CX3CR1 Mediates the Development of Monocyte-Derived Dendritic Cells during Hepatic Inflammation

 ,
,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Mice and Experimental Protocol
2.2. Assessment of Liver Injury
2.3. Flow Cytometry Analysis and Cell Sorting of Liver Leukocytes
2.4. mRNA Extraction and Real-Time PCR
2.5. In Vitro moDC Differentiation from Bone Marrow Myeloid Cells
2.6. Data Analysis and Statistical Calculations
3. Results
3.1. Characterization of Myeloid Dendritic Cells Associated with Acute Liver Inflammation
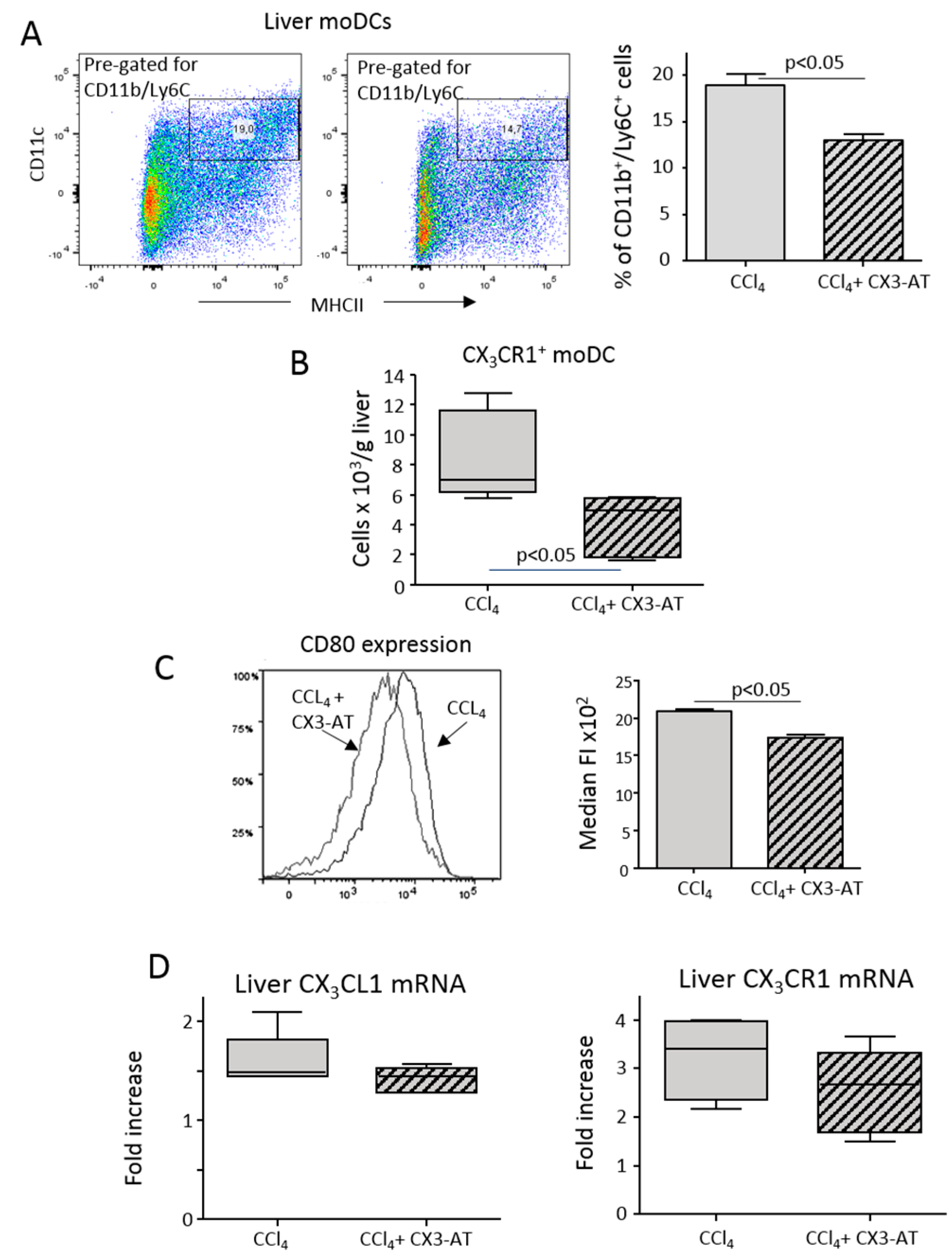
3.2. Interference with CX3CR1 Affects Monocyte Maturation to Dendritic Cells in Injured Livers
3.3. Interference with CX3CR1-Mediated moDC Differentiation Ameliorates Acute Liver Injury and Inflammation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Heymann, F.; Tacke, F. Immunology in the liver-from homeostasis to disease. Nar. Rev. Gastroenterol. Hepatol. 2016, 13, 88–110. [Google Scholar] [CrossRef] [PubMed]
- Koyama, Y.; Benner, D.A. Liver inflammation and fibrosis. J. Clin. Investig. 2017, 127, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Krenker, O.; Tacke, F. Liver macrophages in tissue homeostasis and disease. Nat. Rev. Immunol. 2017, 17, 306–321. [Google Scholar] [CrossRef] [PubMed]
- Rahman, A.H.; Aloman, C. Dendritic cells and liver fibrosis. Biochim. Biophys. Acta 2013, 1832, 998–1004. [Google Scholar] [CrossRef]
- David, B.A.; Rezende, R.M.; Antunes, M.M.; Santos, M.M.; Freitas Lopes, M.A.; Diniz, A.B.; Sousa Pereira, R.V.; Marchesi, S.C.; Alvarenga, D.M.; Nakagaki, B.N.; et al. Combination of mass cytometry and imaging analysis reveals origin, location, and functional repopulation of liver myeloid cells in mice. Gastroenterology 2016, 151, 1176–1191. [Google Scholar] [CrossRef] [PubMed]
- Eckert, C.; Klein, N.; Kormek, M.; Lukacs-Kormek, V. The complex myeloid network of the liver with diverse functional capacity at steady state and in inflammation. Front. Immunol. 2016, 6, 179. [Google Scholar] [CrossRef]
- Doherty, D.G. Antigen-presenting cell function in the tolerogenic liver environment. J. Autoimmun. 2016, 66, 60–75. [Google Scholar] [CrossRef]
- Dou, L.; Ono, Y.; Chen, Y.F.; Thomson, A.W.; Chen, X.P. Hepatic Dendritic Cells, the Tolerogenic Liver Environment, and Liver Disease. Semin. Liver Dis. 2018, 38, 170–180. [Google Scholar] [CrossRef]
- Connolly, M.K.; Bedrosian, A.S.; Mallen-St Clair, J.; Mitchell, A.P.; Ibrahim, J.; Stroud, A.; Pachter, H.L.; Bar-Sagi, D.; Frey, A.B.; Miller, G. In liver fibrosis, dendritic cells govern hepatic inflammation in mice via TNF-alpha. J. Clin. Investig. 2009, 119, 3213–3225. [Google Scholar]
- Henning, J.R.; Graffeo, C.S.; Rehman, A.; Fallon, N.C.; Zambirinis, C.P.; Ochi, A.; Barilla, R.; Jamal, M.; Deutsch, M.; Greco, S.; et al. Dendritic cells limit fibroinflammatory injury in nonalcoholic steatohepatitis in mice. Hepatology 2013, 58, 589–602. [Google Scholar] [CrossRef]
- Sutti, S.; Locatelli, I.; Bruzzì, S.; Jindal, A.; Vacchiano, M.; Bozzola, C.; Albano, E. CX3CR1-expressing inflammatory dendritic cells contribute to the progression of steatohepatitis. Clin. Sci. 2015, 129, 797–808. [Google Scholar] [CrossRef] [PubMed]
- Bamboat, Z.M.; Ocuin, L.M.; Balachandran, V.P.; Obaid, H.; Plitas, G.; DeMatteo, R.P. Conventional DCs reduce liver ischemia/reperfusion injury in mice via IL-10 secretion. J. Clin. Investig. 2010, 120, 559–569. [Google Scholar] [CrossRef] [PubMed]
- Connolly, M.K.; Ayo, D.; Malhotra, A.; Hackman, M.; Bedrosian, A.S.; Ibrahim, J.; Cieza-Rubio, N.E.; Nguyen, A.H.; Henning, J.R.; Dorvil-Castro, M.; et al. Dendritic cell depletion exacerbates acetaminophen hepatotoxicity. Hepatology 2011, 54, 959–968. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Ueki, S.; Kimura, S.; Yoshida, O.; Castellaneta, A.; Ozaki, K.S.; Demetris, A.J.; Ross, M.; Vodovotz, Y.; Thomson, A.W.B.; et al. Roles of dendritic cells in murine hepatic warm and liver transplantation-induced cold ischemia/reperfusion injury. Hepatology 2013, 57, 1585–1596. [Google Scholar] [CrossRef] [PubMed]
- Heier, E.C.; Meier, A.; Julich-Haertel, H.; Djudjaj, S.; Rau, M.; Tschernig, T.; Geier, A.; Boor, P.; Lammert, F.; Lukacs-Kornek, V. Murine CD103+ dendritic cells protect against steatosis progression towards steatohepatitis. J. Hepatol. 2017, 66, 1241–1250. [Google Scholar] [CrossRef] [PubMed]
- Sutti, S.; Heymann, F.; Bruzzì, S.; Peusquens, J.; Trautwein, C.; Albano, E.; Tacke, F. CX3CR1 modulates the anti-inflammatory activity of hepatic dendritic cells in response to acute liver injury. Clin. Sci. 2017, 131, 2289–2301. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, J.; Nguyen, A.H.; Rehman, A.; Ochi, A.; Jamal, M.; Graffeo, C.S.; Henning, J.R.; Zambirinis, C.P.; Fallon, N.C.; Barilla, R.; et al. Dendritic cell populations with different concentrations of lipid regulate tolerance and immunity in mouse and human liver. Gastroenterology 2012, 143, 1061–1072. [Google Scholar] [CrossRef]
- Sumpter, T.L.; Dangi, A.; Matta, B.M.; Huang, C.; Stolz, D.B.; Vodovotz, Y.; Thomson, A.W.; Gandhi, C.R. Hepatic stellate cells undermine the allostimulatory function of liver myeloid dendritic cells via STAT3-dependent induction of IDO. J. Immunol. 2012, 189, 3848–3858. [Google Scholar] [CrossRef]
- Krueger, P.D.; Kim, T.S.; Sung, S.S.; Braciale, T.J.; Hahn, Y.S. Liver-resident CD103+ dendritic cells prime antiviral CD8+ T cells in situ. J. Immunol. 2015, 194, 3213–3222. [Google Scholar] [CrossRef]
- Kelly, A.; Fahey, R.; Fletcher, J.M.; Keogh, C.; Carroll, A.G.; Siddachari, R.; Geoghegan, J.; Hegarty, J.E.; Ryan, E.J.; O’Farrelly, C. CD141(+) myeloid dendritic cells are enriched in healthy human liver. J. Hepatol. 2014, 60, 135–142. [Google Scholar] [CrossRef]
- Segura, E.; Amigorena, S. Inflammatory dendritic cells in mice and humans. Trends Immunol. 2013, 34, 440–445. [Google Scholar] [CrossRef] [PubMed]
- Chow, K.V.; Sutherland, R.M.; Zhan, Y.; Lew, A.M. Heterogeneity, functional specialization and differentiation of monocyte-derived dendritic cells. Immunol. Cell Biol. 2017, 95, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Hochheiser, K.; Kurts, C. Selective Dependence of Kidney Dendritic Cells on CX3CR1. Implications for Glomerulonephritis Therapy. Adv. Exp. Med. Biol. 2015, 850, 55–71. [Google Scholar] [PubMed]
- Mionnet, C.; Buatois, V.; Kanda, A.; Milcent, V.; Fleury, S.; Lair, D.; Langelot, M.; Lacoeuille, Y.; Hessel, E.; Coffman, R.; et al. CX3CR1 is required for airway inflammation by promoting T helper cell survival and maintenance in inflamed lung. Nat. Med. 2010, 16, 1305–1312. [Google Scholar] [CrossRef] [PubMed]
- Staumont-Sallé, D.; Fleury, S.; Lazzari, A.; Molendi-Coste, O.; Hornez, N.; Lavogiez, C.; Kanda, A.; Wartelle, J.; Fries, A.; Pennino, D.; et al. CX3CL1 (fractalkine) and its receptor CX3CR1 regulate atopic dermatitis by controlling effector T cell retention in inflamed skin. J. Exp. Med. 2014, 211, 1185–1196. [Google Scholar] [CrossRef] [PubMed]
- Heymann, F.; Hammerich, L.; Storch, D.; Bartneck, M.; Huss, S.; Rüsseler, V.; Gassler, N.; Lira, S.A.; Luedde, T.; Trautwein, C.; et al. Hepatic macrophage migration and differentiation critical for liver fibrosis is mediated by the chemokine receptor C-C motif chemokine receptor 8 in mice. Hepatology 2012, 55, 898–909. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Quan, N. Immune Cell Isolation from Mouse Femur Bone Marrow. Biol. Protoc. 2015, 20, e1631. [Google Scholar] [CrossRef]
- Nakano, H.; Moran, T.P.; Nakano, K.; Gerrish, K.E.; Bortner, C.D.; Cook, D.N. Complement receptor C5aR1/CD88 and dipeptidyl peptidase-4/CD26 define distinct hematopoietic lineages of dendritic cells. J. Immunol. 2015, 194, 3808–3819. [Google Scholar] [CrossRef]
- Lutz, M.B.; Strobl, H.; Schuler, G.; Romani, N. GM-CSF monocyte-derived cells and Langerhans cells as part of the dendritic cell family. Front. Immunol. 2017, 8, 1388. [Google Scholar] [CrossRef]
- Satpathy, A.T.; Kc, W.; Albring, J.C.; Edelson, B.T.; Kretzer, N.M.; Bhattacharya, D.; Murphy, T.L.; Murphy, K.M. Zbtb46 expression distinguishes classical dendritic cells and their committed progenitors from other immune lineages. J. Exp. Med. 2012, 209, 1135–1152. [Google Scholar] [CrossRef]
- Tamura, T.; Tailor, P.; Yamaoka, K.; Kong, H.J.; Tsujimura, H.; O’Shea, J.J.; Singh, H.; Ozato, K. IFN regulatory factor-4 and -8 govern dendritic cell subset development and their functional diversity. J. Immunol. 2005, 174, 2573–2581. [Google Scholar] [CrossRef] [PubMed]
- Bajaña, S.; Turner, S.; Paul, J.; Ainsua-Enrich, E.; Kovats, S. IRF4 and IRF8 act in CD11c+ cells to regulate terminal differentiation of lung tissue dendritic cells. J. Immunol. 2016, 196, 1666–1677. [Google Scholar] [CrossRef] [PubMed]
- Bleier, J.I.; Katz, S.C.; Chaudhry, U.I.; Pillarisetty, V.G.; Kingham, T.P.; Shah, A.B.; Raab, J.R.; DeMatteo, R.P. Biliary obstruction selectively expands and activates liver myeloid dendritic cells. J. Immunol. 2006, 176, 7189–7195. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.R.; Wohlleber, D.; Reisinger, F.; Jenne, C.N.; Cheng, R.L.; Abdullah, Z.; Schildberg, F.A.; Odenthal, M.; Dienes, H.P.; van Rooijen, N.; et al. Intrahepatic myeloid-cell aggregates enable local proliferation of CD8(+) T cells and successful immunotherapy against chronic viral liver infection. Nat. Immunol. 2013, 14, 574–583. [Google Scholar] [CrossRef] [PubMed]
- Weston, C.J.; Zimmermann, H.W.; Adams, D.H. The Role of Myeloid-Derived Cells in the Progression of Liver Disease. Front. Immunol. 2019, 10, 893. [Google Scholar] [CrossRef] [PubMed]
- Karlmark, K.R.; Zimmermann, H.W.; Roderburg, C.; Gassler, N.; Wasmuth, H.E.; Luedde, T.; Trautwein, C.; Tacke, F. The fractalkine receptor CX3CR1 protects against liver fibrosis by controlling differentiation and survival of infiltrating hepatic monocytes. Hepatology 2010, 52, 1769–1782. [Google Scholar] [CrossRef] [PubMed]
- Riopel, M.; Vassallo, M.; Ehinger, E.; Pattison, J.; Bowden, K.; Winkels, H.; Wilson, M.; de Jong, R.; Patel, S.; Balakrishna, D.; et al. CX3CL1-Fc treatment prevents atherosclerosis in Ldlr KO mice. Mol. Metab. 2019, 20, 89–101. [Google Scholar] [CrossRef]
- Mossanen, J.C.; Krenkel, O.; Ergen, C.; Govaere, O.; Liepelt, A.; Puengel, T.; Heymann, F.; Kalthoff, S.; Lefebvre, E.; Eulberg, D.; et al. Chemokine (C-C motif) receptor 2-positive monocytes aggravate the early phase of acetaminophen-induced acute liver injury. Hepatology 2016, 64, 1667–1682. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sutti, S.; Bruzzì, S.; Heymann, F.; Liepelt, A.; Krenkel, O.; Toscani, A.; Ramavath, N.N.; Cotella, D.; Albano, E.; Tacke, F. CX3CR1 Mediates the Development of Monocyte-Derived Dendritic Cells during Hepatic Inflammation. Cells 2019, 8, 1099. https://doi.org/10.3390/cells8091099
Sutti S, Bruzzì S, Heymann F, Liepelt A, Krenkel O, Toscani A, Ramavath NN, Cotella D, Albano E, Tacke F. CX3CR1 Mediates the Development of Monocyte-Derived Dendritic Cells during Hepatic Inflammation. Cells. 2019; 8(9):1099. https://doi.org/10.3390/cells8091099
Chicago/Turabian StyleSutti, Salvatore, Stefania Bruzzì, Felix Heymann, Anke Liepelt, Oliver Krenkel, Alberto Toscani, Naresh Naik Ramavath, Diego Cotella, Emanuele Albano, and Frank Tacke. 2019. "CX3CR1 Mediates the Development of Monocyte-Derived Dendritic Cells during Hepatic Inflammation" Cells 8, no. 9: 1099. https://doi.org/10.3390/cells8091099
APA StyleSutti, S., Bruzzì, S., Heymann, F., Liepelt, A., Krenkel, O., Toscani, A., Ramavath, N. N., Cotella, D., Albano, E., & Tacke, F. (2019). CX3CR1 Mediates the Development of Monocyte-Derived Dendritic Cells during Hepatic Inflammation. Cells, 8(9), 1099. https://doi.org/10.3390/cells8091099