Effect of Adenomatous Polyposis Coli Loss on Tumorigenic Potential in Pancreatic Ductal Adenocarcinoma

Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Lentiviral Transductions
2.2. Real-Time PCR
2.3. Immunofluorescence
2.4. β-Catenin/TCF Reporter Assays
2.5. Cell Growth Assay
2.6. Cell Migration Assay
2.7. Gemcitabine Assay
2.8. Statistics
3. Results and Discussion
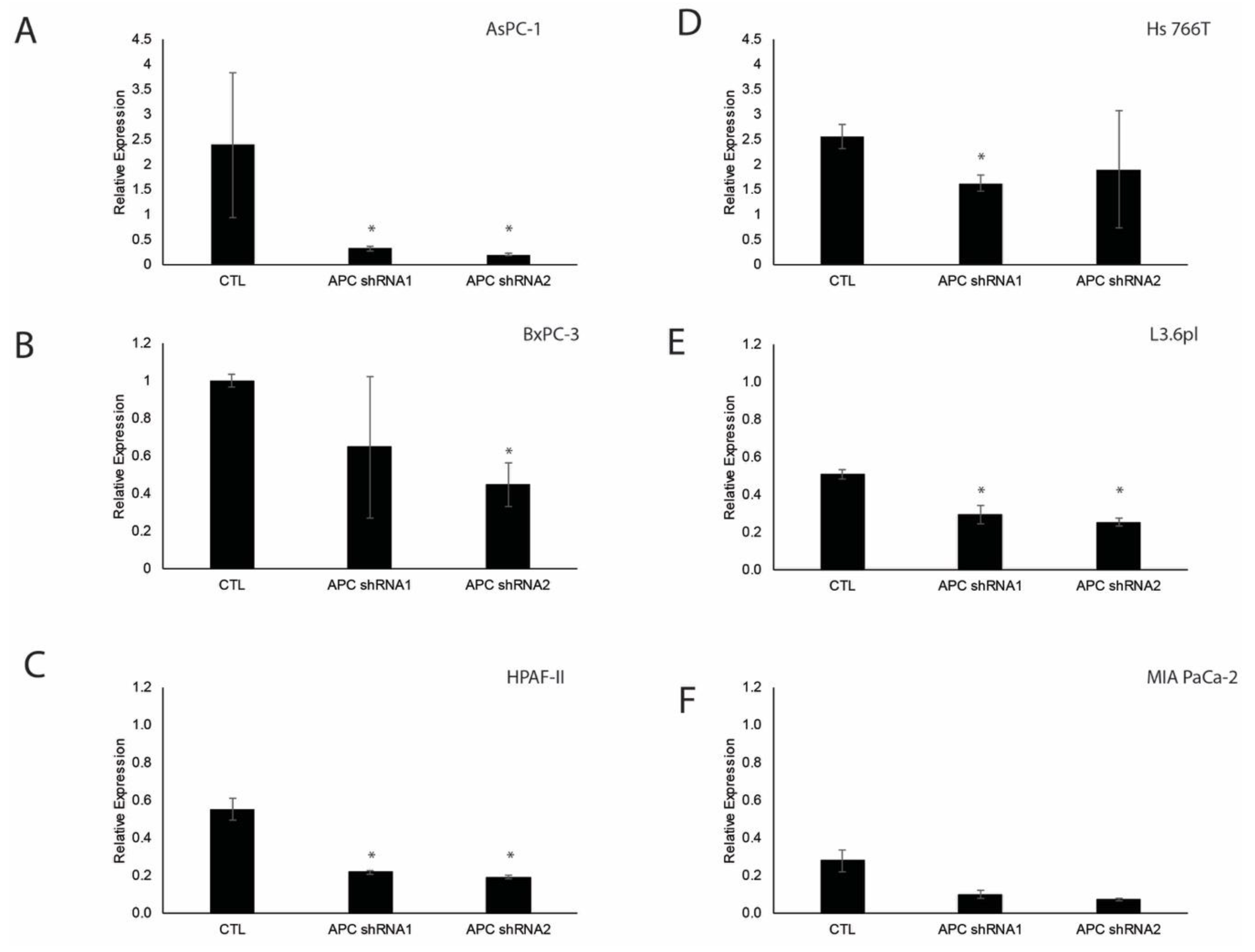
3.1. Verification of APC Knockdown in Pancreatic Ductal Adenocarcinoma Cell Lines
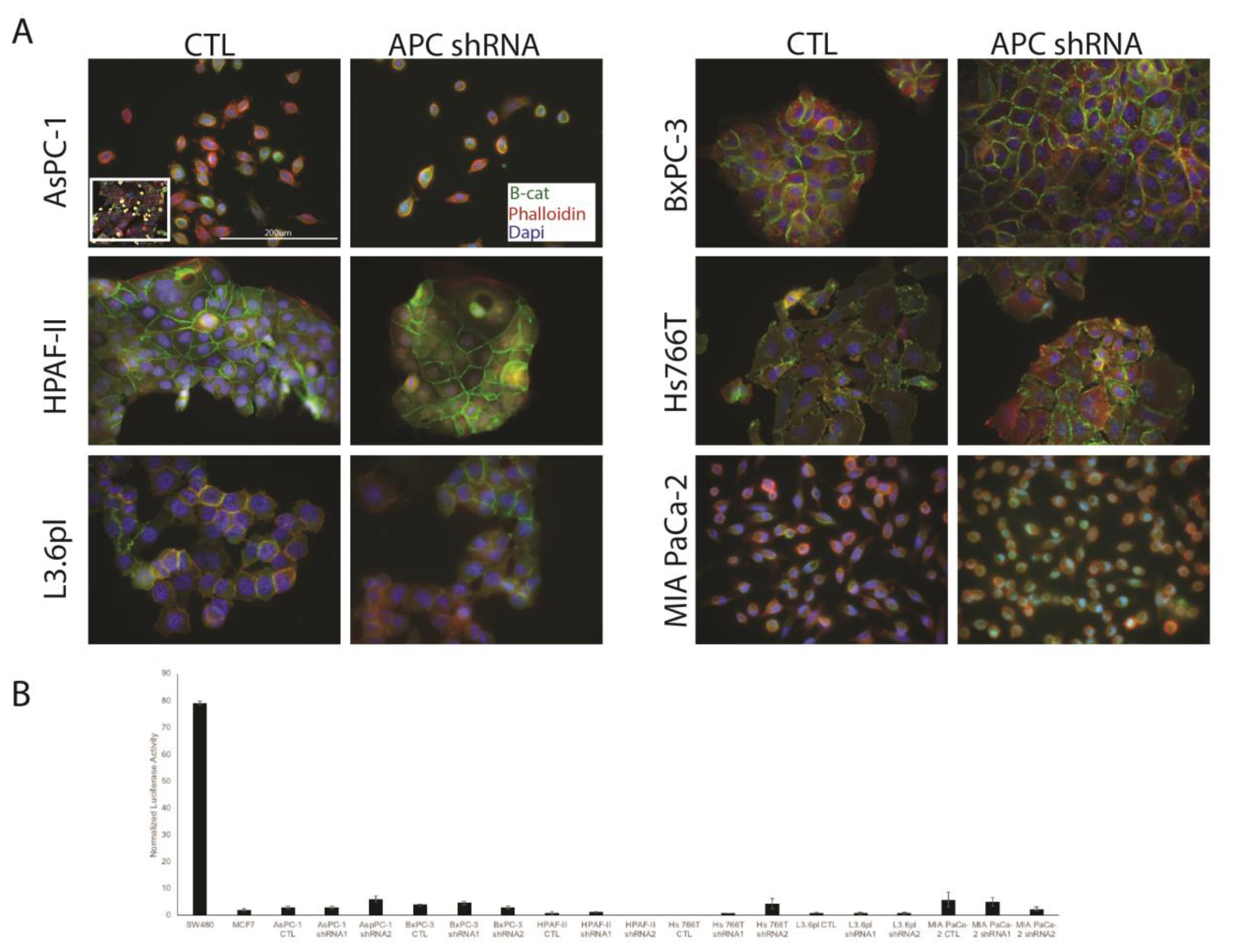
3.2. APC Knockdown in PDAC Cells Does Not Elicit Signaling through Wnt/Β-Catenin
3.3. APC Loss Enhances Proliferation in PDAC Cells
3.4. Effect of APC Knockdown on PDAC Cell Migration
3.5. APC Knockdown Does Not Impact PDAC Cell Response to Gemcitabine
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2015, 65, 5–29. [Google Scholar] [CrossRef]
- Stark, A.P.; Sacks, G.D.; Rochefort, M.M.; Donahue, T.R.; Reber, H.A.; Tomlinson, J.S.; Dawson, D.W.; Eibl, G.; Hines, O.J. Long-term Survival in Patients with Pancreatic Ductal Adenocarcinoma. Surgery 2016, 159, 1520–1527. [Google Scholar] [CrossRef] [PubMed]
- Burris III, H.A.; Moore, M.J.; Andersen, J.; Green, M.R.; Rothenberg, M.L.; Modiano, M.R.; Cripps, M.C.; Portenoy, R.K.; Storniolo, A.M.; Tarassoff, P.; et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: A randomized trial. J. Clin. Oncol. 1997, 15, 2403–2413. [Google Scholar] [CrossRef] [PubMed]
- Herrera, L.; Kakati, S.; Gibas, L.; Pietrzak, E.; Sandberg, A.A.; Reynolds, J.F. Gardner syndrome in a man with an interstitial deletion of 5q. Am. J. Med Genet. 1986, 25, 473–476. [Google Scholar] [CrossRef] [PubMed]
- Bodmer, W.F.; Bailey, C.J.; Bodmer, J.; Bussey, H.J.; Ellis, A.; Gorman, P.; Lucibello, F.C.; Murday, V.A.; Rider, S.H.; Scambler, P.; et al. Localization of the gene for familial adenomatous polyposis on chromosome 5. Nature 1987, 328, 614–616. [Google Scholar] [CrossRef] [PubMed]
- Leppert, M.; Burt, R.; Hughes, J.P.; Samowitz, W.; Nakamura, Y.; Woodward, S.; Gardner, E.; Lalouel, J.M.; White, R. Genetic analysis of an inherited predisposition to colon cancer in a family with a variable number of adenomatous polyps. New England J. Med. 1990, 322, 904–908. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Oshimura, M.; Kikuchi, R.; Seki, M.; Hayashi, T.; Miyaki, M. Suppression of tumorigenicity in human colon carcinoma cells by introduction of normal chromosome 5 or 18. Nature 1991, 349, 340–342. [Google Scholar] [CrossRef] [PubMed]
- Segditsas, S.; Tomlinson, I. Colorectal cancer and genetic alterations in the Wnt pathway. Oncogene 2006, 25, 7531–7537. [Google Scholar] [CrossRef] [PubMed]
- Prosperi, J.R.; Khramtsov, A.I.; Khramtsova, G.F.; Goss, K.H. Apc mutation enhances PyMT-induced mammary tumorigenesis. PLoS ONE 2011, 6, e29339. [Google Scholar] [CrossRef]
- Ginestà, M.M.; Diaz-Riascos, Z.V.; Busquets, J.; Peláez, N.; Serrano, T.; Peinado, M.A.; Jorba, R.; García-Borobia, F.J.; Capellá, G.; Fabregat, J. APC promoter is frequently methylated in pancreatic juice of patients with pancreatic carcinomas or periampullary tumors. Oncol. Lett. 2016, 12, 2210–2216. [Google Scholar] [CrossRef]
- Jäkel, C.; Bergmann, F.; Toth, R.; Assenov, Y.; Van Der Duin, D.; Strobel, O.; Hank, T.; Klöppel, G.; Dorrell, C.; Grompe, M.; et al. Genome-wide genetic and epigenetic analyses of pancreatic acinar cell carcinomas reveal aberrations in genome stability. Nat. Commun. 2017, 8, 1323. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Gerrard, G.; Poskitt, B.; Dawson, K.; Trivedi, P.; Foroni, L.; El-Bahrawy, M. Targeted next generation sequencing of pancreatic solid pseudopapillary neoplasms show mutations in Wnt signaling pathway genes. Pathol. Int. 2019, 69, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Peng, D.-F.; Kanai, Y.; Sawada, M.; Ushijima, S.; Hiraoka, N.; Kitazawa, S.; Hirohashi, S. DNA methylation of multiple tumor-related genes in association with overexpression of DNA methyltransferase 1 (DNMT1) during multistage carcinogenesis of the pancreas. Carcinogenesis 2006, 27, 1160–1168. [Google Scholar] [CrossRef] [PubMed]
- Horii, A.; Nakatsuru, S.; Miyoshi, Y.; Ichii, S.; Nagase, H.; Ando, H.; Yanagisawa, A.; Tsuchiya, E.; Kato, Y.; Nakamura, Y. Frequent somatic mutations of the APC gene in human pancreatic cancer. Cancer Res. 1992, 52. [Google Scholar]
- Giardiello, F.M.; Offerhaus, G.J.; Lee, D.H.; Krush, A.J.; Tersmette, A.C.; Booker, S.V.; Kelley, N.C.; Hamilton, S.R. Increased risk of thyroid and pancreatic carcinoma in familial adenomatous polyposis. Gut 1993, 34, 1394–1396. [Google Scholar] [CrossRef] [PubMed]
- Maire, F.; Hammel, P.; Terris, B.; Olschwang, S.; O’Toole, D.; Sauvanet, A.; Palazzo, L.; Ponsot, P.; Laplane, B.; Levy, P.; et al. Intraductal papillary and mucinous pancreatic tumour: A new extracolonic tumour in familial adenomatous polyposis. Gut 2002, 51, 446–449. [Google Scholar] [CrossRef] [PubMed]
- Arnau, G.; Nemorin, A.; Maledon, E.; Abraham, K. Revision of ploidy status of Dioscorea alata L. (Dioscoreaceae) by cytogenetic and microsatellite segregation analysis. Theor. Appl. Genet. 2009, 118, 1239–1249. [Google Scholar] [CrossRef]
- Chetty, R.; Salahshor, S.; Bapat, B.; Berk, T.; Croitoru, M.; Gallinger, S. Intraductal papillary mucinous neoplasm of the pancreas in a patient with attenuated familial adenomatous polyposis. J. Clin. Pathol. 2005, 58, 97–101. [Google Scholar] [CrossRef]
- Qin, R.-F.; Zhang, J.; Huo, H.-R.; Yuan, Z.-J.; Xue, J.-D. MiR-205 mediated APC regulation contributes to pancreatic cancer cell proliferation. World J. Gastroenterol. 2019, 25, 3775–3786. [Google Scholar] [CrossRef]
- Ikenoue, T.; Ijichi, H.; Kato, N.; Kanai, F.; Masaki, T.; Rengifo, W.; Okamoto, M.; Matsumura, M.; Kawabe, T.; Shiratori, Y.; et al. Analysis of the beta-catenin/T cell factor signaling pathway in 36 gastrointestinal and liver cancer cells. Jpn. J. Cancer Res. 2002, 93, 1213–1220. [Google Scholar] [CrossRef]
- Gebäck, T.; Schulz, M.M.P.; Koumoutsakos, P.; Detmar, M. TScratch: A novel and simple software tool for automated analysis of monolayer wound healing assays. Biotechniques 2009, 46, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Lesko, A.C.; Goss, K.H.; Yang, F.F.; Schwertner, A.; Hulur, I.; Onel, K.; Prosperi, J.R. The APC tumor suppressor is required for epithelial cell polarization and three-dimensional morphogenesis. Biochim. et Biophys. Acta (BBA) - Bioenerg. 2015, 1853, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Odenwald, M.A.; Prosperi, J.R.; Goss, K.H. APC/beta-catenin-rich complexes at membrane protrusions regulate mammary tumor cell migration and mesenchymal morphology. BMC Cancer 2013, 13, 12. [Google Scholar] [CrossRef] [PubMed]
- Chartier, C.; Raval, J.; Axelrod, F.; Bond, C.; Cain, J.; Dee-Hoskins, C.; Ma, S.; Fischer, M.M.; Shah, J.; Wei, J.; et al. Therapeutic Targeting of Tumor-Derived R-Spondin Attenuates beta-Catenin Signaling and Tumorigenesis in Multiple Cancer Types. Cancer Res. 2016, 76, 713–723. [Google Scholar] [CrossRef] [PubMed]
- VanKlompenberg, M.K.; Bedalov, C.O.; Soto, K.F.; Prosperi, J.R. APC selectively mediates response to chemotherapeutic agents in breast cancer. BMC Cancer 2015, 15, 9. [Google Scholar] [CrossRef]
- Sansom, O.J.; Reed, K.R.; Hayes, A.J.; Ireland, H.; Brinkmann, H.; Newton, I.P.; Batlle, E.; Simon-Assmann, P.; Clevers, H.; Näthke, I.S.; et al. Loss of Apc in vivo immediately perturbs Wnt signaling, differentiation, and migration. Genome Res. 2004, 18, 1385–1390. [Google Scholar] [CrossRef] [PubMed]
- Deer, E.L.; Gonzalez-Hernandez, J.; Coursen, J.D.; Shea, J.E.; Ngatia, J.; Scaife, C.L.; Firpo, M.A.; Mulvihill, S.J. Phenotype and Genotype of Pancreatic Cancer Cell Lines. Pancreas 2010, 39, 425–435. [Google Scholar] [CrossRef]
- Chen, W.H.; Horoszewicz, J.S.; Leong, S.S.; Shimano, T.; Penetrante, R.; Sanders, W.H.; Berjian, R.; Douglass, H.O.; Martin, E.W.; Chu, T.M. Human pancreatic adenocarcinoma: In vitro and in vivo morphology of a new tumor line established from ascites. Vitr. Cell. Dev. Biol. Plant 1982, 18, 24–34. [Google Scholar] [CrossRef]
- Tan, M.H.; Nowak, N.J.; Loor, R.; Ochi, H.; Sandberg, A.A.; Lopez, C.; Pickren, J.W.; Berjian, R.; Douglass, H.O.; Chu, T.M. Characterization of a new primary human pancreatic tumor line. Cancer Investig. 1986, 4, 15–23. [Google Scholar] [CrossRef]
- Metzgar, R.S.; Gaillard, M.T.; Levine, S.J.; Tuck, F.L.; Bossen, E.H.; Borowitz, M.J. Antigens of human pancreatic adenocarcinoma cells defined by murine monoclonal antibodies. Cancer Res. 1982, 42, 601–608. [Google Scholar]
- Smith, H.S. In Vitro Properties of Epithelial Cell Lines Established from Human Carcinomas and Nonmalignant Tissue2. J. Natl. Cancer Inst. 1979, 62, 225–230. [Google Scholar]
- Nakamura, T.; Fidler, I.J.; Coombes, K.R. Gene expression profile of metastatic human pancreatic cancer cells depends on the organ microenvironment. Cancer Res. 2007, 67, 139–148. [Google Scholar] [CrossRef]
- Yunis, A.; Arimura, G.K.; Russin, D.J. Human pancreatic carcinoma (MIA PaCa-2) in continuous culture: Sensitivity to asparaginase. Int. J. Cancer 1977, 19, 128–135. [Google Scholar] [CrossRef]
- Sauter, D.R.P.; Novak, I.; Pedersen, S.F.; Larsen, E.H.; Hoffmann, E.K. ANO1 (TMEM16A) in pancreatic ductal adenocarcinoma (PDAC). Pflugers Arch. 2015, 467, 1495–1508. [Google Scholar] [CrossRef]
- Arumugam, T.; Ramachandran, V.; Fournier, K.F.; Wang, H.; Marquis, L.; Abbruzzese, J.L.; Gallick, G.E.; Logsdon, C.D.; McConkey, D.J.; Choi, W. Epithelial to Mesenchymal Transition Contributes to Drug Resistance in Pancreatic Cancer. Cancer Res. 2009, 69, 5820–5828. [Google Scholar] [CrossRef]
- Fryer, R.; Barlett, B.; Galustian, C.; Dalgleish, A.G. Mechanisms underlying gemcitabine resistance in pancreatic cancer and sensitisation by the iMiDTM lenalidomide. Anticancer Res. 2011, 31, 3747–3756. [Google Scholar]
- Kuo, T.L.; Weng, C.C.; Kuo, K.K.; Chen, C.Y.; Wu, D.C.; Hung, W.C.; Cheng, K.H. APC haploinsufficiency coupled with p53 loss sufficiently induces mucinous cystic neoplasms and invasive pancreatic carcinoma in mice. Oncogene 2016, 35, 2223–2234. [Google Scholar] [CrossRef]
- Goehringer, C.; Sutter, C.; Kloor, M.; Gebert, J.; Slater, E.P.; Keller, M.; Treiber, I.; Ganschow, P.; Kadmon, M.; Moog, U. Double germline mutations in APC and BRCA2 in an individual with a pancreatic tumor. Fam. Cancer 2017, 16, 303–309. [Google Scholar] [CrossRef]
- Yurgelun, M.B.; Chittenden, A.B.; Morales-Oyarvide, V.; Rubinson, D.A.; Dunne, R.F.; Kozak, M.M.; Qian, Z.R.; Welch, M.W.; Brais, L.K.; Da Silva, A.; et al. Germline cancer susceptibility gene variants, somatic second hits, and survival outcomes in patients with resected pancreatic cancer. Genet. Med. 2019, 21, 213–223. [Google Scholar] [CrossRef]
- Zhan, W.; Shelton, C.A.; Greer, P.J.; Brand, R.E.; Whitcomb, D.C. Germline Variants and Risk for Pancreatic Cancer: A Systematic Review and Emerging Concepts. Pancreas 2018, 47, 924–936. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| β-Catenin Localization | Proliferation | Wound Healing | Response to Gemcitabine | |
|---|---|---|---|---|
| AsPC-1 CTL | Junctional | Doubling time of approximately 24–48 h. | Wound closed approximately 73% at 48 h. | Significantly decreased total cell number compared to untreated. |
| APC shRNA1 | NC | NC | NC | NC |
| APC shRNA2 | NC | NC | Wound closed significantly slower compared to CTL at 24 and 48 h. | No significance differences compared to untreated. |
| BxPC-3 CTL | Junctional | Doubling time of approximately 24 h. | Wound closed approximately 81% at 48 h. | Significantly decreased total cell number compared to untreated. |
| APC shRNA1 | NC | NC | Wound closed significantly faster compared to CTL at 48 h. | NC |
| APC shRNA2 | NC | Significantly different from CTL at 48 h. | Wound closed significantly faster compared to CTL at 24 and 48 h. | NC |
| HPAF-II CTL | Junctional | Doubling time of approximately 48 h. | Wound closed approximately 92% at 48 h. | No significance differences compared to untreated. |
| APC shRNA1 | NC | NC | NC | Significantly decreased total cell number compared to untreated. |
| APC shRNA2 | NC | NC | NC | Significantly decreased total cell number compared to untreated. |
| Hs 766T CTL | Junctional | Doubling time greater than our 72 h experimental timeframe. | Wound closed approximately 27% at 48 h. | Significantly decreased total cell number compared to CTL. |
| APC shRNA1 | NC | NC | NC | NC |
| APC shRNA2 | NC | Significantly different from CTL at 48 and 72 h. | NC | No significance differences compared to untreated. |
| L3.6pl CTL | Junctional | Doubling time of approximately 24 h. | Wound closed approximately 95% at 48 h. | Significantly decreased total cell number compared to untreated. |
| APC shRNA1 | NC | Significantly different from CTL at 48 h. | Wound closed significantly faster compared to CTL at 24 and 48 h. | NC |
| APC shRNA2 | NC | NC | Wound closed significantly faster compared to CTL at 24 h. | NC |
| MIA-PaCa CTL | Junctional | Doubling time of approximately 30 h. | Wound closed approximately 93% at 48 h. | Significantly decreased total cell number compared to untreated. |
| APC shRNA1 | NC | NC | NC | NC |
| APC shRNA2 | NC | NC | NC | NC |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cole, J.M.; Simmons, K.; Prosperi, J.R. Effect of Adenomatous Polyposis Coli Loss on Tumorigenic Potential in Pancreatic Ductal Adenocarcinoma. Cells 2019, 8, 1084. https://doi.org/10.3390/cells8091084
Cole JM, Simmons K, Prosperi JR. Effect of Adenomatous Polyposis Coli Loss on Tumorigenic Potential in Pancreatic Ductal Adenocarcinoma. Cells. 2019; 8(9):1084. https://doi.org/10.3390/cells8091084
Chicago/Turabian StyleCole, Jennifer M., Kaitlyn Simmons, and Jenifer R. Prosperi. 2019. "Effect of Adenomatous Polyposis Coli Loss on Tumorigenic Potential in Pancreatic Ductal Adenocarcinoma" Cells 8, no. 9: 1084. https://doi.org/10.3390/cells8091084
APA StyleCole, J. M., Simmons, K., & Prosperi, J. R. (2019). Effect of Adenomatous Polyposis Coli Loss on Tumorigenic Potential in Pancreatic Ductal Adenocarcinoma. Cells, 8(9), 1084. https://doi.org/10.3390/cells8091084