Melanoma-Derived Extracellular Vesicles Bear the Potential for the Induction of Antigen-Specific Tolerance

 , , , and
, , , and 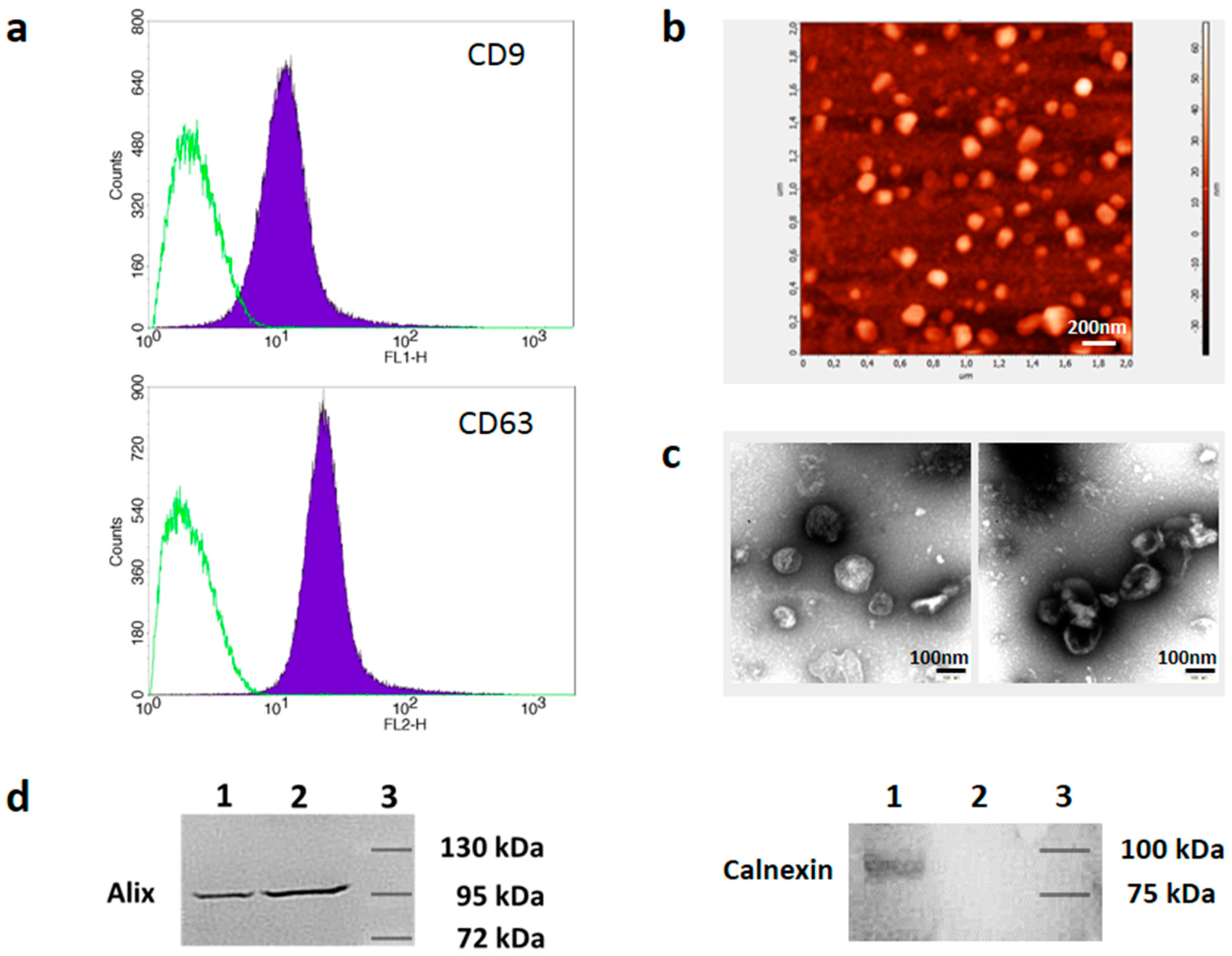{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.1.1. Antigen-Presenting Cells (APCs)
2.1.2. Melanoma Cell Culture for sEV Production
2.2. Isolation and Characterization of Extracellular Vesicles
2.3. Flow Cytometry
2.4. Intercellular Transfer of Peptide-MHC-I Complexes
2.5. RNA Isolation and Real-Time Quantitative Reverse Transcription PCR (qRT-PCR)
2.6. Western Blot Analysis
2.7. Allogeneic Mixed Lymphocyte Reaction
2.8. Statistical Analysis
3. Results
3.1. Isolation and Characterization of Melanoma Cell-Derived sEVs
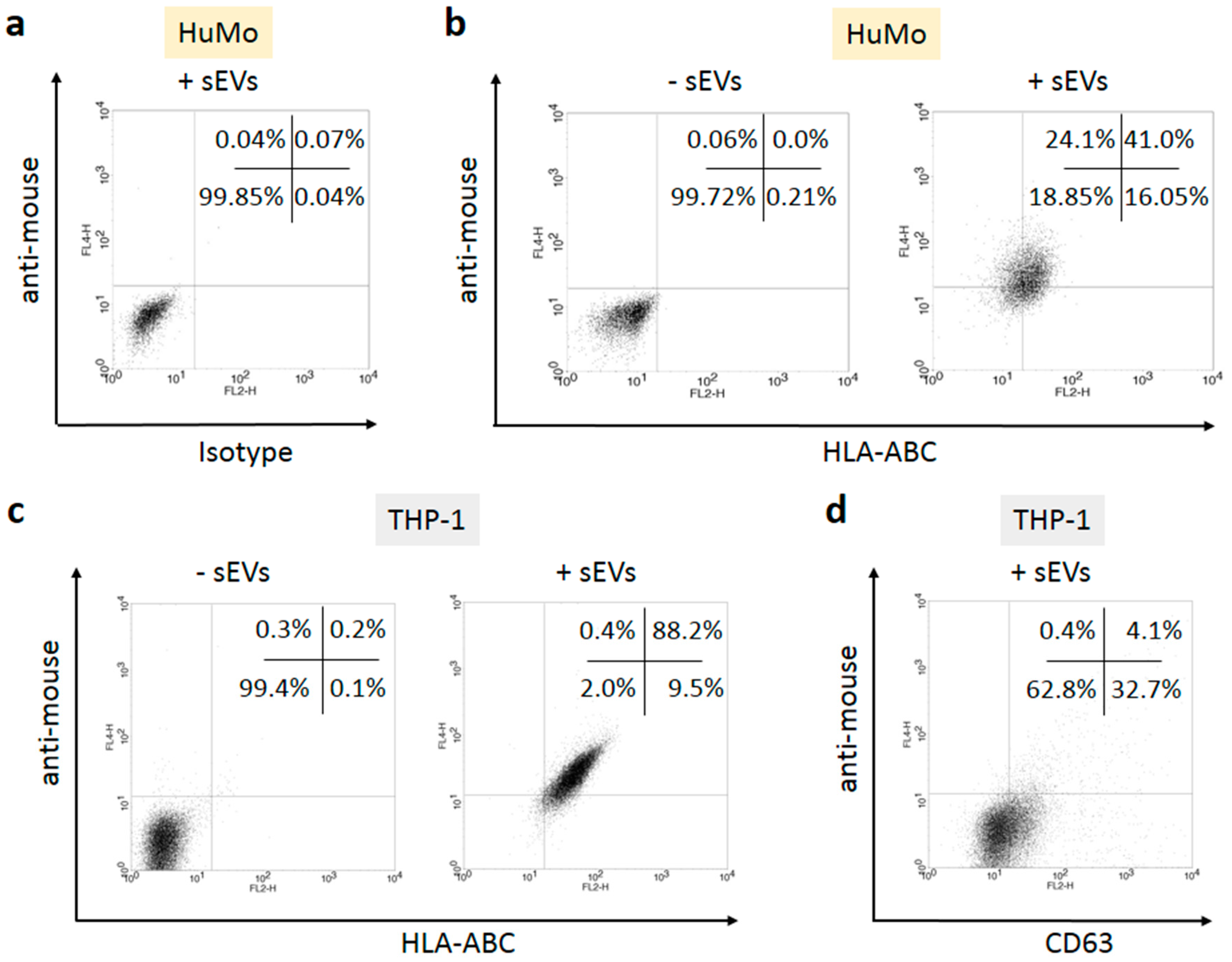
3.2. sEVs Transport MHC Class I Molecules from Melanoma Cells to APCs
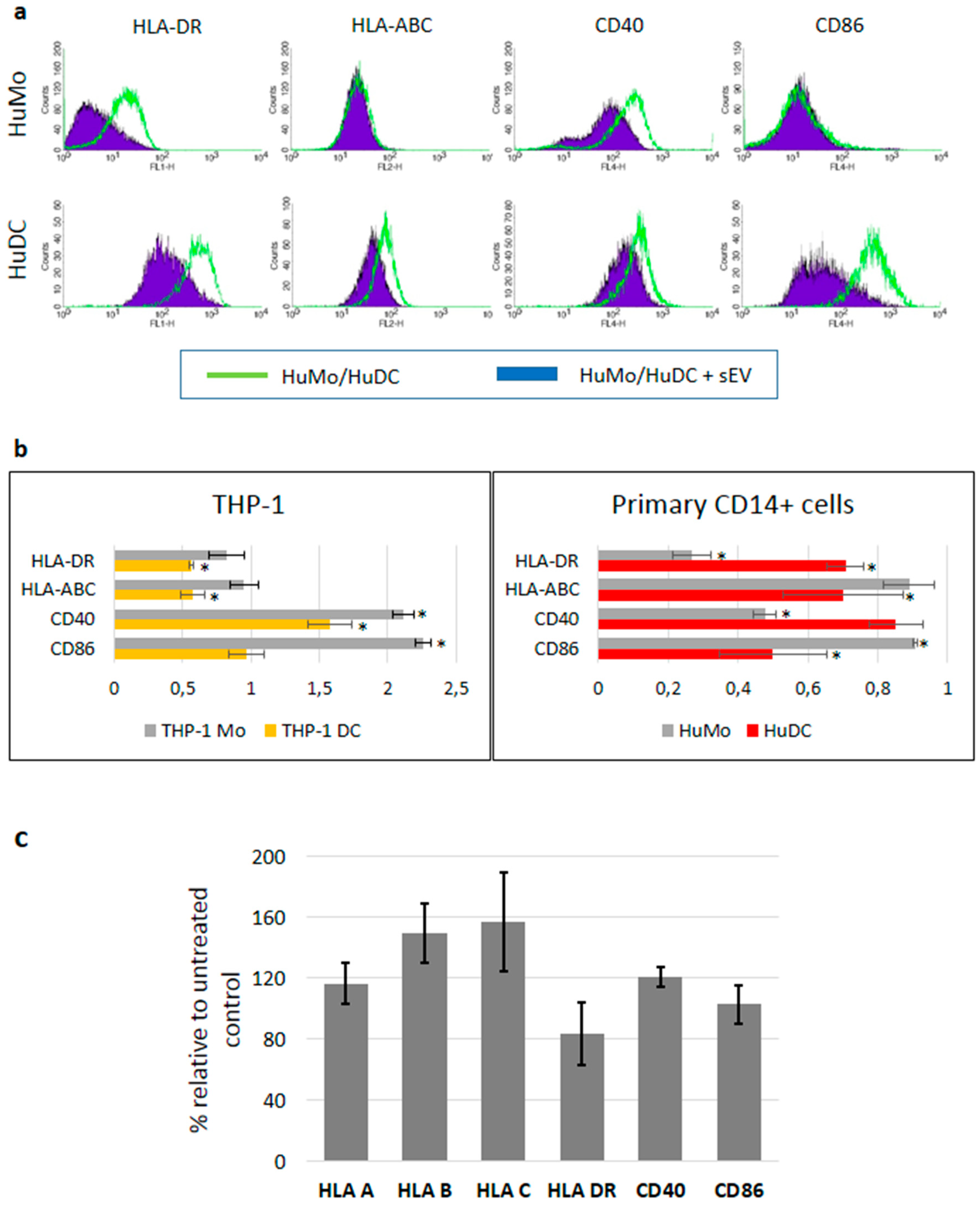
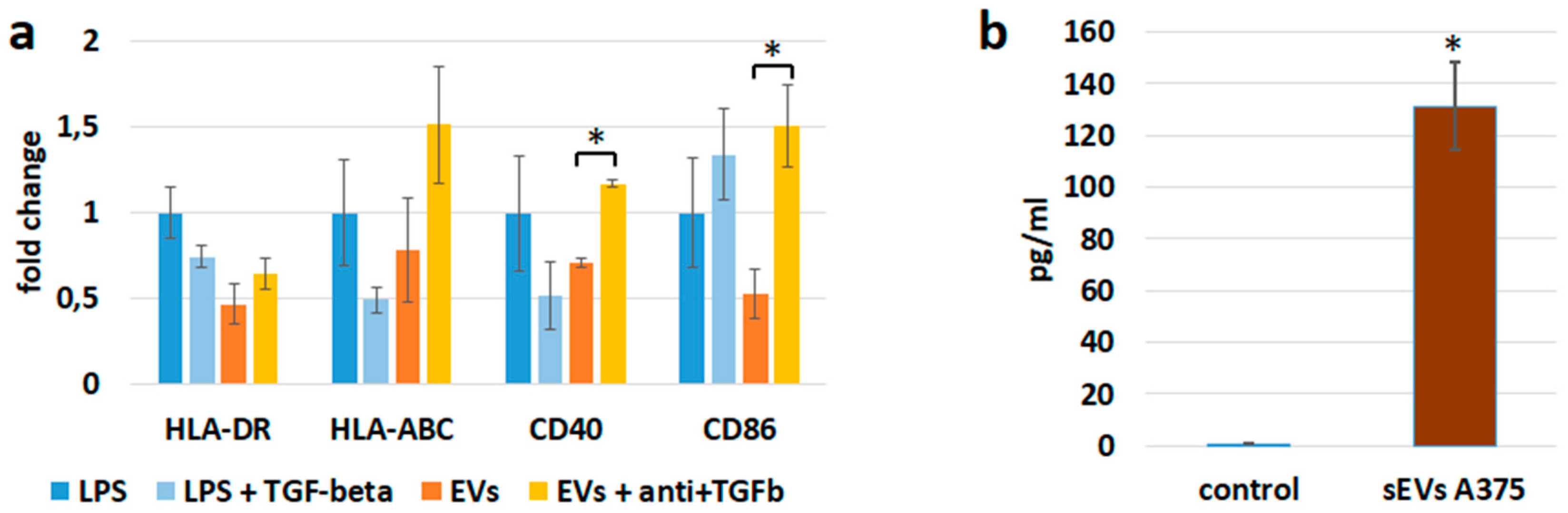
3.3. sEV Mediated Modulation of Immune Receptor Surface Expression on APCs
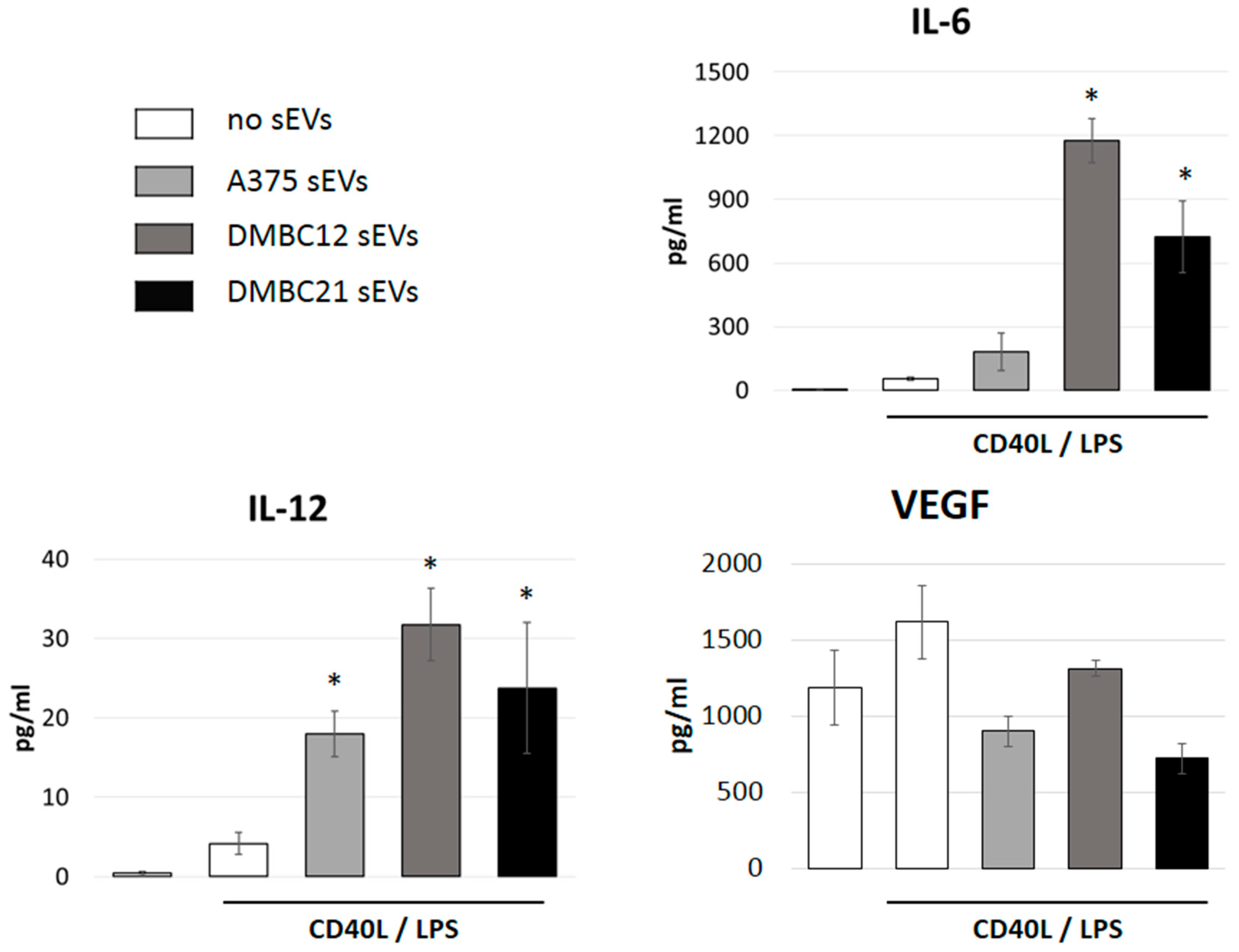
3.4. sEVs Affect the Cytokine Expression of APCs
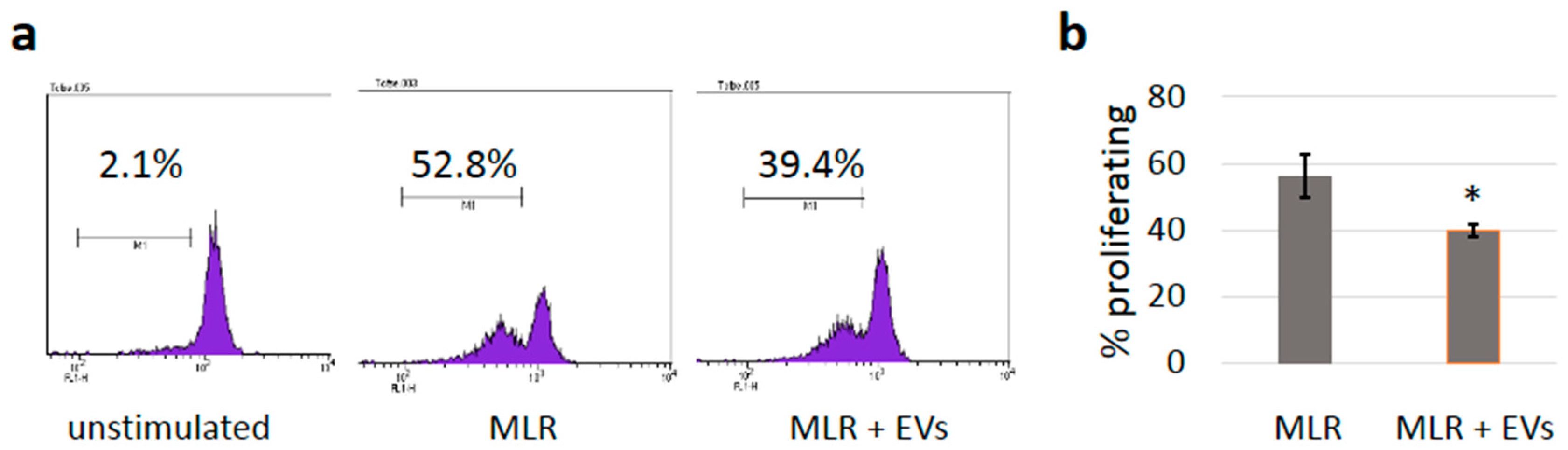
3.5. sEVs Reduce T Cell Proliferation in Allogeneic Mixed Lymphocyte Reactions
3.6. sEVs Transfer TGF-β with Immunosuppressive Activity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jazirehi, A.R.; Lim, A.; Dinh, T. PD-1 inhibition and treatment of advanced melanoma-role of pembrolizumab. Am. J. Cancer Res. 2016, 6, 2117–2128. [Google Scholar] [PubMed]
- Weiss, S.A.; Wolchok, J.D.; Sznol, M. Immunotherapy of Melanoma: Facts and Hopes. Clin. Cancer Res. 2019. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Escors, D. Tumour immunogenicity, antigen presentation and immunological barriers in cancer immunotherapy. New J. Sci. 2014, 2014, 734515. [Google Scholar] [CrossRef] [PubMed]
- Melief, C.J. Regulation of cytotoxic T lymphocyte responses by dendritic cells: peaceful coexistence of cross-priming and direct priming? Eur. J. Immunol. 2003, 33, 2645–2654. [Google Scholar] [CrossRef] [PubMed]
- Nel, A.E. T-cell activation through the antigen receptor. Part 1: signaling components, signaling pathways, and signal integration at the T-cell antigen receptor synapse. J. Allergy Clin. Immunol. 2002, 109, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Mescher, M.F.; Curtsinger, J.M.; Agarwal, P.; Casey, K.A.; Gerner, M.; Hammerbeck, C.D.; Popescu, F.; Xiao, Z. Signals required for programming effector and memory development by CD8+ T cells. Immunol. Rev. 2006, 211, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Manzotti, C.N.; Liu, M.; Burke, F.; Mead, K.I.; Sansom, D.M. CD86 and CD80 differentially modulate the suppressive function of human regulatory T cells. J. Immunol. 2004, 172, 2778–2784. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Hasni, M.S.; Jondal, M.; Yakimchuk, K. Modification of anti-tumor immunity by tolerogenic dendritic cells. Autoimmunity 2017, 50, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Kuwana, M. Induction of anergic and regulatory T cells by plasmacytoid dendritic cells and other dendritic cell subsets. Hum. Immunol. 2002, 63, 1156–1163. [Google Scholar] [CrossRef]
- Wang, L.; Pino-Lagos, K.; de Vries, V.C.; Guleria, I.; Sayegh, M.H.; Noelle, R.J. Programmed death 1 ligand signaling regulates the generation of adaptive Foxp3+CD4+ regulatory T cells. Proc. Natl. Acad. Sci. USA 2008, 105, 9331–9336. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.S.; Sansom, D.M. The emerging role of CTLA4 as a cell-extrinsic regulator of T cell responses. Nat. Rev. Immunol. 2011, 11, 852–863. [Google Scholar] [CrossRef] [PubMed]
- Fritz, J.M.; Lenardo, M.J. Development of immune checkpoint therapy for cancer. J. Exp. Med. 2019, 216, 1244–1254. [Google Scholar] [CrossRef] [PubMed]
- Rabinovich, G.A.; Gabrilovich, D.; Sotomayor, E.M. Immunosuppressive strategies that are mediated by tumor cells. Annu. Rev. Immunol. 2007, 25, 267–296. [Google Scholar] [CrossRef] [PubMed]
- Mittal, S.K.; Roche, P.A. Suppression of antigen presentation by IL-10. Curr. Opin. Immunol. 2015, 34, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Sheng, J.; Chen, W.; Zhu, H.J. The immune suppressive function of transforming growth factor-β (TGF-β) in human diseases. Growth Factors 2015, 33, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Voron, T.; Marcheteau, E.; Pernot, S.; Colussi, O.; Tartour, E.; Taieb, J.; Terme, M. Control of the immune response by pro-angiogenic factors. Front. Oncol. 2014, 4, 70. [Google Scholar] [CrossRef]
- Zhou, G.; Drake, C.G.; Levitsky, H.I. Amplification of tumor-specific regulatory T cells following therapeutic cancer vaccines. Blood 2006, 107, 628–636. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, Y.; Wang, Q.; Liu, Y.; Bao, W.; Wu, S. Exosomes in cancer: Small transporters with big functions. Cancer Lett. 2018, 435, 55–65. [Google Scholar] [CrossRef]
- Kaiser, J. Malignant messengers. Science 2016, 352, 164–166. [Google Scholar] [CrossRef]
- Gurunathan, S.; Kang, M.H.; Jeyaraj, M.; Qasim, M.; Kim, J.H. Review of the Isolation, Characterization, Biological Function, and Multifarious Therapeutic Approaches of Exosomes. Cells 2019, 8, E307. [Google Scholar] [CrossRef] [PubMed]
- Czernek, L.; Düchler, M. Functions of Cancer-Derived Extracellular Vesicles in Immunosuppression. Arch. Immunol. Ther. Exp. (Warsz). 2017, 65, 311–323. [Google Scholar] [CrossRef] [PubMed]
- McConnell, M.J. Extracellular vesicles and immune modulation. Immunol. Cell Biol. 2018, 96, 681–682. [Google Scholar] [CrossRef] [PubMed]
- Anel, A.; Gallego-Lleyda, A.; Miguel, D.; Naval, J.; Martínez-Lostao, L. Role of Exosomes in the Regulation of T-Cell Mediated Immune Responses and in Autoimmune Disease. Cells 2019, 8, 154. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. Immune modulation of T-cell and NK (natural killer) cell activities by TEXs (tumour-derived exosomes). Biochem. Soc. Trans. 2013, 41, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Norbury, C.C. Defining cross-presentation for a wider audience. Curr. Opin. Immunol. 2016, 40, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Sztiller-Sikorska, M.; Hartman, M.L.; Talar, B.; Jakubowska, J.; Zalesna, I.; Czyz, M. Phenotypic diversity of patient-derived melanoma populations in stem cell medium. Lab. Investig. 2015, 95, 672–683. [Google Scholar] [CrossRef]
- Czernek, L.; Chworos, A.; Düchler, M. The Uptake of Extracellular Vesicles is Affected by the Differentiation Status of Myeloid Cells. Scand. J. Immunol. 2015, 82, 506–514. [Google Scholar] [CrossRef]
- Parisse, P.; Rago, I.; Ulloa Severino, L.; Perissinotto, F.; Ambrosetti, E.; Paoletti, P.; Ricci, M.; Beltrami, A.P.; Cesselli, D.; Casalis, L. Atomic force microscopy analysis of extracellular vesicles. Eur. Biophys J. 2017, 46, 813–820. [Google Scholar] [CrossRef]
- Manek, R.; Moghieb, A.; Yang, Z.; Kumar, D.; Kobessiy, F.; Sarkis, G.A.; Raghavan, V.; Wang, K.K.W. Protein Biomarkers and Neuroproteomics Characterization of Microvesicles/Exosomes from Human Cerebrospinal Fluid Following Traumatic Brain Injury. Mol. Neurobiol. 2018, 55, 6112–6128. [Google Scholar] [CrossRef]
- Wieckowski, E.U.; Visus, C.; Szajnik, M.; Szczepanski, M.J.; Storkus, W.J.; Whiteside, T.L. Tumor-derived microvesicles promote regulatory T cell expansion and induce apoptosis in tumor-reactive activated CD8+ T lymphocytes. J. Immunol. 2009, 183, 3720–3730. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Yang, S.C.; Zhu, L.; Reckamp, K.; Gardner, B.; Baratelli, F.; Huang, M.; Batra, R.K.; Dubinett, S.M. Tumor cyclooxygenase-2/prostaglandin E2-dependent promotion of FOXP3 expression and CD4+ CD25+ T regulatory cell activities in lung cancer. Cancer Res. 2005, 65, 5211–5220. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Jin, W.; Hardegen, N.; Lei, K.J.; Li, L.; Marinos, N.; McGrady, G.; Wahl, S.M. Conversion of peripheral CD4+ CD25− naive T cells to CD4+ CD25+ regulatory T cells by TGF-β induction of transcription factor Foxp3. J. Exp. Med. 2003, 198, 1875–1886. [Google Scholar] [CrossRef] [PubMed]
- Ghiringhelli, F.; Puig, P.E.; Roux, S.; Parcellier, A.; Schmitt, E.; Solary, E.; Kroemer, G.; Martin, F.; Chauffert, B.; Zitvogel, L. Tumor cells convert immature myeloid dendritic cells into TGF-β secreting cells inducing CD4+CD25+ regulatory T cell proliferation. J. Exp. Med. 2005, 202, 919–929. [Google Scholar] [CrossRef] [PubMed]
- Xiang, X.; Poliakov, A.; Liu, C.; Liu, Y.; Deng, Z.B.; Wang, J.; Cheng, Z.; Shah, S.V.; Wang, G.J.; Zhang, L.; et al. Induction of myeloid-derived suppressor cells by tumor exosomes. Int. J. Cancer 2009, 124, 2621–2633. [Google Scholar] [CrossRef]
- Montecalvo, A.; Larregina, A.T.; Shufesky, W.J.; Stolz, D.B.; Sullivan, M.L.G.; Karlsson, J.M.; Baty, C.J.; Gibson, G.A.; Erdos, G.; Wang, Z.; et al. Mechanism of transfer of functional microRNAs between mouse dendritic cells via exosomes. Blood 2012, 119, 756–766. [Google Scholar] [CrossRef] [PubMed]
- Chalmin, F.; Ladoire, S.; Mignot, G.; Vincent, J.; Bruchard, M.; Remy-Martin, J.P.; Boireau, W.; Rouleau, A.; Simon, B.; Lanneau, D.; et al. Membrane-associated Hsp72 from tumor-derived exosomes mediates STAT3-dependent immunosuppressive function of mouse and human myeloid-derived suppressor cells. J. Clin. Investig. 2010, 120, 457–471. [Google Scholar] [CrossRef]
- Fernández-Messina, L.; Gutiérrez-Vázquez, C.; Rivas-García, E.; Sánchez-Madrid, F.; de la Fuente, H. Immunomodulatory role of microRNAs transferred by extracellular vesicles. Biol. Cell. 2015, 107, 61–77. [Google Scholar] [CrossRef]
- Greening, D.W.; Gopal, S.K.; Xu, R.; Simpson, R.J.; Chen, W. Exosomes and their roles in immune regulation and cancer. Semin. Cell Dev. Biol. 2015, 40, 72–81. [Google Scholar] [CrossRef]
- Yang, C.; Ruffner, M.A.; Kim, S.H.; Robbins, P.D. Plasma-derived MHC class II+ exosomes from tumor-bearing mice suppress tumor antigen-specific immune responses. Eur. J. Immunol. 2012, 42, 1778–1784. [Google Scholar] [CrossRef]
- Kim, S.H.; Bianco, N.R.; Shufesky, W.J.; Morelli, A.E.; Robbins, P.D. MHC class II+ exosomes in plasma suppress inflammation in an antigen-specific and Fas ligand/Fas-dependent manner. J. Immunol. 2007, 179, 2235–2241. [Google Scholar] [CrossRef] [PubMed]
- Bretz, N.P.; Ridinger, J.; Rupp, A.K.; Rimbach, K.; Keller, S.; Rupp, C.; Marmé, F.; Umansky, L.; Umansky, V.; Eigenbrod, T.; et al. Body fluid exosomes promote secretion of inflammatory cytokines in monocytic cells via Toll-like receptor signaling. J. Biol. Chem. 2013, 288, 36691–36702. [Google Scholar] [CrossRef] [PubMed]
- Gärtner, K.; Battke, C.; Dünzkofer, J.; Hüls, C.; von Neubeck, B.; Kellner, M.K.; Fiestas, E.; Fackler, S.; Lang, S.; Zeidler, R. Tumor-derived extracellular vesicles activate primary monocytes. Cancer Med. 2018, 7, 2013–2020. [Google Scholar] [CrossRef] [PubMed]
- Webber, J.; Steadman, R.; Mason, M.D.; Tabi, Z.; Clayton, A. Cancer exosomes trigger fibroblast to myofibroblast differentiation. Cancer Res. 2010, 70, 9621–9630. [Google Scholar] [CrossRef] [PubMed]
- Yen, E.Y.; Miaw, S.C.; Yu, J.S.; Lai, I.R. Exosomal TGF-β1 is correlated with lymphatic metastasis of gastric cancers. Am. J. Cancer Res. 2017, 7, 2199–2208. [Google Scholar] [PubMed]
- Huang, F.; Wan, J.; Hao, S.; Deng, X.; Chen, L.; Ma, L. TGF-β1-silenced leukemia cell-derived exosomes target dendritic cells to induce potent anti-leukemic immunity in a mouse model. Cancer Immunol. Immunother. 2017, 66, 1321–1331. [Google Scholar] [CrossRef] [PubMed]
- Ning, Y.; Shen, K.; Wu, Q.; Sun, X.; Bai, Y.; Xie, Y.; Pan, J.; Qi, C. Tumor exosomes block dendritic cells maturation to decrease the T cell immune response. Immunol. Lett. 2018, 199, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Gajos-Michniewicz, A.; Düchler, M.; Czyz, M. MiRNA in melanoma-derived exosomes. Cancer Lett. 2014, 347, 29–37. [Google Scholar] [CrossRef]
- Wozniak, M.; Peczek, L.; Czernek, L.; Düchler, M. Analysis of the miRNA Profiles of Melanoma Exosomes Derived Under Normoxic and Hypoxic Culture Conditions. Anticancer Res. 2017, 37, 6779–6789. [Google Scholar] [PubMed]
- Kortylewski, M.; Jove, R.; Yu, H. Targeting STAT3 affects melanoma on multiple fronts. Cancer Metastasis Rev. 2005, 24, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, H.; Ohno, Y.; Toyoshima, Y.; Ohtake, J.; Homma, S.; Kawamura, H.; Takahashi, N.; Taketomi, A. Interleukin-6/STAT3 signaling as a promising target to improve the efficacy of cancer immunotherapy. Cancer Sci. 2017, 108, 1947–1952. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.R.; Kwon, B.E.; Hong, E.H.; Shim, A.; Song, J.H.; Kim, H.M.; Chang, S.Y.; Kim, Y.J.; Kweon, M.N.; Youn, J.I.; et al. Interleukin-10 attenuates tumour growth by inhibiting interleukin-6/signal transducer and activator of transcription 3 signalling in myeloid-derived suppressor cells. Cancer Lett. 2016, 381, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, H.; Nishikata, R.; Senju, S.; Nishimura, Y. Myeloid-derived suppressor cells attenuate TH1 development through IL-6 production to promote tumor progression. Cancer Immunol. Res. 2013, 1, 64–76. [Google Scholar] [CrossRef] [PubMed]
- Horikawa, N.; Abiko, K.; Matsumura, N.; Hamanishi, J.; Baba, T.; Yamaguchi, K.; Yoshioka, Y.; Koshiyama, M.; Konishi, I. Expression of Vascular Endothelial Growth Factor in Ovarian Cancer Inhibits Tumor Immunity through the Accumulation of Myeloid-Derived Suppressor Cells. Clin. Cancer Res. 2017, 23, 587–599. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Düchler, M.; Czernek, L.; Peczek, L.; Cypryk, W.; Sztiller-Sikorska, M.; Czyz, M. Melanoma-Derived Extracellular Vesicles Bear the Potential for the Induction of Antigen-Specific Tolerance. Cells 2019, 8, 665. https://doi.org/10.3390/cells8070665
Düchler M, Czernek L, Peczek L, Cypryk W, Sztiller-Sikorska M, Czyz M. Melanoma-Derived Extracellular Vesicles Bear the Potential for the Induction of Antigen-Specific Tolerance. Cells. 2019; 8(7):665. https://doi.org/10.3390/cells8070665
Chicago/Turabian StyleDüchler, Markus, Liliana Czernek, Lukasz Peczek, Wojciech Cypryk, Malgorzata Sztiller-Sikorska, and Malgorzata Czyz. 2019. "Melanoma-Derived Extracellular Vesicles Bear the Potential for the Induction of Antigen-Specific Tolerance" Cells 8, no. 7: 665. https://doi.org/10.3390/cells8070665
APA StyleDüchler, M., Czernek, L., Peczek, L., Cypryk, W., Sztiller-Sikorska, M., & Czyz, M. (2019). Melanoma-Derived Extracellular Vesicles Bear the Potential for the Induction of Antigen-Specific Tolerance. Cells, 8(7), 665. https://doi.org/10.3390/cells8070665