. [Google Scholar] [CrossRef]
- Nishikawa, T.; Edelstein, D.; Du, X.L.; Yamagishi, S.-I.; Matsumura, T.; Kaneda, Y.; Yorek, M.A.; Beebe, D.; Oates, P.J.; Hammes, H.-P. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000, 404, 787–790. [Google Scholar] [CrossRef]
- Slawson, C.; Copeland, R.; Hart, G. O-GlcNAc Signaling: A Metabolic Link between Diabetes and Cancer? Trends Biochem. Sci. 2010, 35, 547–555. [Google Scholar] [CrossRef]
- Hu, Y.; Suarez, J.; Fricovsky, E.; Wang, H.; Scott, B.T.; Trauger, S.A.; Han, W.; Hu, Y.; Oyeleye, M.O.; Dillmann, W.H. Increased enzymatic o-GlcNAcylation of mitochondrial proteins impairs mitochondrial function in cardiac myocytes exposed to high glucose. J. Biol. Chem. 2009, 284, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Du, X.-L.; Edelstein, D.; Rossetti, L.; Fantus, I.G.; Goldberg, H.; Ziyadeh, F.; Wu, J.; Brownlee, M. Hyperglycemia-induced mitochondrial superoxide overproduction activates the hexosamine pathway and induces plasminogen activator inhibitor-1 expression by increasing Sp1 glycosylation. Proc. Natl. Acad. Sci. USA 2000, 97, 12222–12226. [Google Scholar] [CrossRef] [PubMed]
- Stincone, A.; Prigione, A.; Cramer, T.; Wamelink, M.M.; Campbell, K.; Cheung, E.; Olin-Sandoval, V.; Grüning, N.M.; Krüger, A.; Tauqeer Alam, M. The return of metabolism: Biochemistry and physiology of the pentose phosphate pathway. Biol. Rev. 2015, 90, 927–963. [Google Scholar] [CrossRef] [PubMed]
- Patra, K.C.; Hay, N. The pentose phosphate pathway and cancer. Trends Biochem. Sci. 2014, 39, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Rinnerthaler, M.; Büttner, S.; Laun, P.; Heeren, G.; Felder, T.K.; Klinger, H.; Weinberger, M.; Stolze, K.; Groušl, T.; Hasek, J.; et al. Yno1p/Aim14p, a NADPH-oxidase ortholog, controls extramitochondrial reactive oxygen species generation, apoptosis, and actin cable formation in yeast. Proc. Natl. Acad. Sci. USA 2012, 109, 8658–8663. [Google Scholar] [CrossRef] [PubMed]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lund, J.; Ouwens, D.M.; Wettergreen, M.; Bakke, S.S.; Thoresen, G.H.; Aas, V. Increased Glycolysis and Higher Lactate Production in Hyperglycemic Myotubes. Cells 2019, 8, 1101. https://doi.org/10.3390/cells8091101
Lund J, Ouwens DM, Wettergreen M, Bakke SS, Thoresen GH, Aas V. Increased Glycolysis and Higher Lactate Production in Hyperglycemic Myotubes. Cells. 2019; 8(9):1101. https://doi.org/10.3390/cells8091101
Chicago/Turabian StyleLund, Jenny, D. Margriet Ouwens, Marianne Wettergreen, Siril S. Bakke, G. Hege Thoresen, and Vigdis Aas. 2019. "Increased Glycolysis and Higher Lactate Production in Hyperglycemic Myotubes" Cells 8, no. 9: 1101. https://doi.org/10.3390/cells8091101
APA StyleLund, J., Ouwens, D. M., Wettergreen, M., Bakke, S. S., Thoresen, G. H., & Aas, V. (2019). Increased Glycolysis and Higher Lactate Production in Hyperglycemic Myotubes. Cells, 8(9), 1101. https://doi.org/10.3390/cells8091101