Analysis of Catecholamines and Pterins in Inborn Errors of Monoamine Neurotransmitter Metabolism—From Past to Future


Abstract
1. Introduction
2. Biosynthesis of Catecholamines, Serotonin, and Pterins
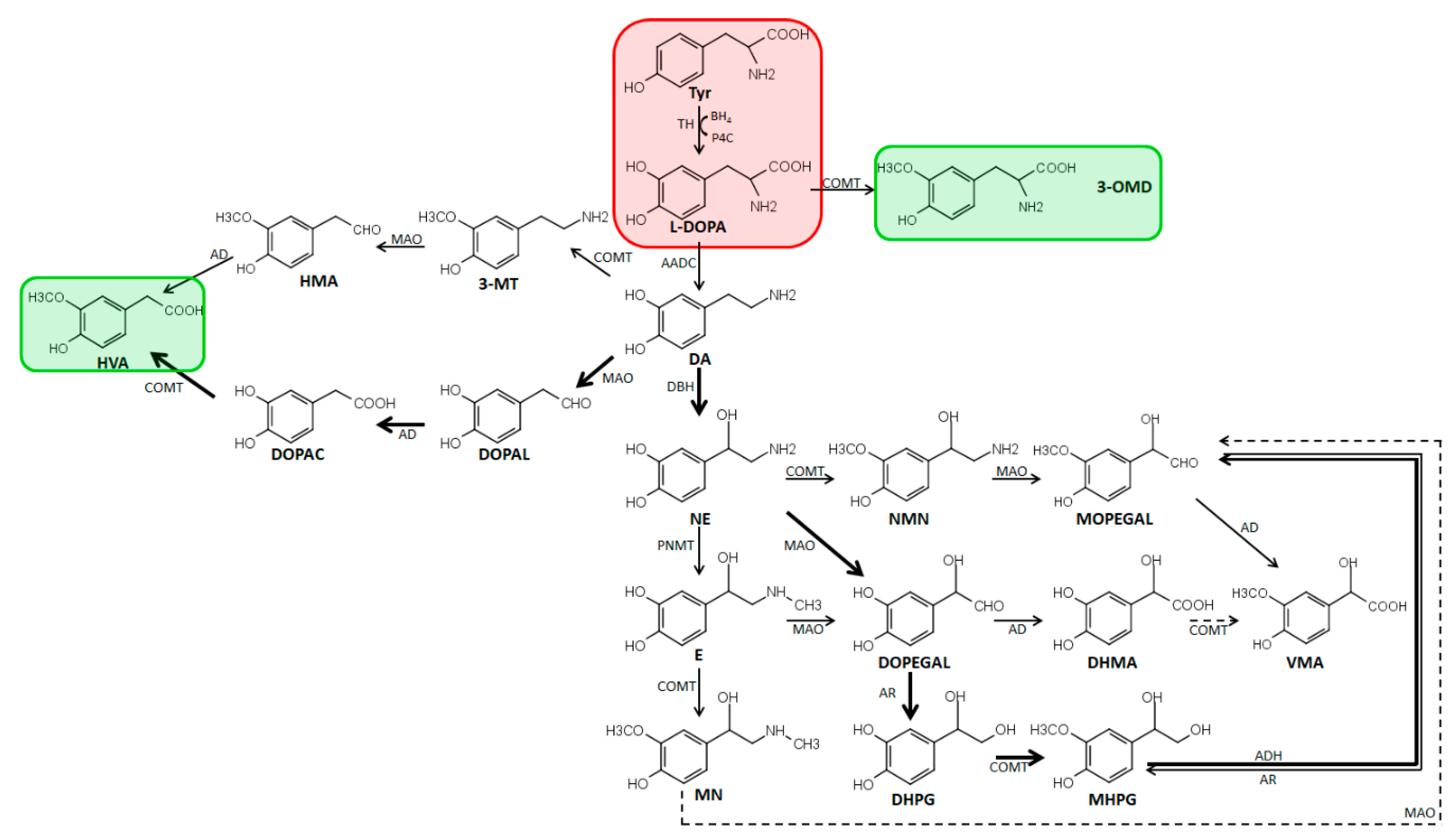
2.1. Metabolism and Biosynthesis of Catecholamines
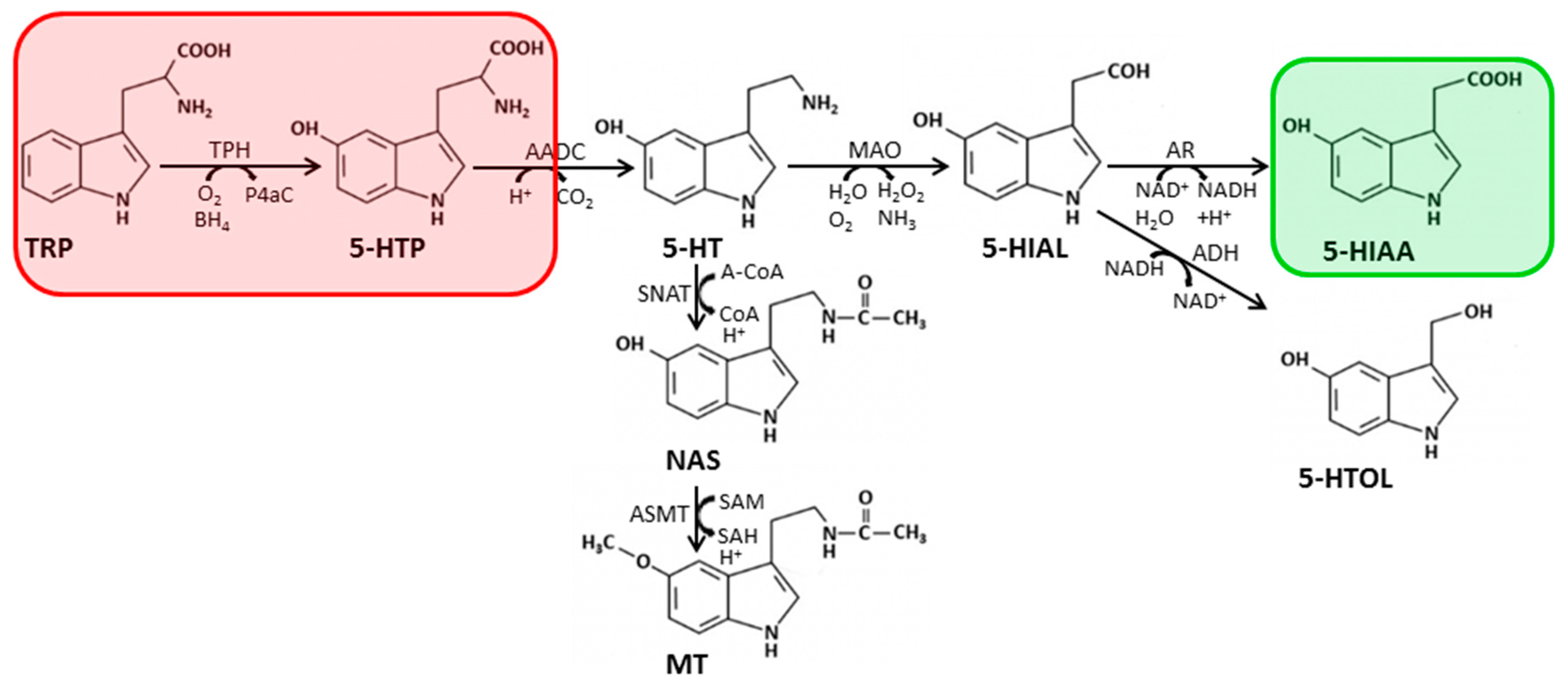
2.2. Biosynthesis and Metabolism of Serotonin
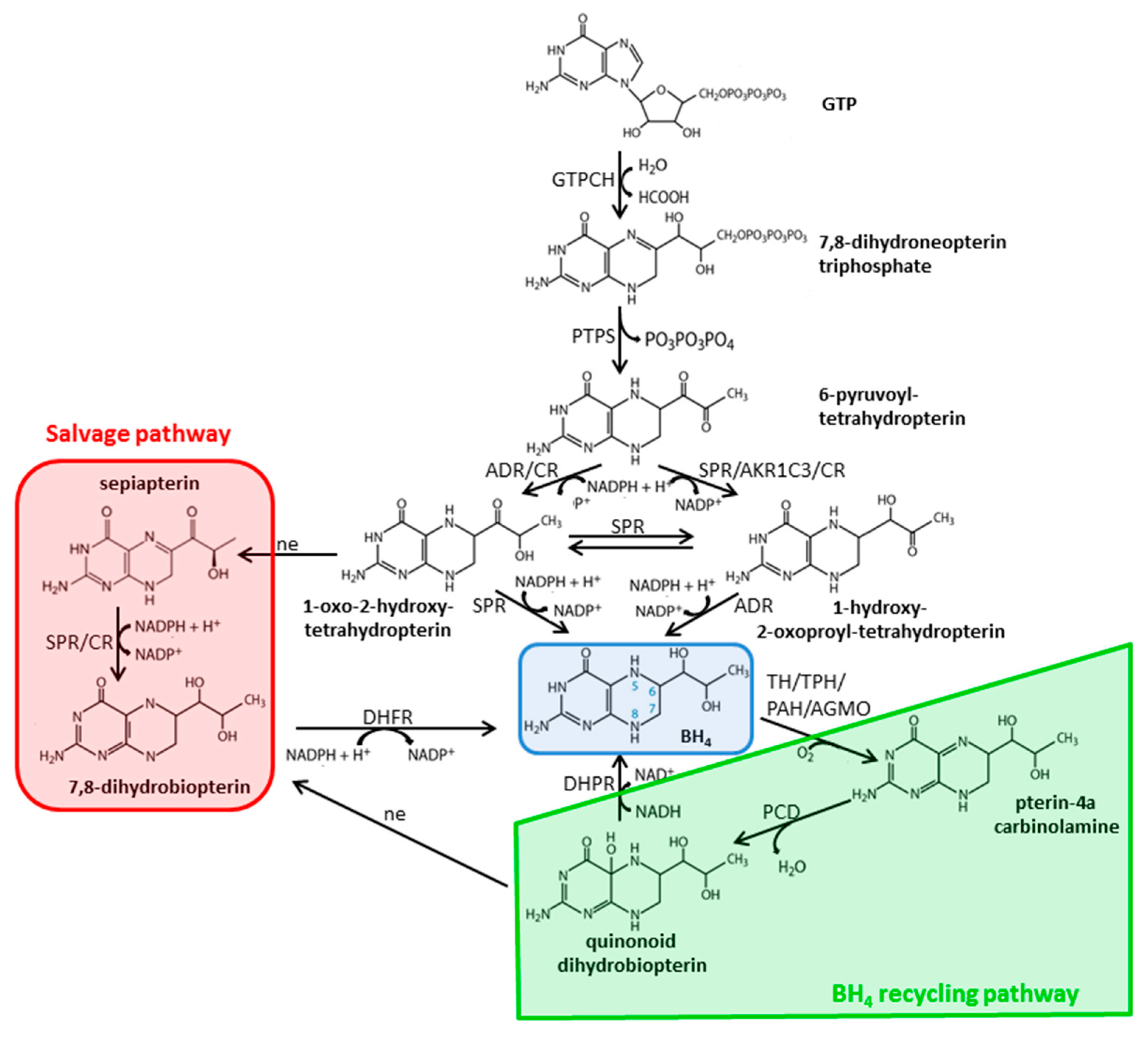
2.3. Biosynthesis and Metabolism of Pterins
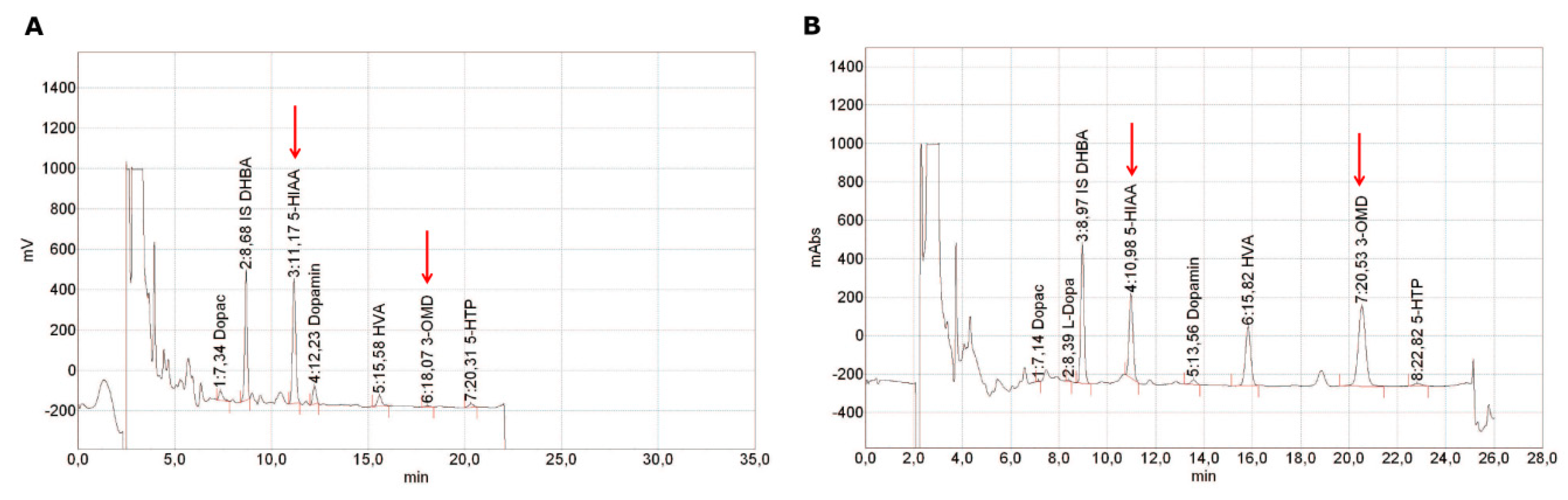
3. Diagnostic Methods
3.1. Quantification of Catecholamines and Catecholamine Metabolites
3.1.1. Pre-processing of Biological Fluids
3.1.2. Pre-Processing of Tissue Samples
3.1.3. Chromatographic Columns, Mobile Phases, and Detection Used
3.1.4. Analysis of Catecholamines in CSF
3.1.5. Analysis of Catecholamines in Urine
3.1.6. Analysis of Catecholamines in Blood Samples
3.1.7. Enzyme Activity Assays
3.2. Quantification of Pterins
3.2.1. Pre-Processing of Biological Fluids
3.2.2. Pre-Processing of Tissue Samples
3.2.3. Chromatographic Columns, Mobile Phases, and Detection Used
3.2.4. Analysis of Pterins in CSF
3.2.5. Analysis of Pterins in Urine
3.2.6. Analysis of Pterins in Blood Samples
3.2.7. Enzyme Activity Assays
4. Pitfalls
4.1. Pre-Analytical and Methodological Pitfalls
4.2. Diagnostic Pitfalls
5. Conclusions and Future Perspective
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A


References
- Brennenstuhl, H.; Jung-Klawitter, S.; Assmann, B.; Opladen, T. Inherited disorders of neurotransmitters: Classification and practical approaches for diagnosis and treatment. Neuropediatrics 2019, 50, 2–14. [Google Scholar] [CrossRef]
- Anikster, Y.; Haack, T.B.; Vilboux, T.; Pode-Shakked, B.; Thony, B.; Shen, N.; Guarani, V.; Meissner, T.; Mayatepek, E.; Trefz, F.K.; et al. Biallelic mutations in dnajc12 cause hyperphenylalaninemia, dystonia, and intellectual disability. Am. J. Hum. Genet. 2017, 100, 257–266. [Google Scholar] [CrossRef]
- Blau, N.; Martinez, A.; Hoffmann, G.F.; Thony, B. Dnajc12 deficiency: A new strategy in the diagnosis of hyperphenylalaninemias. Mol. Genet. Metab. 2018, 123, 1–5. [Google Scholar] [CrossRef]
- Straniero, L.; Guella, I.; Cilia, R.; Parkkinen, L.; Rimoldi, V.; Young, A.; Asselta, R.; Solda, G.; Sossi, V.; Stoessl, A.J.; et al. Dnajc12 and dopa-responsive nonprogressive parkinsonism. Ann. Neurol. 2017, 82, 640–646. [Google Scholar] [CrossRef]
- Bergquist, J.; Sciubisz, A.; Kaczor, A.; Silberring, J. Catecholamines and methods for their identification and quantitation in biological tissues and fluids. J. Neurosci. Methods 2002, 113, 1–13. [Google Scholar] [CrossRef]
- Goldstein, D.S.; Eisenhofer, G.; Kopin, I.J. Sources and significance of plasma levels of catechols and their metabolites in humans. J. Pharmacol. Exp. Ther. 2003, 305, 800–811. [Google Scholar] [CrossRef]
- Tsunoda, M. Recent advances in methods for the analysis of catecholamines and their metabolites. Anal. Bioanal. Chem. 2006, 386, 506–514. [Google Scholar] [CrossRef]
- Eisenhofer, G.; Kopin, I.J.; Goldstein, D.S. Catecholamine metabolism: A contemporary view with implications for physiology and medicine. Pharmacol. Rev. 2004, 56, 331–349. [Google Scholar] [CrossRef]
- Antkiewicz-Michaluk, L.; Ossowska, K.; Romanska, I.; Michaluk, J.; Vetulani, J. 3-methoxytyramine, an extraneuronal dopamine metabolite plays a physiological role in the brain as an inhibitory regulator of catecholaminergic activity. Eur. J. Pharmacol. 2008, 599, 32–35. [Google Scholar] [CrossRef]
- Uutela, P.; Reinila, R.; Harju, K.; Piepponen, P.; Ketola, R.A.; Kostiainen, R. Analysis of intact glucuronides and sulfates of serotonin, dopamine, and their phase i metabolites in rat brain microdialysates by liquid chromatography-tandem mass spectrometry. Anal. Chem. 2009, 81, 8417–8425. [Google Scholar] [CrossRef]
- Bicker, J.; Fortuna, A.; Alves, G.; Falcão, A. Liquid chromatographic methods for the quantification of catecholamines and their metabolites in several biological samples. Anal. Chim. Acta 2013, 768, 12–34. [Google Scholar] [CrossRef]
- Nichol, C.A.; Smith, G.K.; Duch, D.S. Biosynthesis and metabolism of tetrahydrobiopterin and molybdopterin. Annu. Rev. Biochem. 1985, 54, 729–764. [Google Scholar] [CrossRef]
- Milstien, S.; Kaufman, S. Tetrahydro-sepiapterin is an intermediate in tetrahydrobiopterin biosynthesis. Biochem. Biophys. Res. Commun. 1983, 115, 888–893. [Google Scholar] [CrossRef]
- Milstien, S.; Kaufman, S. Immunological studies on the participation of 6-pyruvoyl tetrahydropterin (2’-oxo) reductase, an aldose reductase, in tetrahydrobiopterin biosynthesis. Biochem. Biophys. Res. Commun. 1989, 165, 845–850. [Google Scholar] [CrossRef]
- Iino, T.; Tabata, M.; Takikawa, S.; Sawada, H.; Shintaku, H.; Ishikura, S.; Hara, A. Tetrahydrobiopterin is synthesized from 6-pyruvoyl-tetrahydropterin by the human aldo-keto reductase akr1 family members. Arch. Biochem. Biophys. 2003, 416, 180–187. [Google Scholar] [CrossRef]
- Werner, E.R.; Blau, N.; Thony, B. Tetrahydrobiopterin: Biochemistry and pathophysiology. Biochem. J. 2011, 438, 397–414. [Google Scholar] [CrossRef]
- Peaston, R.T.; Weinkove, C. Measurement of catecholamines and their metabolites. Ann. Clin. Biochem. 2004, 41, 17–38. [Google Scholar] [CrossRef]
- Roberts, N.B.; Higgins, G.; Sargazi, M. A study on the stability of urinary free catecholamines and free methyl-derivatives at different ph, temperature and time of storage. Clin. Chem. Lab. Med. 2010, 48, 81–87. [Google Scholar] [CrossRef]
- Smith, E.A.; Schwartz, A.L.; Lucot, J.B. Measurement of urinary catecholamines in small samples for mice. J. Pharmacol. Toxicol. Methods 2013, 67, 45–49. [Google Scholar] [CrossRef][Green Version]
- Li, W.; Rossi, D.T.; Fountain, S.T. Development and validation of a semi-automated method for l-dopa and dopamine in rat plasma using electrospray lc/ms/ms. J. Pharm. Biomed. Anal. 2000, 24, 325–333. [Google Scholar] [CrossRef]
- Neubecker, T.A.; Coombs, M.A.; Quijano, M.; O’Neill, T.P.; Cruze, C.A.; Dobson, R.L. Rapid and selective method for norepinephrine in rat urine using reversed-phase ion-pair high-performance liquid chromatography-tandem mass spectrometry. J. Chromatogr. B Biomed. Sci. Appl. 1998, 718, 225–233. [Google Scholar] [CrossRef]
- Wang, Y.; Fice, D.S.; Yeung, P.K. A simple high-performance liquid chromatography assay for simultaneous determination of plasma norepinephrine, epinephrine, dopamine and 3,4-dihydroxyphenyl acetic acid. J. Pharm. Biomed. Anal. 1999, 21, 519–525. [Google Scholar] [CrossRef]
- Cakal, C.; Ferrance, J.P.; Landers, J.P.; Caglar, P. Microchip extraction of catecholamines using a boronic acid functional affinity monolith. Anal. Chim. Acta 2011, 690, 94–100. [Google Scholar] [CrossRef]
- Raggi, M.A.; Sabbioni, C.; Casamenti, G.; Gerra, G.; Calonghi, N.; Masotti, L. Determination of catecholamines in human plasma by high-performance liquid chromatography with electrochemical detection. J. Chromatogr. B Biomed. Sci. Appl. 1999, 730, 201–211. [Google Scholar] [CrossRef]
- Rozet, E.; Morello, R.; Lecomte, F.; Martin, G.B.; Chiap, P.; Crommen, J.; Boos, K.S.; Hubert, P. Performances of a multidimensional on-line spe-lc-ecd method for the determination of three major catecholamines in native human urine: Validation, risk and uncertainty assessments. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2006, 844, 251–260. [Google Scholar] [CrossRef]
- De Jong, W.H.; de Vries, E.G.; Wolffenbuttel, B.H.; Kema, I.P. Automated mass spectrometric analysis of urinary free catecholamines using on-line solid phase extraction. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2010, 878, 1506–1512. [Google Scholar] [CrossRef]
- Zhang, G.; Zhang, Y.; Ji, C.; McDonald, T.; Walton, J.; Groeber, E.A.; Steenwyk, R.C.; Lin, Z. Ultra sensitive measurement of endogenous epinephrine and norepinephrine in human plasma by semi-automated spe-lc-ms/ms. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2012, 895, 186–190. [Google Scholar] [CrossRef]
- Raggi, M.A.; Sabbioni, C.; Nicoletta, G.; Mandrioli, R.; Gerra, G. Analysis of plasma catecholamines by liquid chromatography with amperometric detection using a novel spe ion-exchange procedure. J. Sep. Sci. 2003, 26, 1141–1146. [Google Scholar] [CrossRef]
- Sabbioni, C.; Saracino, M.A.; Mandrioli, R.; Pinzauti, S.; Furlanetto, S.; Gerra, G.; Raggi, M.A. Simultaneous liquid chromatographic analysis of catecholamines and 4-hydroxy-3-methoxyphenylethylene glycol in human plasma. Comparison of amperometric and coulometric detection. J. Chromatogr. A 2004, 1032, 65–71. [Google Scholar] [CrossRef]
- Talwar, D.; Williamson, C.; McLaughlin, A.; Gill, A.; O’Reilly, D.S. Extraction and separation of urinary catecholamines as their diphenyl boronate complexes using c18 solid-phase extraction sorbent and high-performance liquid chromatography. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2002, 769, 341–349. [Google Scholar] [CrossRef]
- Hirano, Y.; Tsunoda, M.; Funatsu, T.; Imai, K. Rapid assay for catechol-o-methyltransferase activity by high-performance liquid chromatography-fluorescence detection. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2005, 819, 41–46. [Google Scholar] [CrossRef]
- Masuda, M.; Tsunoda, M.; Imai, K. High-performance liquid chromatography-fluorescent assay of catechol-o-methyltransferase activity in rat brain. Anal. Bioanal. Chem. 2003, 376, 1069–1073. [Google Scholar] [CrossRef]
- Chi, J.D.; Odontiadis, J.; Franklin, M. Simultaneous determination of catecholamines in rat brain tissue by high-performance liquid chromatography. J. Chromatogr. B Biomed. Sci. Appl. 1999, 731, 361–367. [Google Scholar] [CrossRef]
- Vaarmann, A.; Kask, A.; Maeorg, U. Novel and sensitive high-performance liquid chromatographic method based on electrochemical coulometric array detection for simultaneous determination of catecholamines, kynurenine and indole derivatives of tryptophan. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2002, 769, 145–153. [Google Scholar] [CrossRef]
- Yoshitake, T.; Kehr, J.; Yoshitake, S.; Fujino, K.; Nohta, H.; Yamaguchi, M. Determination of serotonin, noradrenaline, dopamine and their metabolites in rat brain extracts and microdialysis samples by column liquid chromatography with fluorescence detection following derivatization with benzylamine and 1,2-diphenylethylenediamine. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2004, 807, 177–183. [Google Scholar] [CrossRef]
- Tornkvist, A.; Sjoberg, P.J.; Markides, K.E.; Bergquist, J. Analysis of catecholamines and related substances using porous graphitic carbon as separation media in liquid chromatography-tandem mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2004, 801, 323–329. [Google Scholar] [CrossRef]
- Hubbard, K.E.; Wells, A.; Owens, T.S.; Tagen, M.; Fraga, C.H.; Stewart, C.F. Determination of dopamine, serotonin, and their metabolites in pediatric cerebrospinal fluid by isocratic high performance liquid chromatography coupled with electrochemical detection. Biomed. Chromatogr. 2010, 24, 626–631. [Google Scholar] [CrossRef]
- Verbeek, M.M.; Blom, A.M.; Wevers, R.A.; Lagerwerf, A.J.; van de Geer, J.; Willemsen, M.A. Technical and biochemical factors affecting cerebrospinal fluid 5-mthf, biopterin and neopterin concentrations. Mol. Genet. Metab. 2008, 95, 127–132. [Google Scholar] [CrossRef]
- Batllori, M.; Molero-Luis, M.; Ormazabal, A.; Casado, M.; Sierra, C.; Garcia-Cazorla, A.; Kurian, M.; Pope, S.; Heales, S.J.; Artuch, R. Analysis of human cerebrospinal fluid monoamines and their cofactors by hplc. Nat. Protoc. 2017, 12, 2359–2375. [Google Scholar] [CrossRef]
- Lo, A.; Guibal, P.; Doummar, D.; Rodriguez, D.; Hautem, J.Y.; Couderc, R.; Billette De Villemeur, T.; Roze, E.; Chaminade, P.; Moussa, F. Single-step rapid diagnosis of dopamine and serotonin metabolism disorders. ACS Omega 2017, 2, 5962–5972. [Google Scholar] [CrossRef]
- He, X.; Kozak, M. Development of a liquid chromatography-tandem mass spectrometry method for plasma-free metanephrines with ion-pairing turbulent flow online extraction. Anal. Bioanal. Chem. 2012, 402, 3003–3010. [Google Scholar] [CrossRef]
- Petteys, B.J.; Graham, K.S.; Parnas, M.L.; Holt, C.; Frank, E.L. Performance characteristics of an lc-ms/ms method for the determination of plasma metanephrines. Clin. Chim. Acta 2012, 413, 1459–1465. [Google Scholar] [CrossRef]
- He, X.; Gabler, J.; Yuan, C.; Wang, S.; Shi, Y.; Kozak, M. Quantitative measurement of plasma free metanephrines by ion-pairing solid phase extraction and liquid chromatography-tandem mass spectrometry with porous graphitic carbon column. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2011, 879, 2355–2359. [Google Scholar] [CrossRef]
- Peaston, R.T.; Graham, K.S.; Chambers, E.; van der Molen, J.C.; Ball, S. Performance of plasma free metanephrines measured by liquid chromatography-tandem mass spectrometry in the diagnosis of pheochromocytoma. Clin. Chim. Acta 2010, 411, 546–552. [Google Scholar] [CrossRef]
- Tsunoda, M.; Nagayama, M.; Funatsu, T.; Hosoda, S.; Imai, K. Catecholamine analysis with microcolumn LC-peroxyoxylate chemiluminescence reaction detection. Clin. Chim. Acta 2005, 366, 168–173. [Google Scholar] [CrossRef]
- Karimi, M.; Carl, J.L.; Loftin, S.; Perlmutter, J.S. Modified high-performance liquid chromatography with electrochemical detection method for plasma measurement of levodopa, 3-o-methyldopa, dopamine, carbidopa and 3,4-dihydroxyphenyl acetic acid. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2006, 836, 120–123. [Google Scholar] [CrossRef]
- Machida, M.; Sakaguchi, A.; Kamada, S.; Fujimoto, T.; Takechi, S.; Kakinoki, S.; Nomura, A. Simultaneous analysis of human plasma catecholamines by high-performance liquid chromatography with a reversed-phase triacontylsilyl silica column. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2006, 830, 249–254. [Google Scholar] [CrossRef]
- Ragab, G.H.; Nohta, H.; Zaitsu, K. Chemiluminescence determination of catecholamines in human blood plasma using 1,2-bis(3-chlorophenyl)ethylenediamine as pre-column derivatizing reagent for liquid chromatography. Anal. Chim. Acta 2000, 403, 155–160. [Google Scholar] [CrossRef]
- Hollenbach, E.; Schulz, C.; Lehnert, H. Rapid and sensitive determination of catecholamines and the metabolite 3-methoxy-4-hydroxyphen-ethyleneglycol using hplc following novel extraction procedures. Life Sci. 1998, 63, 737–750. [Google Scholar] [CrossRef]
- Clark, Z.D.; Frank, E.L. Urinary metanephrines by liquid chromatography tandem mass spectrometry: Using multiple quantification methods to minimize interferences in a high throughput method. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2011, 879, 3673–3680. [Google Scholar] [CrossRef]
- Kumar, A.; Hart, J.P.; McCalley, D.V. Determination of catecholamines in urine using hydrophilic interaction chromatography with electrochemical detection. J. Chromatogr. A 2011, 1218, 3854–3861. [Google Scholar] [CrossRef]
- Sakaguchi, Y.; Yoshida, H.; Hayama, T.; Itoyama, M.; Todoroki, K.; Yamaguchi, M.; Nohta, H. Selective liquid-chromatographic determination of native fluorescent biogenic amines in human urine based on fluorous derivatization. J. Chromatogr. A 2011, 1218, 5581–5586. [Google Scholar] [CrossRef]
- Thomas, D.H.; Taylor, J.D.; Barnaby, O.S.; Hage, D.S. Determination of free catecholamines in urine by tandem affinity/ion-pair chromatography and flow injection analysis. Clin. Chim. Acta 2008, 398, 63–69. [Google Scholar] [CrossRef]
- Fotopoulou, M.A.; Ioannou, P.C. Post-column terbium complexation and sensitized fluorescence detection for the determination of norepinephrine, epinephrine and dopamine using high-performance liquid chromatography. Anal. Chim. Acta 2002, 462, 179–185. [Google Scholar] [CrossRef]
- Kushnir, M.M.; Urry, F.M.; Frank, E.L.; Roberts, W.L.; Shushan, B. Analysis of catecholamines in urine by positive-ion electrospray tandem mass spectrometry. Clin. Chem. 2002, 48, 323–331. [Google Scholar]
- Chan, E.C.; Ho, P.C. High-performance liquid chromatography/atmospheric pressure chemical ionization mass spectrometric method for the analysis of catecholamines and metanephrines in human urine. Rapid Commun. Mass Spectrom. 2000, 14, 1959–1964. [Google Scholar] [CrossRef]
- Chan, E.C.; Wee, P.Y.; Ho, P.C. Evaluation of degradation of urinary catecholamines and metanephrines and deconjugation of their sulfoconjugates using stability-indicating reversed-phase ion-pair hplc with electrochemical detection. J. Pharm. Biomed. Anal. 2000, 22, 515–526. [Google Scholar] [CrossRef]
- Blau, N.; Kierat, L.; Matasovic, A.; Leimbacher, W.; Heizmann, C.W.; Guardamagna, O.; Ponzone, A. Antenatal diagnosis of tetrahydrobiopterin deficiency by quantification of pterins in amniotic fluid and enzyme activity in fetal and extrafetal tissue. Clin. Chim. Acta 1994, 226, 159–169. [Google Scholar] [CrossRef]
- Gabler, J.; Miller, A.; Wang, S. A simple liquid chromatography-tandem mass spectrometry method for measuring metanephrine and normetanephrine in urine. Clin. Chem. Lab. Med. 2011, 49, 1213–1216. [Google Scholar] [CrossRef]
- Nalewajko, E.; Wiszowata, A.; Kojlo, A. Determination of catecholamines by flow-injection analysis and high-performance liquid chromatography with chemiluminescence detection. J. Pharm. Biomed. Anal. 2007, 43, 1673–1681. [Google Scholar] [CrossRef]
- Gu, Q.S.; Shi, Y.; Yin, P.; Gao, P.; Lu, X.; Xu, G. Analysis of catecholamines and their metabolites in adrenal gland by liquid chromatography tandem mass spectrometry. Anal. Chim. Acta 2008, 609, 192–200. [Google Scholar] [CrossRef]
- Tsunoda, M.A.; Aoyama, C.; Nomura, H.; Toyoda, T.; Masuki, N.; Funatsu, T. Simultaneous determination of dopamine and 3,4-dihydroxyphenylacetic acid in mouse striatum using mixed-mode reversed-phase and cation-exchange high-performance liquid chromatography. J. Pharm. Biomed. Anal. 2010, 51, 712–715. [Google Scholar] [CrossRef]
- Heidbreder, C.A.; Lacroix, L.; Atkins, A.R.; Organ, A.J.; Murray, S.; West, A.; Shah, A.J. Development and application of a sensitive high performance ion-exchange chromatography method for the simultaneous measurement of dopamine, 5-hydroxytryptamine and norepinephrine in microdialysates from the rat brain. J. Neurosci. Methods 2001, 112, 135–144. [Google Scholar] [CrossRef]
- Okumura, T.; Nakajima, Y.; Matsuoka, M.; Takamatsu, T. Study of salivary catecholamines using fully automated column-switching high-performance liquid chromatography. J. Chromatogr. B Biomed. Sci. Appl. 1997, 694, 305–316. [Google Scholar] [CrossRef]
- Tsunoda, M.; Funatsu, T. Catecholamine analysis with strong cation exchange column liquid chromatography-peroxyoxalate chemiluminescence reaction detection. Anal. Bioanal. Chem. 2012, 402, 1393–1397. [Google Scholar] [CrossRef]
- Ji, C.; Walton, J.; Su, Y.; Tella, M. Simultaneous determination of plasma epinephrine and norepinephrine using an integrated strategy of a fully automated protein precipitation technique, reductive ethylation labeling and uplc-ms/ms. Anal. Chim. Acta 2010, 670, 84–91. [Google Scholar] [CrossRef]
- Parrot, S.; Neuzeret, P.C.; Denoroy, L. A rapid and sensitive method for the analysis of brain monoamine neurotransmitters using ultra-fast liquid chromatography coupled to electrochemical detection. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2011, 879, 3871–3878. [Google Scholar] [CrossRef]
- Chaurasia, C.S.; Chen, C.E.; Ashby, C.R., Jr. In vivo on-line hplc-microdialysis: Simultaneous detection of monoamines and their metabolites in awake freely-moving rats. J. Pharm. Biomed. Anal. 1999, 19, 413–422. [Google Scholar] [CrossRef]
- Ormazabal, A.; García-Cazorla, A.; Fernández, Y.; Fernández-Álvarez, E.; Campistol, J.; Artuch, R. Hplc with electrochemical and fluorescence detection procedures for the diagnosis of inborn errors of biogenic amines and pterins. J. Neurosci. Methods 2005, 142, 153–158. [Google Scholar] [CrossRef]
- Patel, B.A.; Arundell, M.; Allen, M.C.; Gard, P.; O’Hare, D.; Parker, K.; Yeoman, M.S. Changes in the properties of the modulatory cerebral giant cells contribute to aging in the feeding system of lymnaea. Neurobiol. Aging 2006, 27, 1892–1901. [Google Scholar] [CrossRef]
- Manica, D.P.; Mitsumori, Y.; Ewing, A.G. Characterization of electrode fouling and surface regeneration for a platinum electrode on an electrophoresis microchip. Anal. Chem. 2003, 75, 4572–4577. [Google Scholar] [CrossRef]
- Yoshitake, M.; Nohta, H.; Yoshida, H.; Yoshitake, T.; Todoroki, K.; Yamaguchi, M. Selective determination of native fluorescent bioamines through precolumn derivatization and liquid chromatography using intramolecular fluorescence resonance energy transfer detection. Anal. Chem. 2006, 78, 920–927. [Google Scholar] [CrossRef]
- Todoroki, K.; Yoshida, H.; Hayama, T.; Itoyama, M.; Nohta, H.; Yamaguchi, M. Highly sensitive and selective derivatization-lc method for biomolecules based on fluorescence interactions and fluorous separations. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2011, 879, 1325–1337. [Google Scholar] [CrossRef]
- Chan, E.C.; Wee, P.Y.; Ho, P.Y.; Ho, P.C. High-performance liquid chromatographic assay for catecholamines and metanephrines using fluorimetric detection with pre-column 9-fluorenylmethyloxycarbonyl chloride derivatization. J. Chromatogr. B Biomed. Sci. Appl. 2000, 749, 179–189. [Google Scholar] [CrossRef]
- Fujino, K.; Yoshitake, T.; Kehr, J.; Nohta, H.; Yamaguchi, M. Simultaneous determination of 5-hydroxyindoles and catechols by high-performance liquid chromatography with fluorescence detection following derivatization with benzylamine and 1,2-diphenylethylenediamine. J. Chromatogr. A 2003, 1012, 169–177. [Google Scholar] [CrossRef]
- Umegae, Y.; Nohta, H.; Lee, M.; Ohkura, Y. 1,2-diarylethylenediamines as pre-column fluorescence derivatization reagents in high-performance liquid chromatographic determination of catecholamines in urine and plasma. Chem. Pharm. Bull. (Tokyo) 1990, 38, 2293–2295. [Google Scholar] [CrossRef]
- Jeon, H.K.; Nohta, H.; Ohkura, Y. High-performance liquid chromatographic determination of catecholamines and their precursor and metabolites in human urine and plasma by postcolumn derivatization involving chemical oxidation followed by fluorescence reaction. Anal. Biochem. 1992, 200, 332–338. [Google Scholar] [CrossRef]
- Tsunoda, M.; Mitsuhashi, K.; Masuda, M.; Imai, K. Simultaneous determination of 3,4-dihydroxyphenylacetic acid and homovanillic acid using high performance liquid chromatography-fluorescence detection and application to rat kidney microdialysate. Anal. Biochem. 2002, 307, 153–158. [Google Scholar] [CrossRef]
- Aoyama, N.; Tsunoda, M.; Nakagomi, K.; Imai, K. A rapid assay method for catechol-o-methyltransferase activity by flow injection analysis. Biomed. Chromatogr. 2002, 16, 255–260. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Yoshitake, T.; Fujino, K.; Kawano, K.; Kehr, J.; Ishida, J. Determination of norepinephrine in microdialysis samples by microbore column liquid chromatography with fluorescence detection following derivatization with benzylamine. Anal. Biochem. 1999, 270, 296–302. [Google Scholar] [CrossRef]
- Tsunoda, M.; Takezawa, K.; Masuda, M.; Imai, K. Rat liver and kidney catechol-o-methyltransferase activity measured by high-performance liquid chromatography with fluorescence detection. Biomed. Chromatogr. 2002, 16, 536–541. [Google Scholar] [CrossRef]
- Tsunoda, M.; Takezawa, K.; Santa, T.; Imai, K. Simultaneous automatic determination of catecholamines and their 3-o-methyl metabolites in rat plasma by high-performance liquid chromatography using peroxyoxalate chemiluminescence reaction. Anal. Biochem. 1999, 269, 386–392. [Google Scholar] [CrossRef]
- Zhang, M.Y.; Beyer, C.E. Measurement of neurotransmitters from extracellular fluid in brain by in vivo microdialysis and chromatography-mass spectrometry. J. Pharm. Biomed. Anal. 2006, 40, 492–499. [Google Scholar] [CrossRef]
- Zhang, W.; Xie, Y.; Ai, S.; Wan, F.; Wang, J.; Jin, L.; Jin, J. Liquid chromatography with amperometric detection using functionalized multi-wall carbon nanotube modified electrode for the determination of monoamine neurotransmitters and their metabolites. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2003, 791, 217–225. [Google Scholar] [CrossRef]
- Forster, C.D.; Macdonald, I.A. The assay of the catecholamine content of small volumes of human plasma. Biomed. Chromatogr. 1999, 13, 209–215. [Google Scholar] [CrossRef]
- Liu, L.; Li, Q.; Li, N.; Ling, J.; Liu, R.; Wang, Y.; Sun, L.; Chen, X.H.; Bi, K. Simultaneous determination of catecholamines and their metabolites related to alzheimer’s disease in human urine. J. Sep. Sci. 2011, 34, 1198–1204. [Google Scholar] [CrossRef]
- Hyland, K. Clinical utility of monoamine neurotransmitter metabolite analysis in cerebrospinal fluid. Clin. Chem. 2008, 54, 633–641. [Google Scholar] [CrossRef]
- Strawn, J.R.; Ekhator, N.N.; Geracioti, T.D., Jr. In-use stability of monoamine metabolites in human cerebrospinal fluid. J. Chromatogr. B Biomed. Sci. Appl. 2001, 760, 301–306. [Google Scholar] [CrossRef]
- Willemsen, J.J.; Ross, H.A.; Lenders, J.W.; Sweep, F.C. Stability of urinary fractionated metanephrines and catecholamines during collection, shipment, and storage of samples. Clin. Chem. 2007, 53, 268–272. [Google Scholar] [CrossRef]
- Boomsma, F.; Alberts, G.; van Eijk, L.; Man in’t Veld, A.J.; Schalekamp, M.A. Optimal collection and storage conditions for catecholamine measurements in human plasma and urine. Clin. Chem. 1993, 39, 2503–2508. [Google Scholar]
- Moleman, P. Preservation of urine samples for assay of catecholamines and their metabolites. Clin. Chem. 1985, 31, 653–654. [Google Scholar]
- Ellingson, T.; Duddempudi, S.; Greenberg, B.D.; Hooper, D.; Eisenhofer, G. Determination of differential activities of soluble and membrane-bound catechol-o-methyltransferase in tissues and erythrocytes. J. Chromatogr. B Biomed. Sci. Appl. 1999, 729, 347–353. [Google Scholar] [CrossRef]
- Nagatsu, T.; Levitt, M.; Udenfriend, S. Tyrosine hydroxylase. The initial step in norepinephrine biosynthesis. J. Biol. Chem. 1964, 239, 2910–2917. [Google Scholar]
- Vermeer, L.M.; Higgins, C.A.; Roman, D.L.; Doorn, J.A. Real-time monitoring of tyrosine hydroxylase activity using a plate reader assay. Anal. Biochem. 2013, 432, 11–15. [Google Scholar] [CrossRef]
- Fossbakk, A.; Kleppe, R.; Knappskog, P.M.; Martinez, A.; Haavik, J. Functional studies of tyrosine hydroxylase missense variants reveal distinct patterns of molecular defects in dopa-responsive dystonia. Hum. Mutat. 2014, 35, 880–890. [Google Scholar] [CrossRef]
- Nagatsu, T.; Levitt, M.; Udenfriend, S. A rapid and simple radioassay for tyrosine hydroxylase activity. Anal. Biochem. 1964, 9, 122–126. [Google Scholar] [CrossRef]
- Nagatsu, T.; Levitt, M.; Udenfriend, S. Conversion of l-tyrosine to 3,4-dihydroxyphenylalanine by cell-free preparations of brain and sympathetically innervated tissues. Biochem. Biophys. Res. Commun. 1964, 14, 543–549. [Google Scholar] [CrossRef]
- Nagatsu, T.; Ichinose, H.; Kojima, K.; Kameya, T.; Shimase, J.; Kodama, T.; Shimosato, Y. Aromatic l-amino acid decarboxylase activities in human lung tissues: Comparison between normal lung and lung carcinomas. Biochem. Med. 1985, 34, 52–59. [Google Scholar] [CrossRef]
- Hoekstra, R.; Fekkes, D. Pteridines and affective disorders. Acta Neuropsychiatr. 2002, 14, 120–126. [Google Scholar] [CrossRef]
- Perry, M.; Li, Q.; Kennedy, R.T. Review of recent advances in analytical techniques for the determination of neurotransmitters. Anal. Chim. Acta 2009, 653, 1–22. [Google Scholar] [CrossRef]
- Blau, N.; Thony, B. Pterins and related enzymes. In Laboratory Guide to the Methods in Biochemical Genetics; Blau, N.D.M., Gibson, K.M., Eds.; Springer: Heidelberg, Germany, 2008; pp. 665–702. [Google Scholar]
- Opladen, T.; Abu Seda, B.; Rassi, A.; Thony, B.; Hoffmann, G.F.; Blau, N. Diagnosis of tetrahydrobiopterin deficiency using filter paper blood spots: Further development of the method and 5 years experience. J. Inherit. Metab. Dis. 2011, 34, 819–826. [Google Scholar] [CrossRef]
- Carducci, C.; Santagata, S.; Friedman, J.; Pasquini, E.; Carducci, C.; Tolve, M.; Angeloni, A.; Leuzzi, V. Urine sepiapterin excretion as a new diagnostic marker for sepiapterin reductase deficiency. Mol. Genet. Metab. 2015, 115, 157–160. [Google Scholar] [CrossRef]
- Bourcier, S.; Benoist, J.F.; Clerc, F.; Rigal, O.; Taghi, M.; Hoppilliard, Y. Detection of 28 neurotransmitters and related compounds in biological fluids by liquid chromatography/tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2006, 20, 1405–1421. [Google Scholar] [CrossRef]
- Zhao, Y.; Cao, J.; Chen, Y.S.; Zhu, Y.; Patrick, C.; Chien, B.; Cheng, A.; Foehr, E.D. Detection of tetrahydrobiopterin by lc-ms/ms in plasma from multiple species. Bioanalysis 2009, 1, 895–903. [Google Scholar] [CrossRef]
- Fiege, B.; Ballhausen, D.; Kierat, L.; Leimbacher, W.; Goriounov, D.; Schircks, B.; Thony, B.; Blau, N. Plasma tetrahydrobiopterin and its pharmacokinetic following oral administration. Mol. Genet. Metab. 2004, 81, 45–51. [Google Scholar] [CrossRef]
- Fekkes, D.; Voskuilen-Kooijman, A. Quantitation of total biopterin and tetrahydrobiopterin in plasma. Clin. Biochem. 2007, 40, 411–413. [Google Scholar] [CrossRef]
- Arning, E.; Bottiglieri, T. Lc-ms/ms analysis of cerebrospinal fluid metabolites in the pterin biosynthetic pathway. JIMD Rep. 2016, 29, 1–9. [Google Scholar]
- Fukushima, T.; Nixon, J.C. Analysis of reduced forms of biopterin in biological tissues and fluids. Anal. Biochem. 1980, 102, 176–188. [Google Scholar] [CrossRef]
- Fukushima, T.; Nixon, J.C. Chromatographic analysis of pteridines. Methods Enzymol. 1980, 66, 429–436. [Google Scholar]
- Werner, E.R.; Werner-Felmayer, G.; Wachter, H. High-performance liquid chromatographic methods for the quantification of tetrahydrobiopterin biosynthetic enzymes. J. Chromatogr. B Biomed. Appl. 1996, 684, 51–58. [Google Scholar] [CrossRef]
- Espinosa-Mansilla, A.M.; de la Pena, A.M.; Canada-Canada, F.; Mancha de Llanos, A. Lc determination of biopterin reduced forms by uv-photogeneration of biopterin and fluorimetric detection. Talanta 2008, 77, 844–851. [Google Scholar] [CrossRef]
- Canada-Canada, F.; Espinosa-Mansilla, A.; Munoz de la Pena, A.; Mancha de Llanos, A. Determination of marker pteridins and biopterin reduced forms, tetrahydrobiopterin and dihydrobiopterin, in human urine, using a post-column photoinduced fluorescence liquid chromatographic derivatization method. Anal. Chim. Acta 2009, 648, 113–122. [Google Scholar] [CrossRef]
- Milstien, S.; Kaufman, S.; Sakai, N. Tetrahydrobiopterin biosynthesis defects examined in cytokine-stimulated fibroblasts. J. Inherit. Metab. Dis. 1993, 16, 975–981. [Google Scholar] [CrossRef]
- Bonafe, L.; Thony, B.; Penzien, J.M.; Czarnecki, B.; Blau, N. Mutations in the sepiapterin reductase gene cause a novel tetrahydrobiopterin-dependent monoamine-neurotransmitter deficiency without hyperphenylalaninemia. Am. J. Hum. Genet. 2001, 69, 269–277. [Google Scholar] [CrossRef]
- Hibiya, M.; Ichinose, H.; Ozaki, N.; Fujita, K.; Nishimoto, T.; Yoshikawa, T.; Asano, Y.; Nagatsu, T. Normal values and age-dependent changes in gtp cyclohydrolase i activity in stimulated mononuclear blood cells measured by high-performance liquid chromatography. J. Chromatogr. B Biomed. Sci. Appl. 2000, 740, 35–42. [Google Scholar] [CrossRef]
- Fismen, L.; Eide, T.; Djurhuus, R.; Svardal, A.M. Simultaneous quantification of tetrahydrobiopterin, dihydrobiopterin, and biopterin by liquid chromatography coupled electrospray tandem mass spectrometry. Anal. Biochem. 2012, 430, 163–170. [Google Scholar] [CrossRef]
- Guibal, P.; Leveque, N.; Doummar, D.; Giraud, N.; Roze, E.; Rodriguez, D.; Couderc, R.; Billette De Villemeur, T.; Moussa, F. Simultaneous determination of all forms of biopterin and neopterin in cerebrospinal fluid. ACS Chem. Neurosci. 2014, 5, 533–541. [Google Scholar] [CrossRef]
- Yuan, T.F.; Huang, H.Q.; Gao, L.; Wang, S.T.; Li, Y. A novel and reliable method for tetrahydrobiopterin quantification: Benzoyl chloride derivatization coupled with liquid chromatography-tandem mass spectrometry analysis. Free Radic. Biol. Med. 2018, 118, 119–125. [Google Scholar] [CrossRef]
- Ponzone, A.; Guardamagna, O.; Spada, M.; Ponzone, R.; Sartore, M.; Kierat, L.; Heizmann, C.W.; Blau, N. Hyperphenylalaninemia and pterin metabolism in serum and erythrocytes. Clin. Chim. Acta 1993, 216, 63–71. [Google Scholar] [CrossRef]
- Slazyk, W.E.; Spierto, F.W. Liquid-chromatographic measurement of biopterin and neopterin in serum and urine. Clin. Chem. 1990, 36, 1364–1368. [Google Scholar]
- Tornero, E.M.; Meras, I.; Espinosa-Mansilla, A. HPLC determination of serum pteridine pattern as biomarkers. Talanta 2014, 128, 319–326. [Google Scholar] [CrossRef]
- Zurfluh, M.R.; Giovannini, M.; Fiori, L.; Fiege, B.; Gokdemir, Y.; Baykal, T.; Kierat, L.; Gartner, K.H.; Thony, B.; Blau, N. Screening for tetrahydrobiopterin deficiencies using dried blood spots on filter paper. Mol. Genet. Metab. 2005, 86 (Suppl. 1), S96–S103. [Google Scholar] [CrossRef]
- Santagata, S.; Di Carlo, E.; Carducci, C.; Leuzzi, V.; Angeloni, A.; Carducci, C. Development of a new uplc-esi-ms/ms method for the determination of biopterin and neopterin in dried blood spot. Clin. Chim. Acta 2017, 466, 145–151. [Google Scholar] [CrossRef]
- Allegri, G.; Costa Netto, H.J.; Ferreira Gomes, L.N.; Costa de Oliveira, M.L.; Scalco, F.B.; de Aquino Neto, F.R. Determination of six pterins in urine by lc-ms/ms. Bioanalysis 2012, 4, 1739–1746. [Google Scholar] [CrossRef]
- Burton, C.; Shi, H.; Ma, Y. Simultaneous detection of six urinary pteridines and creatinine by high-performance liquid chromatography-tandem mass spectrometry for clinical breast cancer detection. Anal. Chem. 2013, 85, 11137–11145. [Google Scholar] [CrossRef]
- Koslinski, P.; Jarzemski, P.; Markuszewski, M.J.; Kaliszan, R. Determination of pterins in urine by hplc with uv and fluorescent detection using different types of chromatographic stationary phases (hilic, rp c8, rp c18). J. Pharm. Biomed. Anal. 2014, 91, 37–45. [Google Scholar] [CrossRef]
- Burton, C.; Shi, H.; Ma, Y. Development of a high-performance liquid chromatography—tandem mass spectrometry urinary pterinomics workflow. Anal. Chim. Acta 2016, 927, 72–81. [Google Scholar] [CrossRef]
- Antonozzi, I.; Carducci, C.; Vestri, L.; Pontecorvi, A.; Moretti, F. Rapid and sensitive method for high-performance liquid chromatographic analysis of pterins in biological fluids. J. Chromatogr. 1988, 459, 319–324. [Google Scholar] [CrossRef]
- Dhondt, J.L.; Largilliere, C.; Ardouin, P.; Farriaux, J.P.; Dautrevaux, M. Diagnosis of variants of hyperphenylalaninemia by determination of pterins in urine. Clin. Chim. Acta 1981, 110, 205–214. [Google Scholar] [CrossRef]
- Niederwieser, A.; Staudenmann, W.; Wetzel, E. High-performance liquid chromatography with column switching for the analysis of biogenic amine metabolites and pterins. J. Chromatogr. 1984, 290, 237–246. [Google Scholar] [CrossRef]
- Tomsikova, H.; Solich, P.; Novakova, L. Sample preparation and uhplc-fd analysis of pteridines in human urine. J. Pharm. Biomed. Anal. 2014, 95, 265–272. [Google Scholar] [CrossRef]
- Dhondt, J.C.; Hayte, J.M.; Forzy, G.; Delcroix, M.; Farriaux, J.P. Unconjugated pteridins in amniotic fluid during gestation. Clin. Chim. Acta 1986, 161, 269–273. [Google Scholar] [CrossRef]
- Niederwieser, A.; Shintaku, H.; Hasler, T.; Curtius, H.C.; Lehmann, H.; Guardamagna, O.; Schmidt, H. Prenatal diagnosis of "dihydrobiopterin synthetase" deficiency, a variant form of phenylketonuria. Eur. J. Pediatr. 1986, 145, 176–178. [Google Scholar] [CrossRef]
- Guroff, G.; Rhoads, C.A.; Abramowitz, A. A simple radioisotope assay for phenylalanine hydroxylase cofactor. Anal. Biochem. 1967, 21, 273–278. [Google Scholar] [CrossRef]
- Niederwieser, A.; Curtius, H.C.; Bettoni, O.; Bieri, J.; Schircks, B.; Viscontini, M.; Schaub, J. Atypical phenylketonuria caused by 7, 8-dihydrobiopterin synthetase deficiency. Lancet 1979, 1, 131–133. [Google Scholar] [CrossRef]
- Schlesinger, P.; Watson, B.M.; Cotton, R.G.; Danks, D.M. Urinary dihydroxanthopterin in the diagnosis of malignant hyperphenylalaninemia and phenylketonuria. Clin. Chim. Acta 1979, 92, 187–195. [Google Scholar] [CrossRef]
- Leeming, R.J.; Blair, J.A.; Green, A.; Raine, D.N. Biopterin derivatives in normal and phenylketonuric patients after oral loads of l-phenylalanine, l-tyrosine, and l-tryptophan. Arch. Dis. Child. 1976, 51, 771–777. [Google Scholar] [CrossRef][Green Version]
- Hyland, K. Estimation of tetrahydro, dihydro and fully oxidised pterins by high-performance liquid chromatography using sequential electrochemical and fluorometric detection. J. Chromatogr. 1985, 343, 35–41. [Google Scholar] [CrossRef]
- Hyland, K.; Surtees, R.A.; Heales, S.J.; Bowron, A.; Howells, D.W.; Smith, I. Cerebrospinal fluid concentrations of pterins and metabolites of serotonin and dopamine in a pediatric reference population. Pediatr. Res. 1993, 34, 10–14. [Google Scholar] [CrossRef]
- Howells, D.W.; Smith, I.; Hyland, K. Estimation of tetrahydrobiopterin and other pterins in cerebrospinal fluid using reversed-phase high-performance liquid chromatography with electrochemical and fluorescence detection. J. Chromatogr. 1986, 381, 285–294. [Google Scholar] [CrossRef]
- Guibal, P.; Lo, A.; Maitre, P.; Moussa, F. Pterin determination in cerebrospinal fluid: State of the art. Pteridines 2017, 28, 83–89. [Google Scholar] [CrossRef]
- Blau, N.; Niederwieser, A. Guanosine triphosphate cyclohydrolase i assay in human and rat liver using high-performance liquid chromatography of neopterin phosphates and guanine nucleotides. Anal. Biochem. 1983, 128, 446–452. [Google Scholar] [CrossRef]
- Werner, E.R.; Werner-Felmayer, G.; Fuchs, D.; Hausen, A.; Reibnegger, G.; Yim, J.J.; Pfleiderer, W.; Wachter, H. Tetrahydrobiopterin biosynthetic activities in human macrophages, fibroblasts, thp-1, and t 24 cells. Gtp-cyclohydrolase i is stimulated by interferon-gamma, and 6-pyruvoyl tetrahydropterin synthase and sepiapterin reductase are constitutively present. J. Biol. Chem. 1990, 265, 3189–3192. [Google Scholar]
- Viveros, O.H.; Lee, C.L.; Abou-Donia, M.M.; Nixon, J.C.; Nichol, C.A. Biopterin cofactor biosynthesis: Independent regulation of gtp cyclohydrolase in adrenal medulla and cortex. Science 1981, 213, 349–350. [Google Scholar] [CrossRef]
- Hatakeyama, K.; Yoneyama, T. A sensitive assay for the enzymatic activity of GTP cyclohydrolase I. Methods Mol. Biol. 1998, 100, 265–272. [Google Scholar]
- Laboratory Guide to the Methods in Biochemical Genetics; Blau, N., Duran, M., Gibson, K.M., Eds.; Springer: Bwelin, Germany, 2008. [Google Scholar]
- Ferre, J.; Naylor, E.W. Sepiapterin reductase in human amniotic and skin fibroblasts, chorionic villi, and various blood fractions. Clin. Chim. Acta 1988, 174, 271–282. [Google Scholar] [CrossRef]
- Arai, N.; Narisawa, K.; Hayakawa, H.; Tada, K. Hyperphenylalaninemia due to dihydropteridine reductase deficiency: Diagnosis by enzyme assays on dried blood spots. Pediatrics 1982, 70, 426–430. [Google Scholar]
- Werner, E.R.; Werner-Felmayer, G.; Fuchs, D.; Hausen, A.; Reibnegger, G.; Wachter, H. Parallel induction of tetrahydrobiopterin biosynthesis and indoleamine 2,3-dioxygenase activity in human cells and cell lines by interferon-gamma. Biochem. J. 1989, 262, 861–866. [Google Scholar] [CrossRef]
- Shintaku, H.; Niederwieser, A.; Leimbacher, W.; Curtius, H.C. Tetrahydrobiopterin deficiency: Assay for 6-pyruvoyl-tetrahydropterin synthase activity in erythrocytes, and detection of patients and heterozygous carriers. Eur. J. Pediatr. 1988, 147, 15–19. [Google Scholar] [CrossRef]
- Firgaira, F.A.; Cotton, R.G.; Danks, D.M. Dihydropteridine reductase deficiency diagnosis by assays on peripheral blood cells. Lancet 1980, 1, 160. [Google Scholar] [CrossRef]
- Lipson, A.; Yu, J.; O’Halloran, M.; Potter, M.; Wilken, B. Dihydropteridine reductase deficiency: Non-response to oral tetrahydrobiopterin load test. J. Inherit. Metab. Dis. 1984, 7, 69–71. [Google Scholar] [CrossRef]
- Blau, N.; Hennermann, J.B.; Langenbeck, U.; Lichter-Konecki, U. Diagnosis, classification, and genetics of phenylketonuria and tetrahydrobiopterin (bh4) deficiencies. Mol. Genet. Metab. 2011, 104, S2–S9. [Google Scholar] [CrossRef]
- Rebrin, I.; Bailey, S.W.; Boerth, S.R.; Ardell, M.D.; Ayling, J.E. Catalytic characterization of 4a-hydroxytetrahydropterin dehydratase. Biochemistry 1995, 34, 5801–5810. [Google Scholar] [CrossRef]
- Bailey, S.W.; Rebrin, I.; Boerth, S.R.; Auling, J.E. Synthesis of 4a-hydroxytetrahydropterins and the mechanism of their nonenzymatic dehydration to quinoid dihydropterins. J. Am. Chem. Soc. 1995, 117, 10203–10211. [Google Scholar] [CrossRef]
- Thony, B.; Neuheiser, F.; Kierat, L.; Blaskovics, M.; Arn, P.H.; Ferreira, P.; Rebrin, I.; Ayling, J.; Blau, N. Hyperphenylalaninemia with high levels of 7-biopterin is associated with mutations in the pcbd gene encoding the bifunctional protein pterin-4a-carbinolamine dehydratase and transcriptional coactivator (dcoh). Am. J. Hum. Genet. 1998, 62, 1302–1311. [Google Scholar] [CrossRef]
- Rebrin, I.; Bailey, S.W.; Ayling, J.E. Activity of the bifunctional protein 4a-hydroxy-tetrahydropterin dehydratase/dcoh during human fetal development: Correlation with dihydropteridine reductase activity and tetrahydrobiopterin levels. Biochem. Biophys. Res. Commun. 1995, 217, 958–965. [Google Scholar] [CrossRef]
- Ayling, J.E.; Bailey, S.W.; Boerth, S.R.; Giugliani, R.; Braegger, C.P.; Thony, B.; Blau, N. Hyperphenylalaninemia and 7-pterin excretion associated with mutations in 4a-hydroxy-tetrahydrobiopterin dehydratase/dcoh: Analysis of enzyme activity in intestinal biopsies. Mol. Genet. Metab. 2000, 70, 179–188. [Google Scholar] [CrossRef]
- Schwarz, M.A. Enzyme-catalyzed amperometric oxidation of neurotransmitters in chip-capillary electrophoresis. Electrophoresis 2004, 25, 1916–1922. [Google Scholar] [CrossRef]
- Cosentino, M.; Bombelli, R.; Ferrari, M.; Marino, F.; Rasini, E.; Maestroni, G.J.; Conti, A.; Boveri, M.; Lecchini, S.; Frigo, G. Hplc-ed measurement of endogenous catecholamines in human immune cells and hematopoietic cell lines. Life Sci. 2000, 68, 283–295. [Google Scholar] [CrossRef]
- Garcia-Cazorla, A.; Duarte, S.; Serrano, M.; Nascimento, A.; Ormazabal, A.; Carrilho, I.; Briones, P.; Montoya, J.; Garesse, R.; Sala-Castellvi, P.; et al. Mitochondrial diseases mimicking neurotransmitter defects. Mitochondrion 2008, 8, 273–278. [Google Scholar] [CrossRef]
- Molero-Luis, M.; Serrano, M.; Ormazabal, A.; Perez-Duenas, B.; Garcia-Cazorla, A.; Pons, R.; Artuch, R.; Neurotransmitter Working, G. Homovanillic acid in cerebrospinal fluid of 1388 children with neurological disorders. Dev. Med. Child. Neurol. 2013, 55, 559–566. [Google Scholar] [CrossRef]
- Shaywitz, B.A.; Cohen, D.J.; Bowers, M.B. Reduced cerebrospinal fluid 5-hydroxyindoleacetic acid and homovanillic acid in children with epilepsy. Neurology 1975, 25, 72–79. [Google Scholar] [CrossRef]
- Devinsky, O.; Emoto, S.; Goldstein, D.S.; Stull, R.; Porter, R.J.; Theodore, W.H.; Nadi, N.S. Cerebrospinal fluid and serum levels of dopa, catechols, and monoamine metabolites in patients with epilepsy. Epilepsia 1992, 33, 263–270. [Google Scholar] [CrossRef]
- Giroud, M.; Dumas, R.; Dauvergne, M.; D’Athis, P.; Rochette, L.; Beley, A.; Bralet, J. 5-hydroxyindoleacetic acid and homovanillic acid in cerebrospinal fluid of children with febrile convulsions. Epilepsia 1990, 31, 178–181. [Google Scholar] [CrossRef]
- Papeschi, R.; Molina-Negro, P.; Sourkes, T.L.; Erba, G. The concentration of homovanillic and 5-hydroxyindoleacetic acids in ventricular and lumbar csf. Studies in patients with extrapyramidal disorders, epilepsy, and other diseases. Neurology 1972, 22, 1151–1159. [Google Scholar] [CrossRef]
- Tabaddor, K.; Wolfson, L.I.; Sharpless, N.S. Diminished ventricular fluid dopamine metabolites in adult-onset dystonia. Neurology 1978, 28, 1254–1258. [Google Scholar] [CrossRef]
- Dobyns, W.B.; Ozelius, L.J.; Kramer, P.L.; Brashear, A.; Farlow, M.R.; Perry, T.R.; Walsh, L.E.; Kasarskis, E.J.; Butler, I.J.; Breakefield, X.O. Rapid-onset dystonia-parkinsonism. Neurology 1993, 43, 2596–2602. [Google Scholar] [CrossRef]
- Banki, C.M.; Molnar, G.; Vojnik, M. Cerebrospinal fluid amine metabolites, tryptophan and clinical parameters in depression. Part 2. Psychopathological symptoms. J. Affect. Disord. 1981, 3, 91–99. [Google Scholar] [CrossRef]
- Banki, C.M.; Vojnik, M.; Molnar, G. Cerebrospinal fluid amine metabolites, tryptophan and clinical parameters in depression. Part 1. Background variables. J. Affect. Disord. 1981, 3, 81–89. [Google Scholar] [CrossRef]
- Sjogren, M.; Minthon, L.; Passant, U.; Blennow, K.; Wallin, A. Decreased monoamine metabolites in frontotemporal dementia and alzheimer’s disease. Neurobiol. Aging 1998, 19, 379–384. [Google Scholar] [CrossRef]
- Van Der Heyden, J.C.; Rotteveel, J.J.; Wevers, R.A. Decreased homovanillic acid concentrations in cerebrospinal fluid in children without a known defect in dopamine metabolism. Eur. J. Paediatr. Neurol. 2003, 7, 31–37. [Google Scholar] [CrossRef]
- Mercimek-Mahmutoglu, S.; Sidky, S.; Hyland, K.; Patel, J.; Donner, E.J.; Logan, W.; Mendoza-Londono, R.; Moharir, M.; Raiman, J.; Schulze, A.; et al. Prevalence of inherited neurotransmitter disorders in patients with movement disorders and epilepsy: A retrospective cohort study. Orphanet J. Rare Dis. 2015, 10, 12. [Google Scholar] [CrossRef]
- Blau, N.; Bonafe, L.; Krageloh-Mann, I.; Thony, B.; Kierat, L.; Hausler, M.; Ramaekers, V. Cerebrospinal fluid pterins and folates in aicardi-goutieres syndrome: A new phenotype. Neurology 2003, 61, 642–647. [Google Scholar] [CrossRef]
- Hagberg, L.; Cinque, P.; Gisslen, M.; Brew, B.J.; Spudich, S.; Bestetti, A.; Price, R.W.; Fuchs, D. Cerebrospinal fluid neopterin: An informative biomarker of central nervous system immune activation in hiv-1 infection. AIDS Res. Ther. 2010, 7, 15. [Google Scholar] [CrossRef]
- Millner, M.M.; Franthal, W.; Thalhammer, G.H.; Berghold, A.; Aigner, R.M.; Fuger, G.F.; Reibnegger, G. Neopterin concentrations in cerebrospinal fluid and serum as an aid in differentiating central nervous system and peripheral infections in children. Clin. Chem. 1998, 44, 161–167. [Google Scholar]
- Sheline, Y.; Bardgett, M.E.; Csernansky, J.G. Correlated reductions in cerebrospinal fluid 5-hiaa and mhpg concentrations after treatment with selective serotonin reuptake inhibitors. J. Clin. Psychopharmacol. 1997, 17, 11–14. [Google Scholar] [CrossRef]
- Potter, W.Z.; Scheinin, M.; Golden, R.N.; Rudorfer, M.V.; Cowdry, R.W.; Calil, H.M.; Ross, R.J.; Linnoila, M. Selective antidepressants and cerebrospinal fluid. Lack of specificity on norepinephrine and serotonin metabolites. Arch. Gen. Psychiatry 1985, 42, 1171–1177. [Google Scholar] [CrossRef]
- Burlina, A.B.; Celato, A.; Polo, G.; Edini, C.; Burlina, A.P. The utility of csf for the diagnosis of primary and secondary monoamine neurotransmitter deficiencies. EJIFCC 2017, 28, 64–76. [Google Scholar]
- Bowden, C.L.; Koslow, S.H.; Hanin, I.; Maas, J.W.; Davis, J.M.; Robins, E. Effects of amitriptyline and imipramine on brain amine neurotransmitter metabolites in cerebrospinal fluid. Clin. Pharmacol. Ther. 1985, 37, 316–324. [Google Scholar] [CrossRef]
- Brun, L.; Ngu, L.H.; Keng, W.T.; Ch’ng, G.S.; Choy, Y.S.; Hwu, W.L.; Lee, W.T.; Willemsen, M.A.; Verbeek, M.M.; Wassenberg, T.; et al. Clinical and biochemical features of aromatic l-amino acid decarboxylase deficiency. Neurology 2010, 75, 64–71. [Google Scholar] [CrossRef]
- Aitkenhead, H.; Heales, S.J. Establishment of paediatric age-related reference intervals for serum prolactin to aid in the diagnosis of neurometabolic conditions affecting dopamine metabolism. Ann. Clin. Biochem. 2013, 50, 156–158. [Google Scholar] [CrossRef] [PubMed]
- Capozzi, A.; Scambia, G.; Pontecorvi, A.; Lello, S. Hyperprolactinemia: Pathophysiology and therapeutic approach. Gynecol. Endocrinol. 2015, 31, 506–510. [Google Scholar] [CrossRef] [PubMed]
- Verhelst, J.; Abs, R. Hyperprolactinemia: Pathophysiology and management. Treat. Endocrinol. 2003, 2, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Paul, P.; Talib, V.J. Serum prolactin in seizure disorders. Indian Pediatr. 2004, 41, 827–831. [Google Scholar] [PubMed]
- Leucht, S.; Cipriani, A.; Spineli, L.; Mavridis, D.; Orey, D.; Richter, F.; Samara, M.; Barbui, C.; Engel, R.R.; Geddes, J.R.; et al. Comparative efficacy and tolerability of 15 antipsychotic drugs in schizophrenia: A multiple-treatments meta-analysis. Lancet 2013, 382, 951–962. [Google Scholar] [CrossRef]
- Liu, H.; Li, N.; Zhang, H.; Zhang, F.; Su, X. A simple and convenient fluorescent strategy for the highly sensitive detection of dopamine and ascorbic acid based on graphene quantum dots. Talanta 2018, 189, 190–195. [Google Scholar] [CrossRef]
- Kanagasubbulakshmi, S.; Kadirvelu, K. Photoinduced holes transfer based visual determination of dopamine in human serum. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2019, 206, 512–519. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.X.; Li, Y.F. Cu (ii)-based metal-organic xerogels as a novel nanozyme for colorimetric detection of dopamine. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2019, 207, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Jafarinejad, S.; Ghazi-Khansari, M.; Ghasemi, F.; Sasanpour, P.; Hormozi-Nezhad, M.R. Colorimetric fingerprints of gold nanorods for discriminating catecholamine neurotransmitters in urine samples. Sci. Rep. 2017, 7, 8266. [Google Scholar] [CrossRef]
- Ghasemi, F.; Hormozi-Nezhad, M.R.; Mahmoudi, M. Identification of catecholamine neurotransmitters using fluorescence sensor array. Anal. Chim. Acta 2016, 917, 85–92. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
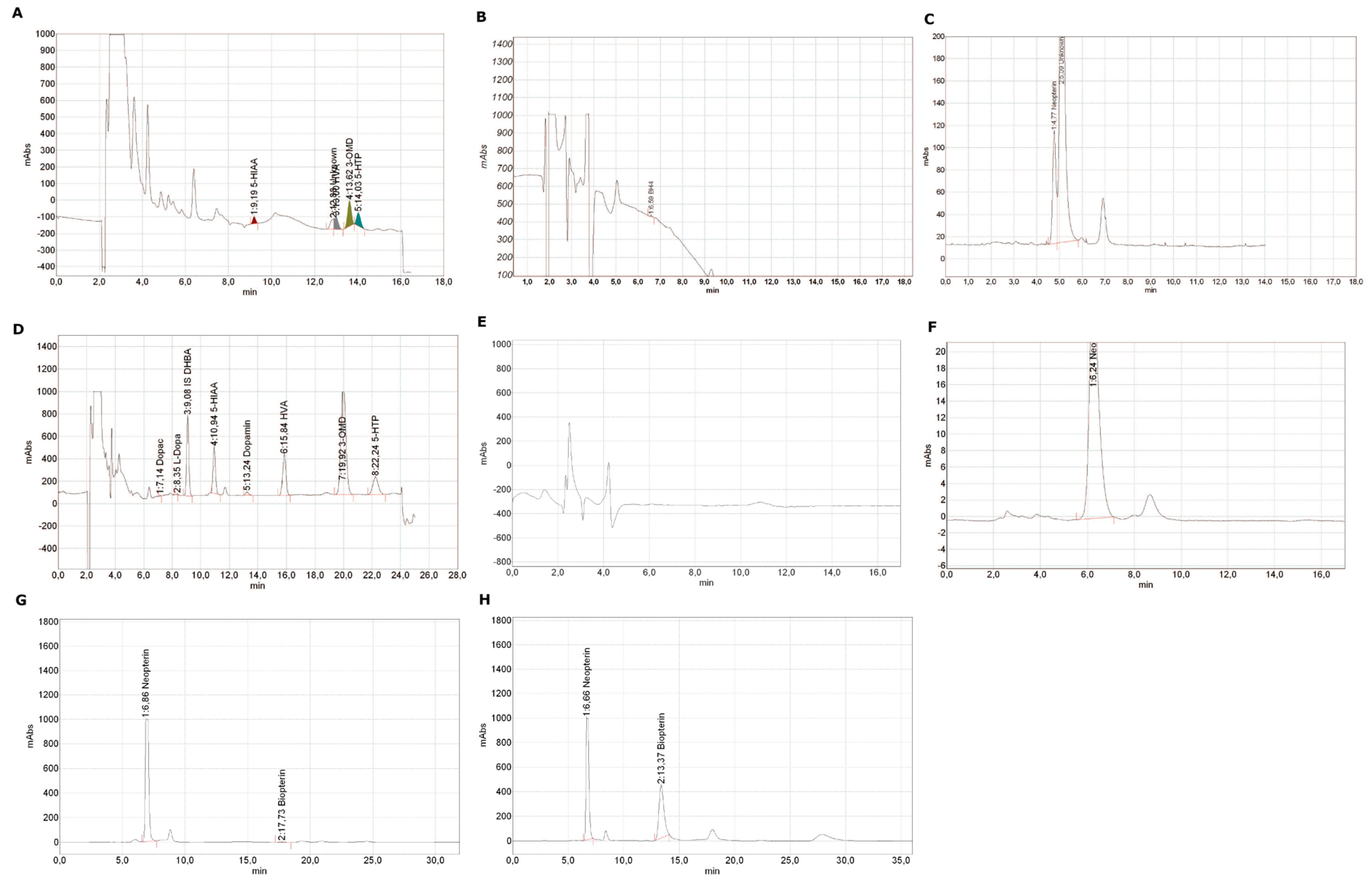{kind=link}
| Matrix | Volume (mL) | Sample Preparation | Solvent | Analytes | Ref. |
|---|---|---|---|---|---|
| Human CSF | 1.0 | Dilution | 6 mM L-cysteine/2 mM oxalic acid/1.3% glacial acetic acid | DA; DOPAC; HVA; HT; HIAA | [37] |
| 6th–8th mL | Dilution | 0.03% formic acid | L-Dopa; 3-MT; HVA; HIAA; MHPG; 5-HTP | [38] | |
| 2nd tube (400th–800th µL) | Dilution; filtration | NA | HVA; 5-HIAA; 3-OMD; MHPG; 5-HTP | [39] | |
| 50 µL | Dilution; filtration | 250 nM 2,5-dihdroxybenzoic acid; 5000 MWCO PES Vivaspin 500 filter | HVA; 5-HIAA; 3-OMD; 5-HTP; MHPG | [40] | |
| Human plasma | 0.5 | SPE | Water/acetonitrile (40:60, v/v) with 2.5% formic acid | NE; E | [27] |
| 0.5 | PP, TFC | 10% TCA; A: Water/0.1% perfluoroheptanoic acid; B: Water/acetonitrile (40:60, v/v); C: Isopropanol/acetone/acetonitrile (1:1:1) with 0.3% formic acid; D: Water/5 mM ammonium acetate/50% acetonitrile | NMN/MN | [41] | |
| 0.2 | SPE | Acetonitrile with 2% formic acid | NMN; MN | [42] | |
| 0.5 | SPE | Water/acetonitrile (40:60, v/v) | NMN; MN | [43] | |
| 0.1 | SPE | Water/acetonitrile (5:95, v/v) with 2% formic acid | NMN; MN; 3-MT | [44] | |
| 0.015 | Dilution | 10 mM glutathione/10 mM citric acid/100 mgL−1 EDTA pH4.5 | DA; NE; E; NMN; MN; 3-MT | [45] | |
| 0.5 | PP/filtration | 1.2 M perchloric acid | L-Dopa; DA; DOPAC | [46] | |
| 0.5 | SPE | Aqueous solution (10.5 g L−1 citric acid/20 mg L−1 EDTA)/acetonitrile (98:2, v/v); pH 2.8, 1 M NaOH | L-Dopa; DA; NE; E; DHPG | [47] | |
| 0.02 | SPE | 0.6 M potassium chloride/acetonitrile (2:3, v/v) | DA; NE; E | [48] | |
| 0.5 | SPE | 10.5 g L−1 citric acid/20 mg L−1 OSA/20 mg L−1 EDTA/methanol (97.5:2.5, v/v) pH 2.9, 1 M NaOH | DA; NE; E; | [24] | |
| 0.5 | SPE | 10.5 g L−1 citric acid/20 mg L−1 OSA/20 mg L−1 EDTA/methanol (95:5, v/v) pH 3.5, 1 M NaOH; MHPG: methanol | DA; NE/E; MHPG | [29] | |
| 0.1/0.5 | LLE | Ammonia buffer/heptane mixture; 80 mM acetic acid/octanol; MHPG: Ethyl acetate | DA; NE; E; MHPG (free and conjugated) | [49] | |
| Human urine | 0.25 | SPE | Water/methanol (95:5, v/v) with 2% formic acid | NMN; MN | [50] |
| 3.0 | SPE | Water/acetonitrile (20:80, v/v) with 1% formic acid | DA; NE; E | [51] | |
| 0.04 | Filtration | NR | L-Dopa; DA; NE; E, MN | [52] | |
| 20 | PBA affinity column | 0.1 M phosphate buffer/1 mM EDTA/300 mg L-1 SOS/ methanol (10:1, v/v), pH 2.5 | DA/NE/E | [53] | |
| 0.02 | SPE | 50 mM potassium dihydrogenphosphate/2.5 mM OSA/0.1 g L-1 EDTA/acetonitrile (96.5:3.5, v/v); pH 3.5, phosphoric acid | DA; NE; E | [25] | |
| 5.0 | SPE | 6 M acetic acid | DA/NE/E | [54] | |
| 0.3 | LLE | Ammonia buffer/heptane mixture; 166 mM aqueous acetic acid/1-octanol | DA/NE/E | [55] | |
| 1.0 | SPE | 1 M acetic acid | DA; NE; E | [30] | |
| 5.0 | Bio-Rex 70 resin | 4 M formic acid | DA; NE; E; NMN; MN | [56] | |
| 5.0 | Bio-Rex 70 resin | 4 M formic acid | DA; NE; E; NMN; MN | [57] | |
| 0.5 | LLE | Ammonia buffer/heptane mixture; 80 mM acetic acid/1-octanol; MHPG: Ethyl acetate | DA; NE; E; MHPG (free and conjugated) | [49] | |
| Amniotic fluid | 0.2 | Dilution | 20 mM phosphate buffer; pH 3.0; 0.5 mM heptasulfonic acid; 0.12 mM EDTA; 0.28% perchloric acid; 15% methanol | HIAA; HVA | [58] |
| Matrix | Technique | Analytes | Sample Preparation | Internal Standard | Column | Mobile Phase | Elution | Detection | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| Human CSF | HPLC | DA; DOPAC; HVA; 5-HT; 5-HIAA | Dilution | NR | ESA MD-150 C18 | 75 mM monobasic sodium phosphate buffer/0.5 mM EDTA/0.81 mM OSA/5% tetrahydrofuran/acetonitrile (95:5, v/v) pH 3.1; phosphoric acid | Isocratic | Coulometric | [37] |
| HPLC | DA; NE; DOPAC; HVA; MHPG; 5-HT; 5-HIAA | Direct injection dialysate | NR | Luna C18 | 0.2 M phosphate buffer pH 5.0 | Isocratic | Amperometric | [84] | |
| HPLC | 3-OMD; HVA; 5-HIAA; MHPG; 5-HTP | Dilution, filtration | 3-OMD, HVA; 5-HIAA; MHPG; 5-HTP | ODS (C18) | 0.1 M sodium acetate; 0.1 M citric acid; 1.2 mmol/l EDTA, 1.2 mmol/l 1-heptanosulfonic acid; 75 mL methanol; adjusted to pH 4.0 | Isocratic | Coulometric | [39] | |
| UHPLC | HVA; 5-HIAA; 3-OMD; 5-HTP; MHPG | Direct injection | HVA; 5-HIAA; MHPG; 3-OMD; 5-HTP | ACQUITY UPLC HSS T3 | 0.05 M citrate buffer; pH 5.2; methanol (97:3, v/v) | Isocratic | Coulometric; FL ex: 350 nm; em: 450 nm | [40] | |
| Human plasma | LC | NE; E | SPE | d6-NE; d6-E | C18 | A: 10 mM ammonium formate in water; B: Methanol | Gradient | MS/MS; [positive ionization electrospray] | [27] |
| LC | NMN; MN | PP; TFC | d3-NMN; d3-MN | Hypercarb PGC | A: 50 mM ammonium formate/1% formic acid in water; B: 0.1% formic acid in acetonitrile; C: Isopropanol/acetone/acetonitrile (9:2:9, v/v/v); D: 0.1% perfluoroheptanoic acid in water | Gradient | MS/MS; [positive ionization electrospray] | [41] | |
| UHPLC | NMN; MN | SPE | d3-NMN; d3-MN | Atlantis HILIC | A: Acetonitrile; B: 200 mM ammonium formate pH 3.0 | Gradient | MS/MS; [positive ionization electrospray] | [42] | |
| LC | NMN; MN | SPE | d3-NMN; d3-MN | Hypercarb PGC Hypersil Gold HILIC | A: 50 mM ammonium formate/1% formic acid in water; B: 0.1% formic acid in acetonitrile; C: Isopropanol/acetone/acetonitrile (9:2:9, v/v/v) A: 100 mM ammonium formate/acetonitrile (5:95, v/v) pH 3.2; B: Acetonitrile/water/100 mM ammonium formate (50:45:5, v/v/v) pH 3.2 | Gradient | MS/MS; [positive ionization electrospray] | [43] | |
| HPLC | NMN; MN; 3-MT | SPE | d3-NMN; d3-MN; d4-3-MT | Atlantis HILIC | A: 100 mM ammonium formate in water pH 3.0; formic acid; B: Acetonitrile | Gradient | MS/MS; [positive ionization electrospray] | [44] | |
| HPLC | DA; NE; E; NMN; MN; 3-MT | Dilution | MHBA | Unison UK-C18 | 75 mM potassium acetate buffer/100 mM potassium phosphate buffer/8 mM sodium 1-hexanesulfonate/acetonitrile (93.1:4.9:2, v/v/v); pH 3.2 | Isocratic | CL; [TDPO/H2O2] | [45] | |
| HPLC | L-DOPA; DA; DOPAC; 3-O-MD; Carbidopa | PP/Filtration | DHBA | ESA HR-80 C18 | Modified CAT-A-PHASE buffer: Phosphate buffer/patented ion-pairing agent/methanol/acetonitrile (99.7:0.3, v/v); pH 3.2; 2 N NaOH | Isocratic | Coulometric | [46] | |
| HPLC | L-DOPA; DA; NE; E; DHPG | SPE | DHBA | Deverosil RPAQUEOUS-AR-5 C30 | Aqueous solution of 10.5 g L−1 citric acid/20 mg L−1 EDTA/acetonitrile (98:2, v/v); pH 2.8; 1 M NaOH | Isocratic | Amperometric | [47] | |
| HPLC | DA; NE; E; MHPG | SPE | DHBA | Microsorb C8 | 10.5 g L−1 citric acid/20 mg L−1 EDTA/20 mg L−1 OSA/methanol (95:5, v/v); pH 3.5; 1 M NaOH | Isocratic | Amperometric | [29] | |
| HPLC | DA; NE; E | SPE | DHBA | Rainin C8 | 25 mM citric acid/20 mg L−1 EDTA/20 mg L−1 OSA/methanol (97:3, v/v) pH 2.9; 1 M NaOH | Isocratic | Amperometric | [28] | |
| HPLC | DA; NE; E | SPE | IPT | TSK gel ODS-120 T | 120 mM imidazole buffer/methanol/acetonitrile (13:4:18, v/v/v) pH 5.8 | Isocratic | CL | [48] | |
| HPLC | DA; NE; E | SPE | DHBA | Jones Apex C8 | 10.5 g L−1 citric acid/20 mg L−1 EDTA/20 mg L−1 OSA/methanol (97.5:2.5, v/v); pH 2.9; 1 M NaOH | Isocratic | Coulometric | [24] | |
| HPLC | NE; E | LLE | DHBA | Hypersil ODS | 9.02 g sodium acetate/0.372 g EDTA/100 mg SDS/methanol (85:15) or (80:20) pH 5.1; glacial acetic acid | Isocratic | Amperometric | [85] | |
| Human urine | LC | NMN; MN | SPE | d3-NMN d3-MN | Ultra II PFP propyl | 0.2% formic acid/methanol (95:5, v/v) | Isocratic | MS/MS; [positive ionization electrospray] | [50] |
| HPLC | L-DOPA; DA; NE; E; MN; 5-HT; tryptophan; andderivative | Filtration | NR | Fluofix-II 120E | Water/acetonitrile/trifluoroacetic acid (40:60:0.05, v/v/v) | Isocratic | FL; [PFOEI]; ex: 280 nm; em: 320 nm | [52] | |
| HPLC | L-DOPA; DA; NE; E; DOPAC | Dilution | IPT | Kromasil C18 | A: Methanol; B: 0.1 M sodium acetate buffer pH 5.0; acetic acid | Gradient | FL; [DPE]; ex: 350 nm; em: 480 nm | [86] | |
| HPLC | DA; NE; E | SPE | NR | ZIC-HILIC; BEH-amide | 6.5 mM ammonium formate/acetonitrile (25:75, v/v) pH 3.0; 6.5 mM ammonium formate/acetonitrile (15:85, v/v) pH 3.0 | Isocratic | Coulometric | [51] | |
| LC | NMN; MN | SPE | d3-NMN d3-MN | Atlantis T3 C18 | A: 10 mM ammonium formate/1% formic acid; B: Methanol | Gradient | MS/MS; [positive ionization electrospray] | [59] | |
| HPLC | DA; NE; E | SPE | d4-DA; d3-NE; d3-E | Allure PFP propyl | A: 25 mM ammonium formate in water pH 3.0; formic acid; B: Methanol | Gradient | MS/MS; [positive ionization electrospray] | [26] | |
| HPLC | DA; NE; E | PBA affinity column | NR | Nucleosil C18 | 0.1 M phosphate buffer/1 mM EDTA/300 mg L−1 SOS/methanol (10:1, v/v) pH 2.5 | Isocratic | Amperometric | [53] | |
| HPLC | DA; NE; E | SPE | NR | Lichrosorb LC-8 C8 | 50 mM potassium dihydrogen phosphate/500 mg L−1 SDS/250 mg L−1 EDTA/100 mL L−1 methanol/ 200 mL L−1 acetonitrile pH 3.5; orthophosphoric acid | Isocratic | CL; [luminol–I2] | [60] | |
| HPLC | DA; NE; E | SPE | DHBA | RECIPE reversed-phase | 50 mM potassium dihydrogen phosphate/2.5 mM OSA/0.1 g L−1 EDTA/acetonitrile (96.5:3.5, v/v) pH 3.5; phosphoric acid | Isocratic | Amperometric | [25] | |
| HPLC | DA; NE; E | SPE | DHBA | Hypersil-BDS | 50 mM acetate buffer/0.11 mM EDTA/1.1 mM OSA/methanol (85:15, v/v) pH 4.7; 8.5 M acetic acid | Isocratic | FL [TbCl3]; ex: 300 nm; em: 545 nm | [54] | |
| HPLC | DA; NE; E | LLE | d4-DA; d3-NE; d3-E | Allure Basix | 6.5 mM aqueous formic acid/tetrahydrofuran (2:3, v/v) | Isocratic | MS/MS; [positive ionization electrospray] | [55] | |
| HPLC | DA; NE; E | SPE | DHBA | Luna C18 | 50 mM dihydrogen phosphate buffer/500 mg L−1 SDS/250 mg L−1 EDTA/100 mL L−1 methanol/200 mL L−1 acetonitrile pH 2.9; 6 M orthophosphoric acid | Isocratic | Coulometric | [30] | |
| HPLC | DA; NE; E; NMN; MN | LLE | DHBA | Spherisorb C8 | A: Acetonitrile; B: 3.0 g L−1 acetic acid solution | Gradient | FL; [FMOC-Cl]; ex: 263 nm; em: 313 nm | [74] | |
| HPLC | DA; NE; E; NMN; MN | SPE | DHBA | Nova-Pak C18 | 50 mM ammonium formate; pH 3.0; formic acid | Isocratic | APcI-MS | [56] | |
| HPLC | DA; NE; E; NMN; MN | Cation exchange resin | DHBA | Nova-Pak C18 | 200 mM NaH2PO4·H2O/0.2 g L−1 EDTA/4 mM sodium 1-heptanesulfonate/acetonitrile (97.8:2.2, v/v) pH 3.0; 1 M orthophosphoric acid | Isocratic | Amperometric | [57] | |
| HPLC | NE; E; 5-HIs | Dilution/filtration | 5-HIA | Cosmosil 5C18 | 10 mM acetate buffer/acetonitrile (65:35, v/v) pH 6.0 | Isocratic | FL; [benzylamine]; ex: 345 nm; em: 480 nm | [80] | |
| Amniotic fluid | HPLC | 5-HIAA; HVA | NA | NA | Hypersil 3MOS | 20 mM phosphate buffer; pH 3.0; 0.5 mM heptasulfonic acid; 0.12 mM EDTA; 0.28% perchloric acid; 15% methanol | Isocratic | Amperometric | [58] |
| Deficiency | HVA | 5-HIAA | 3-OMD |
|---|---|---|---|
| Tyrosine hydroxylase | low | normal | normal |
| AADC | low | low | high |
| DBH | high | normal | normal |
| Pterin deficiency (recessive) | low | low | normal |
| Pterin deficiency (dominant) | normal to low | normal to low | normal |
| DAT | high | normal | NR |
| VMAT2 | normal | normal | NR |
| Matrix | Volume (mL) | Sample Preparation | Solvent | Analytes | Ref. |
|---|---|---|---|---|---|
| Human CSF | 1 mL | Precipitation; oxidation | 33 mg TCA/1 mg DTT per mL CSF; 0.1 mL HCl (0.1 M)/0.2% iodione/0.4% potassium iodide; 1% ascorbic acid; 1 M HCl/1 mg MnO2 per 200 µL CSF | N; B | [38] |
| 30 µL | Stabilization; oxidation | DTT; DETAPAC | BH4; BH2; N; S | [108] | |
| 100 µL | Filtration | NA | BH4; BH2; DHN; B; N | [118] | |
| 3rd tube (800th–1200th µL) | Dilution; filtration | NA | BH4; BH2; N | [39] | |
| 50 µL | Dilution; filtration | 250 nM 2,5-dihdroxybenzoic acid; 5000 MWCO PES Vivaspin 500 filter | BH4; BH2; B; N; DHN | [40] | |
| Human plasma | 400 µL | Precipitation; oxidation | 1 M TCA; 0.5% iodine/1% potassium iodide/0.2 M TCA, 1% ascorbic acid (biopterin); 6; sodium hydroxide/0.5% iodine/1% potassium iodide/0.2 M TCA; 1% ascorbic acid/6 M sodium hydroxide (BH4) | B; BH4 | [107] |
| 100 µL | Protein precipitation; derivatization; liquid phase extraction; drying; reconstitution | Ice-cold acetonitrile; 500 mM ammonium carbonate; benzoyl chloride; ethyl acetate; hexane; acetonitrile | BH4 | [119] | |
| 4 mL | Precipitation; oxidation; purification | 2N TCA/0.5% iodione/1% potassium iodide in 0.2 N TCA; Dowex 50 column | B; N | [110] | |
| Human serum | 200 µL | Oxidation; deproteinization | 1 M HCl with 1 mg MnO2; Ultrafree (NMWL 10000) | B; N | [120] |
| 2 mL | Oxidation; ion exchange | I2 (5 g/L) in 0.2 M TCA or I2 (5 g/L) and KI (10 g/L) in 0.1 M NaOH; AGMP-50 (200–400 mesh (H+)) | B; N | [121] | |
| 3 mL | Filtration; oxidation; ion exchange; evaporation | 0.22 µM nylon mesh; 3 M TCA/2% iodione/4% potassium iodide; ISOLUTEENV; elution in acetonitrile/water (80/29, v/v); dissolved in mobile phase | PCA; X; N; M; ISO; P; 6-B; 7-B; 6-HMP | [122] | |
| Dried blood spots | 4 blood spots | Extraction; sonication; ultrafiltration | 250 µL 20 mM HCl; Ultrafree (NMWL 10000) | N; B; ISO; P | [123] |
| 2 blood spots | Extraction; sonication; ultrafiltration | 250 µL 20 mM HCl; Ultrafree Nanosep 10 Ω | B; N | [124] | |
| Erythrocytes | NA | Washing; lysis; deproteinization; oxidation | 154 mM NaCl; water; 1.84 M TCA; 1 M HCl with 1 mg MnO2 | B; N | [120] |
| Human urine | 500 µL | Oxidation; filtration | 6 M HCl/10 mg MnO2 | P; ISO; 6-B; 7-B; 6-N; 7-N | [125] |
| 100 µL | Oxidation; filtration | 4% potassium iodide/2% iodine solution (w/v) | 6-B; 6-HMP; N; P; ISO; X | [126] | |
| 400 µL | Oxidation; filtration | A: 2 M NaOH, iodide/iodine solution; B: 5 mM KMnO4 | B; N; P; PCA; 6,7 DMP; ISO; X; 6-HMP | [127] | |
| 100 µL | Filtration | A: Lugol’s solution (4% iodide/2% iodine solution (w/v)), B: MnO2; C: Potassium permanganate | P; X; 7,8-DX; ISO; 6-B; S; N; M; 6-CP; 6-HMP; 6,7-DMP; 6-MP; 6-HLU; 7-HLU; 6-FP; L | [128] | |
| 100 µL | Acidification of urine; oxidation | 0.5% iodine/1% iodide in alkaline and acidic solution | B; N; P; BH4; BH2 | [129] | |
| 360 µL | Stabilization; filtration | 1% ascorbic acid; Nanosep 10Ω | S | [103] | |
| 500 µL | Dilution | Citrate buffer 10 mM; pH 5.5 | N; B; P; ISO | [113] | |
| 1 mL | Acidification; oxidation; extraction | 6N HCl; iodide/iodine solution (in 0.1 N NaOH or 0.1 N HCl); Dowex 50W X8, elution with 0.5 M NH4OH; Dowex 1 X8, elution in 1 N acetic acid | X; N; B; BH4 | [130] | |
| 500 µL | Acidification; oxidation | 6 M hydrochloric acid; MnO2 (10 mg); | B; N; M | [131] | |
| 1 µL (injection volume) | Dilution; SPE | 20× in 1% DTT | BH4; BH2; N; DHN | [132] | |
| Amniotic fluid | 200 µL | Acidic oxidation; deproteinization | MnO2; 30% TCA | N; M; ISO; B; PR; P | [58] |
| Oxidation; clean-up with ion exchange resin | Iodide/iodine at pH 1.0 | B; N | [133] | ||
| 200 µL | Oxidation; precipitation | 1 M hydrochloric acid; 2 m MnO2; 30% TCA | B; N | [134] | |
| Cell lysates | 80 µL | Lysis and sonication | 0.2 M TCA, 50 mM ascorbic acid; 1 mM EDTA; 6.5 mM DTE | BH4; BH2; B | [117] |
| 150 µL | Lysis; oxidation; deproteinization | 50 mM Tris-HCl; pH 7.5; 1 mM DTT (lysis); acidic iodine (10 g/L) | B; N | [115] | |
| 350 µl per pellet | Lysis; sonication; protein isolation | 50 mM potassium phosphate buffer; pH 7.0; 0.2 mM PMSF; fast desalting column | N | [116] | |
| NA | Lysis; sonication; deproteinization; oxidation | Extraction buffer (20 mM Tris-HCl, pH 7.4, 0.1 mM EDTA, 1 mM DTT, 10% glycerol, 0.1% Tween 20); 30% TCA; MnO2 (10 mg) with 0.2 M H3PO4 | B; N | [114] |
| Matrix | Technique | Analytes | Sample Preparation | Internal Standard | Column | Mobile Phase | Elution | Detection | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| Human CSF | HPLC | B; N | Dilution | NA | UltraPure Torsic Acid | 4.6 g/L NH4HPO4; pH 3.5 | Isocratic | FL; ex: 350 nm; em: 450 nm | [38] |
| HPLC | N; BH4; BH2; S | Dilution | 15N-BH4; 15N-BH2; 15N-N | AAA-MS column | A: 0.1% formic acid/0.1% heptafluorobutyric acid in water; B: 0.1% formic acid in methanol | Gradient | MS/MS; [positive ionization electrospray] | [108] | |
| HPLC | BH4; BH2; DHN; B; N | Dilution | LU | Atlantis dC18 | 0.005 M sodium citrate/methanol (97:3; v/v) | Isocratic | Post-column oxidation; FL: ex: 350 nm; em: 450 nm | [118] | |
| HPLC | BH4; BH2; N | Dilution, filtration | BH4; BH2; N | ODS (C18) | 50 mM sodium acetate; 5 mM citric acid; 48 µM EDTA; 160 µM DTE | Isocratic | FL; ex: 360 nm; em: 440 nm (oxidized pterins); coulometric (reduced pterins) | [39] | |
| UHPLC | BH4; BH2; B; N; DHN | Direct injection | BH2; B; N; DHN | ACQUITY UPLC HSS T3 | 0.05 M citrate buffer; pH 5.2 (pH 7.4 for BH4); methanol (97:3, v/v) | Isocratic | Coulometric, FL ex: 350 nm; em: 450 nm | [40] | |
| Human plasma | HPLC | B; BH4 | Dilution | NA | Hypersil C18 | A: 15 mM potassium phosphate buffer; pH 6.45; B: Methanol | Gradient | FL; ex: 360 nm; em: 440 nm | [107] |
| HPLC | BH4 | Derivatization | BH4-benzoyl chloride-d5 | HILIC polar imidazole column | A: Acetonitrile/water (15%/85%; v/v) with 0.2% formic acid; B: Acetonitrile with 0.2% formic acid | Gradient | MS/MS; [positive ionization electrospray] | [119] | |
| HPLC | B; N | Dilution | N | Whatman 10 µm ODS; Partisil 10; µBondapak C18 | 5% or 20% methanol in water | Isocratic | FL; ex: 350 nm; em: 410 nm | [110] | |
| Human serum | HPLC | B; N | Cation exchange resin | BH4; BH2 | Excalibur ODS | Methanol/water (15/85, v/v) | Isocratic | FL; ex: 370 nm; em: 418–700 nm | [121] |
| HPLC | PCA; X; N; M; ISO; P; 6-B; 7-B; 6-HMP | Cation exchange | PCA; X; N; M; ISO; P; 6-B; 7-B; 6-HMP | Zorbax-Eclipse XDB C18 and Poroshell 120 | 2 mM ammonium formiate; pH 7.1 | Isocratic | FL; ex: 272 nm; em: 410 nm, 445 nm, 465 nm | [122] | |
| Dried blood spots | HPLC | B; N; ISO; P | None | B; N; P | Pre column C8 Spherisorb; ODS-1 Spherisorb | 1.5mM potassium hydrogen phosphate buffer; pH 4.6/8% methanol (v/v) | Isocratic | FL; ex: 350 nm; em: 450 nm | [123] |
| UPLC | B; N | Dilution | 13C5-N; D3-B | ACQUITY UPLC HSS T3 | A: 0.2% formic acid in water; B: 0.2% formic acid in methanol | Gradient | MS/MS; [positive ionization electrospray] | [124] | |
| Human urine | HPLC | P; ISO; 6-B; 7-B; 6-N; 7-N | Dilution | BH4; P; ISO; 6,7-DMP | ZORBAX C18; LUNA amino; HILIC; AQUA C18 | Methanol/0.1% formic acid; acetonitrile/0.1% formic acid; water/0.1% formic acid/10 mM ammonium formiate | Isocratic | MS/MS; [negative ionization electrospray] | [125] |
| HPLC | 6-B; 6-HMP; N; P; ISO; X | Dilution | 6-B; 6-HMP; N; P; ISO; X | Phenyl-hexyl column | A: 0.1% formic acid in H2O; B: 0.2% formic acid in acetonitrile | Gradient | MS/MS; [positive ionization electrospray] | [126] | |
| HPLC | B; N; P; PCA; 6,7 DMP; ISO; X; 6-HMP | Dilution | B; N; P; PCA; 6,7 DMP; ISO; X; 6-HMP | LiChrospher C8 60 RP; Aquasil C18; HILIC Luna | A: 10 mM phosphate buffer, pH 7.0; B: Methanol (LiCrospher); A: 10 mM Tris-HCl, pH 6.8; B: Methanol (Aquasil); A: 100 mM ammonium acetate buffer pH 5.8; B: Acetonitrile (HILIC) | Gradient (LiCrospher; HILIC); isocratic (Aquasil) | FL; ex: 280 nm; em: 444 nm; UV: 215 nm and 254 nm | [127] | |
| HPLC | P; X; 7,8-DX; ISO; 6-B; S; N; M; 6-CP; 6-HMP; 6,7-DMP; 6-MP, L; 6-HLU; 7-HLU; 6-FP; L | Dilution | P; X; 7,8-DX; ISO; 6-B; S; N; M; 6-CP; 6-HMP; 6,7-DMP; 6-MP; L; 6-HLU; 7-HLU; 6-FP; L | Luna | A: 0.025% (v/v) formic acid in 99% water/1% acetonitrile; B: Methanol | Gradient | MS/MS; [positive ionization electrospray] | [128] | |
| HPLC | B, N, P, BH4, BH2 | 6-MP | RP 18 | Water/methanol (97/3, v/v) | Gradient | FL; ex: 350 nm; em: 410 nm | [129] | ||
| HPLC | N; B; P; ISO | Post-column photo derivatization | BH4; BH2 | Pre-column: XDB-C18; Zorbax Eclipse XDB-C18 | Citrate buffer; pH 5.5/acetonitrile (98/2, v/v) | Isocratic | FL; ex: 272 nm; em: 445 nm; photometric: 256 nm | [113] | |
| HPLC | S | X | Spherisorb S5 ODS1250 | A: 24 mM K2HPO4, pH 5.0; B: Methanol/water (70/30; v/v) | Gradient | FL; ex: 425 nm; em: 530 nm | [103] | ||
| HPLC | X; N; B; BH4 | None | 6-MP | Partisil-10 SCX | 1 mM ammonium dihydrogen phosphate; pH 2.8/7% methanol/5% acetonitrile | Isocratic | FL; ex: 360 nm; em: 420 nm | [130] | |
| HPLC | B; N; M | Dilution | B; M; N; P | Spherisorb S5 ODS | A: Methanol/water (3:97, v/v); B: Isopropanol/methanol/acetic acid (49:49:2, v/v/v); C: Isopropanol-methanol-water (1:1:8, v/v/v); D: 6.6 mM Na2HPO4/13.3 mM citric acid/0.06 mM Na2EDTA/1.4 mM octanesulphonic acid/10% methanol, pH 3.3; E: 6.6 mM Na2HPO4/13.3 mM citric acid/0.06 mM Na2EDTA/1.4 mM octanesulphonic acid/10% isopropanol, pH 3.3; F: 20 mM KH2PO4/0.85 mM octanesulphonic acid/0.1 mM Na2EDTA/1% methanol, pH 3.0 | Gradient (A, B, C for oxidized pterins); Isocratic (E, F for reduced pterins) | FL; ex: 350 nm; em: 450 nm (oxidized pterins); amperometric (reduced pterins) | [131] | |
| UHPLC | BH4; BH2; N; DHN | SPE | BH4; BH2; N; DHN | BEH Amide column | 50 mM ammonium acetate; pH 6.8; acetonitrile (15:85, v/v) | Isocratic | FL; ex: 353 nm; em: 438 nm; UV detection (PDA detector) at 253 nm | [132] | |
| Amniotic fluid | HPLC | B; N | Ion exchange resin | 6-MP | Partisil-10 SCX | 1 mM ammonium dihydrogen phosphate; pH 2.8/7% methanol/5% acetonitrile | Isocratic | FL; ex: 360 nm; em: 420 nm | [133] |
| Cell lysates | HPLC | BH4; BH2; B | None | 15N-BH4; 15N-BH2; 15N-B | Poroshell 120 SB-C18 column | A: 150 mM acetic acid, 12 mM HFBA; B: 12 mM HFBA, methanol | Gradient | MS/MS; [positive ionization electrospray] | [117] |
| HPLC | B; N | Deproteinization | B | Spherisorb C18 | ND | Gradient | FL; ex: 350 nm; em: 450 nm | [115] | |
| HPLC | N | GTPCH activity assay | N | Inartsil ODS-3 | 10 mM sodium phosphate; pH 7.0/1 mM EDTA | Isocratic | FL; ex: 365 nm; em: 475 nm | [116] | |
| HPLC | B; N | NA | NA | Econosphere C18 | 0.1 M sodium phosphate; pH 3.0/5% methanol | Isocratic | FL; ex: 350 nm; em: 450 nm | [114] |
| Matrix | Analytes | arGTPCHD | adGTPCHD | PTPSD | DHPRD | SPRD | PCDD |
|---|---|---|---|---|---|---|---|
| Human urine | biopterin | low | low to normal | low | normal (to high) | normal | low to normal |
| neopterin | low | low to normal | high | normal (to high) | normal | normal to high | |
| xanthopterin | NR | NR | NR | (high) | NR | NR | |
| primapterin | NR | NR | NR | normal | normal | high | |
| sepiapterin | NR | NR | NR | NR | high | NR | |
| Human CSF | biopterin | low | low | low | normal (to high) | normal (to high) | normal |
| BH4 | low | low to normal | low | low to normal | low | NR | |
| neopterin | low | low | high | normal | normal | normal | |
| BH2 | (low) | NR | NR | normal (to high) | high | NR | |
| primapterin | (normal) | NR | NR | NR | NR | NR | |
| sepiapterin | (normal) | NR | NR | NR | high | NR | |
| Dried blood spot | biopterin | low | low to normal | low | normal to high | normal | NR |
| neopterin | low | low to normal | high | normal to high | normal | NR | |
| primapterin | NR | NR | NR | normal | normal | NR |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jung-Klawitter, S.; Kuseyri Hübschmann, O. Analysis of Catecholamines and Pterins in Inborn Errors of Monoamine Neurotransmitter Metabolism—From Past to Future. Cells 2019, 8, 867. https://doi.org/10.3390/cells8080867
Jung-Klawitter S, Kuseyri Hübschmann O. Analysis of Catecholamines and Pterins in Inborn Errors of Monoamine Neurotransmitter Metabolism—From Past to Future. Cells. 2019; 8(8):867. https://doi.org/10.3390/cells8080867
Chicago/Turabian StyleJung-Klawitter, Sabine, and Oya Kuseyri Hübschmann. 2019. "Analysis of Catecholamines and Pterins in Inborn Errors of Monoamine Neurotransmitter Metabolism—From Past to Future" Cells 8, no. 8: 867. https://doi.org/10.3390/cells8080867
APA StyleJung-Klawitter, S., & Kuseyri Hübschmann, O. (2019). Analysis of Catecholamines and Pterins in Inborn Errors of Monoamine Neurotransmitter Metabolism—From Past to Future. Cells, 8(8), 867. https://doi.org/10.3390/cells8080867