Genome-Wide Profiling of Laron Syndrome Patients Identifies Novel Cancer Protection Pathways

 ,
, 


Abstract
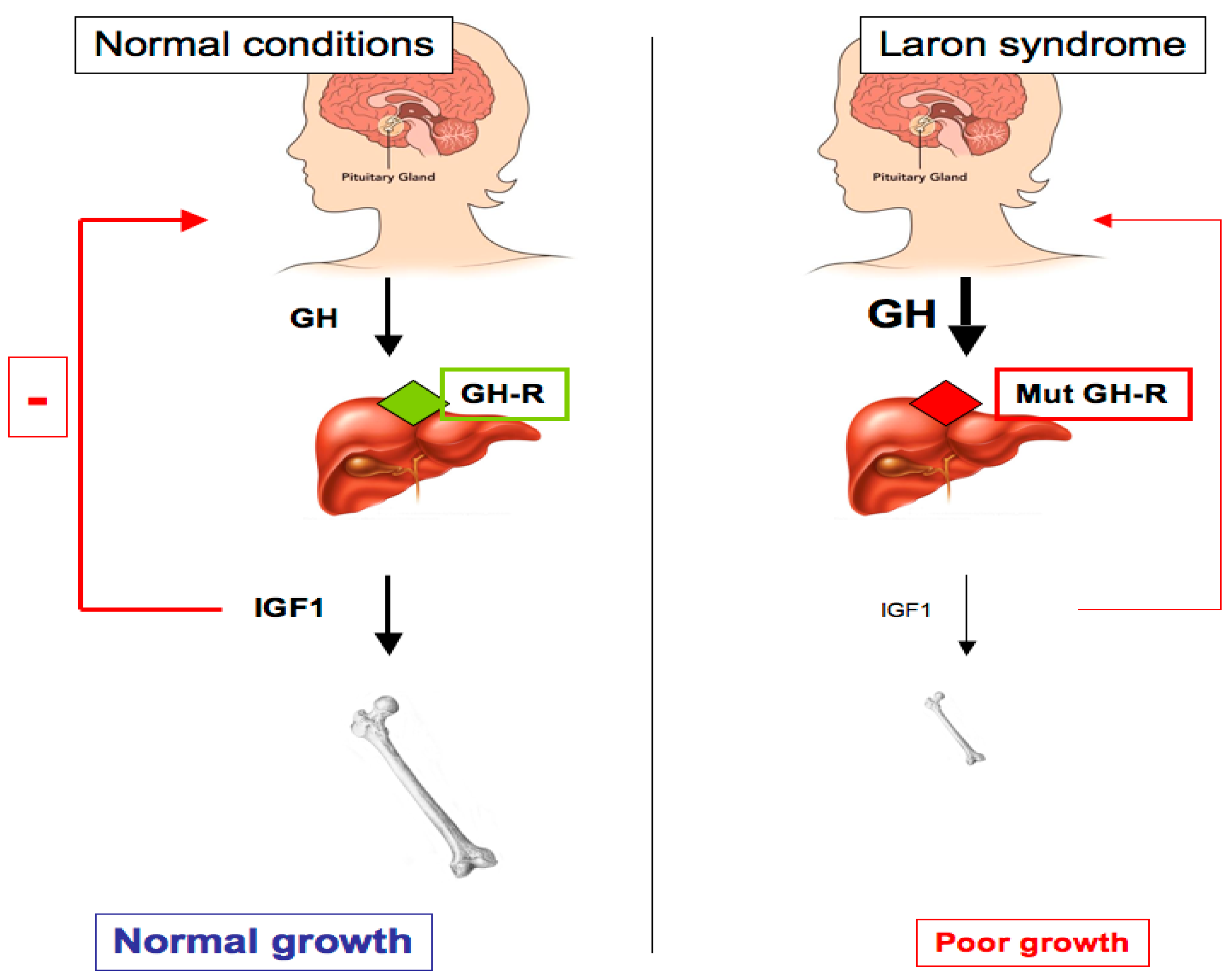
1. The Somatotropic Axis and Its Role in Growth Retardation
2. Laron Syndrome: A Classical Paradigm of Congenital IGF1 Deficiency
3. Congenital IGF1 Deficiency Confers Protection from Cancer Development
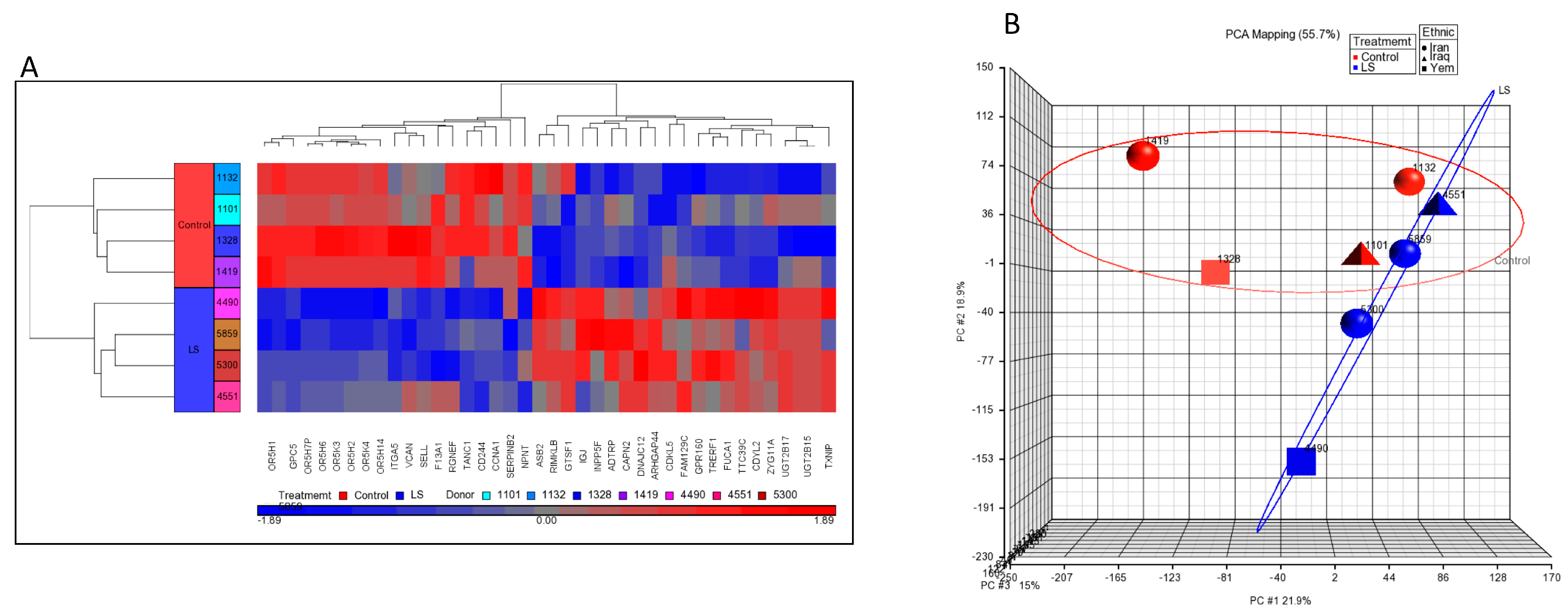
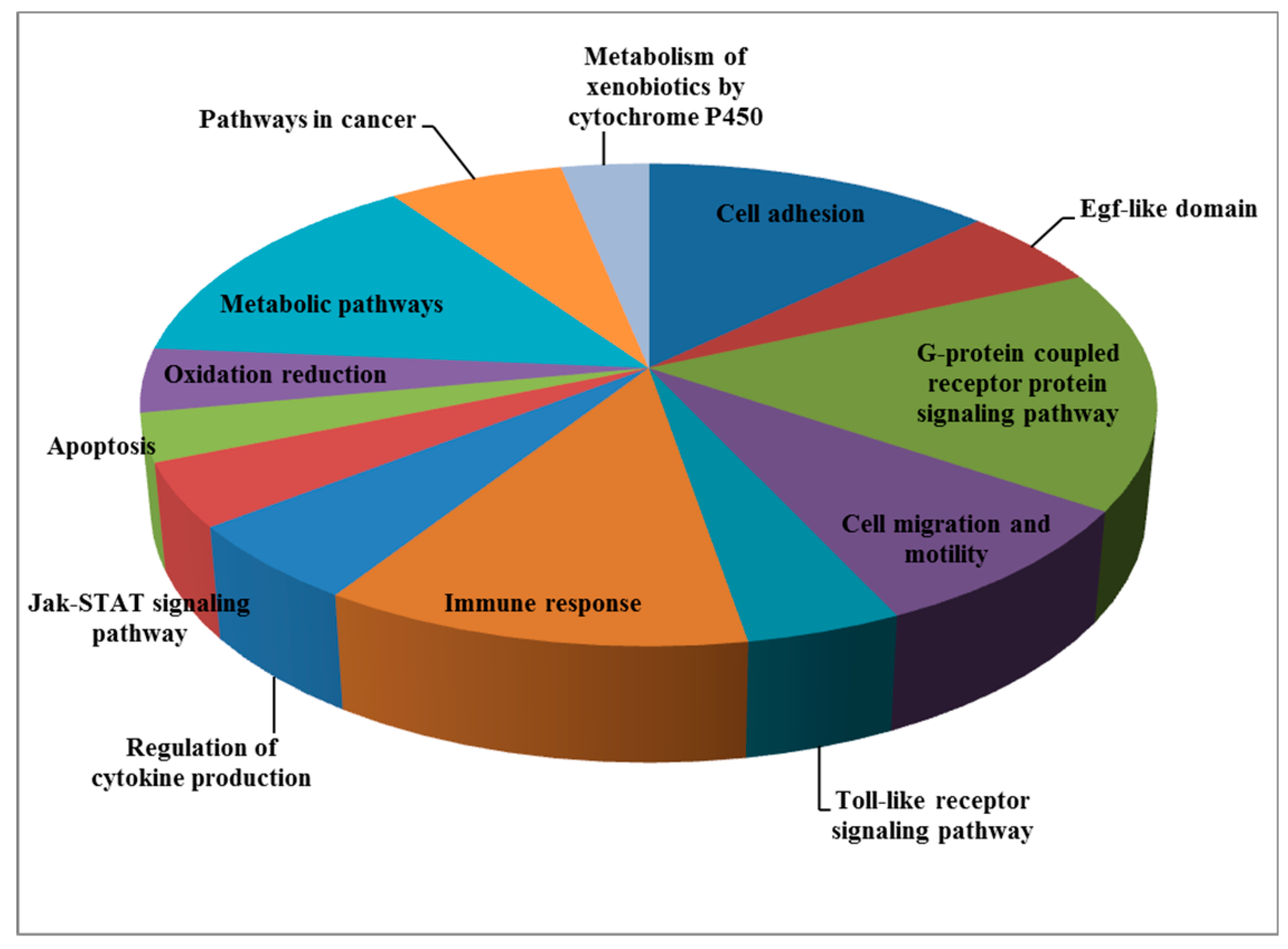
4. Genome-Wide Profiling of Laron Syndrome Patients Identifies Pathways Associated with Cancer Evasion
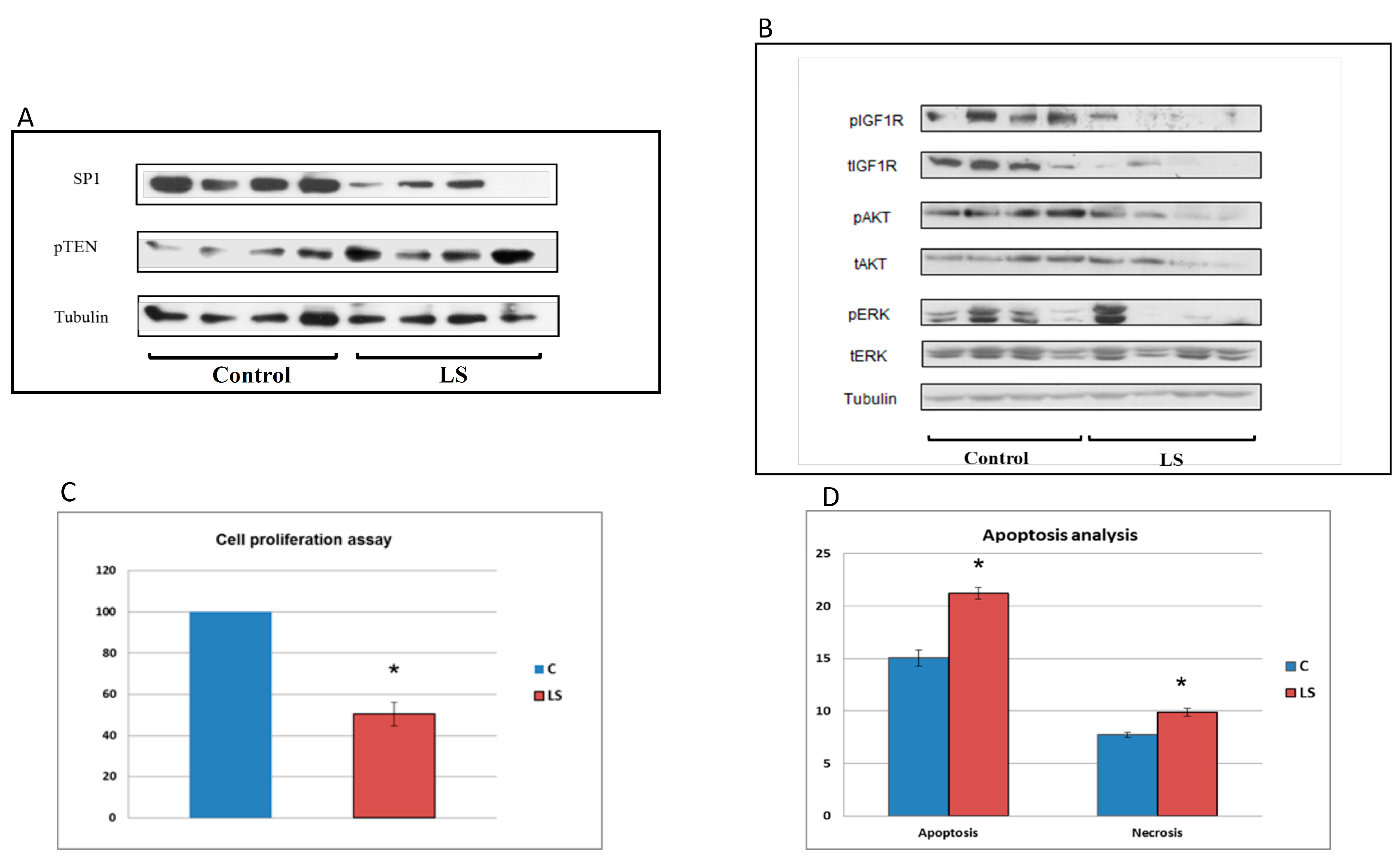
5. Differential Regulation of Oncogenes and Anti-Oncogenes in Laron Syndrome
6. Identification of Novel Metabolic Targets for IGF1 Action
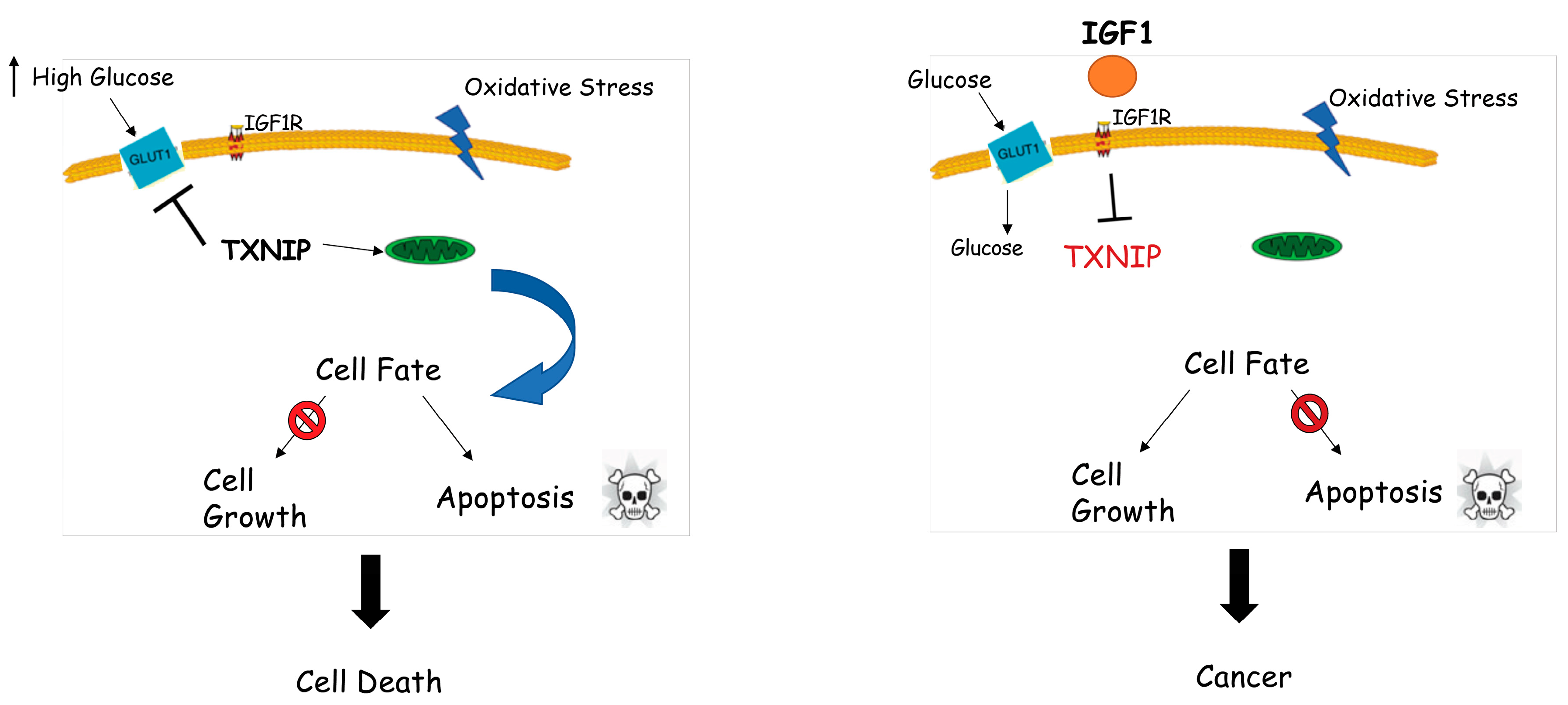
7. Identification of Thioredoxin-Interacting Protein (TXNIP) as a New Target of IGF1
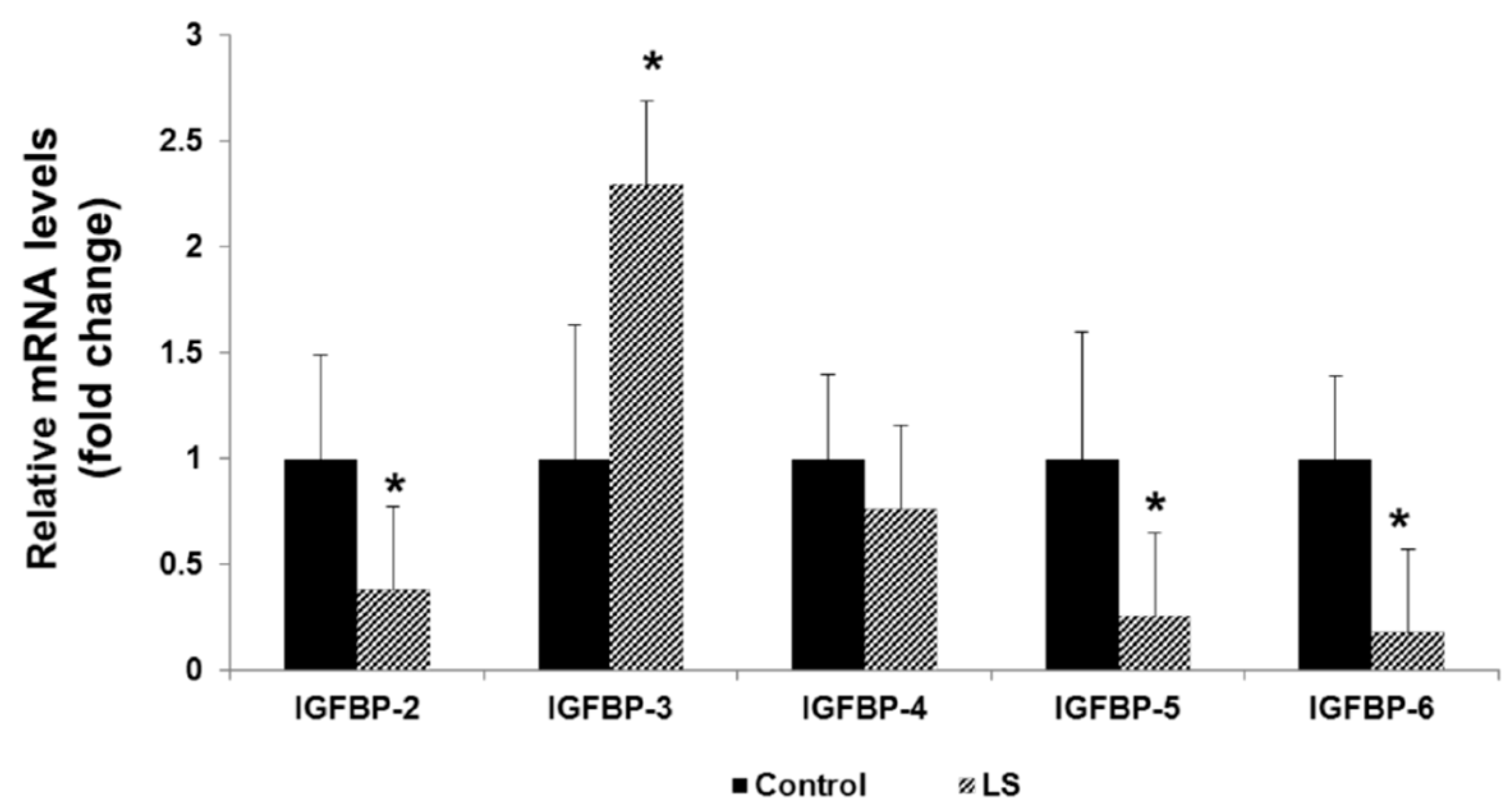
8. IGFBPs are Differentially Expressed in Laron Syndrome
9. Implications in Personalized Medicine
10. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- LeRoith, D.; Bondy, C.; Yakar, S.; Liu, J.-L.; Butler, A. The Somatomedin Hypothesis: 2001. Endocr. Rev. 2001, 22, 53–74. [Google Scholar] [CrossRef] [PubMed]
- Yakar, S.; Adamo, M.L. Insulin-like growth factor 1 physiology: Lessons from mouse models. Endocr. Metab. Clin. North Am. 2012, 41, 231–247. [Google Scholar] [CrossRef] [PubMed]
- LeRoith, D.; Yakar, S. Mechanisms of disease: Metabolic effects of growth hormone and insulin-like growth factor-1. Nat. Clin. Pract. Endocr. Metab. 2007, 3, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, R.G. Insulin-like growth factors and the basis of growth. N. Engl. J. Med. 2003, 349, 2184–2186. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, R.G. Molecular mechanisms of IGF-I deficiency. Horm. Res. 2006, 65 (Suppl. 1), 15–20. [Google Scholar] [CrossRef]
- Grimberg, A.; Hawkes, C.; Phillip, M. The physiology and mechanisms of growth. World Rev. Nutr. Diet 2018, 117, 1–14. [Google Scholar] [PubMed]
- Werner, H.; Weinstein, D.; Bentov, I. Similarities and differences between insulin and IGF-I: Structures, receptors, and signaling pathways. Arch. Physiol. Biochem. 2008, 114, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P. Controversy in clinical endocrinology: Problems with reclassification of insulin-like growth factor I production and action disorders. J. Clin. Endocr. Metab. 2008, 91, 4235–4236. [Google Scholar] [CrossRef]
- Pollak, M. The insulin and insulin-like growth factor receptor family in neoplasia: An update. Nat. Rev. Cancer 2012, 12, 159–169. [Google Scholar] [CrossRef]
- Wit, J.M.; Oostdijk, W.; Losekoot, M. Spectrum of insulin-like growth factor deficiency. Endocr. Dev. 2012, 23, 30–41. [Google Scholar]
- Klammt, J.; Pfaffle, R.; Werner, H.; Kiess, W. IGF signaling defects as causes of growth failure and IUGR. Trends Endocr. Metab. 2008, 19, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Domené, S.; Domené, H.M. Genetic mutations in the GH/IGF axis. Pediatr. Endocr. Rev. 2018, 16, 39–62. [Google Scholar]
- Rodriguez, S.; Gaunt, T.R.; Day, I.N. Molecular genetics of human growth hormone, insulin-like growth factors and their pathways in common disease. Hum. Genet. 2007, 122, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.; Rogol, A.D.; Deal, C.L.; Saenger, P.; Reiter, E.O.; Ross, J.L.; Chernausek, S.D.; Savage, M.O.; Wit, J.M. Consensus statement on the diagnosis and treatment of children with idiopathis short stature: A summary of the Growth Hormone Research Society, The Lawson Wilkins Pediatric Endocrine Society and the European Society for Pediatric Endocrinology Workshop. J. Clin. Endocr. Metab. 2008, 93, 4210–4217. [Google Scholar] [CrossRef] [PubMed]
- Aguiar-Oliveira, M.H.; Davalos, C.; Campos, V.C.; Oliveira Neto, L.A.; Marinho, C.G.; Oliveira, C.R.P. Hypothalamic abnormalities: Growth failure due to defects of the GHRH receptor. Growth Horm. IGF Res. 2018, 38, 14–18. [Google Scholar] [CrossRef]
- Domené, H.M.; Fierro-Carrión, G. Genetic disorders of GH action pathway. Growth Horm. IGF Res. 2018, 38, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Laron, Z.; Kopchik, J.J. Laron Syndrome—From Man to Mouse; Springer-Verlag: Berlin, Germany, 2011. [Google Scholar]
- Woods, K.A.; Camacho-Hubner, C.; Savage, M.O.; Clark, A.J.L. Intrauterine growth retardation and postnatal growth failure associated with deletion of the insulin-like growth factor I gene. N. Engl. J. Med. 1996, 335, 1363–1367. [Google Scholar] [CrossRef]
- Fang, P.; Riedl, S.; Amselem, S.; Pratt, K.L.; Little, B.; Haeusler, G.; Hwa, V.; Frisch, H.; Rosenfeld, R.G. Primary growth hormone insensitivity and insulin-like growth factor deficiency caused by novel compound heterozygous mutations of the GH receptor gene: Genetic and functional studies of simple and compound heterozygous states. J. Clin. Endocr. Metab. 2007, 92, 2223–2231. [Google Scholar] [CrossRef]
- Domene, H.; Bengolea, S.V.; Martinez, A.S.; Ropelato, M.G.; Pennisi, P.; Scaglia, P.; Heinrich, J.; Jasper, H. Deficiency of the circulating IGF system associated with inactivation of the acid-labile subunit gene. N. Engl. J. Med. 2004, 350, 570–577. [Google Scholar] [CrossRef]
- Argente, J.; Chowen, J.A.; Perez-Jurada, L.A.; Frystyk, J.; Oxvig, C. One level up: Abnormal proteolytic regulation of IGF activity plays a role in human pathophysiology. EMBO Mol. Med. 2017, 9, 1338–1345. [Google Scholar] [CrossRef]
- Abuzzahab, M.J.; Schneider, A.; Goddard, A.; Grigorescu, F.; Lautier, C.; Keller, E.; Kiess, W.; Klammt, J.; Kratzsch, J.; Osgood, D.; et al. IGF-I receptor mutations resulting in intrauterine and postnatal growth retardation. N. Engl. J. Med. 2003, 349, 2211–2222. [Google Scholar] [CrossRef] [PubMed]
- Walenkamp, M.J.; Karperien, M.; Pereira, A.M.; Hilhorst-Hofstee, Y.; van Doorn, J.; Chen, J.W.; Mohan, S.; Denley, A.; Forbes, B.; van Duyvenvoorde, H.A.; et al. Homozygous and heterozygous expression of a novel insulin-like growth factor-I mutation. J. Clin. Endocr. Metab. 2005, 90, 2855–2864. [Google Scholar] [CrossRef] [PubMed]
- Solomon-Zemler, R.; Basel-Vanagaite, L.; Steier, D.; Yakar, S.; Mel, E.; Phillip, M.; Bazak, L.; Bercovich, D.; Werner, H.; de Vries, L. A novel heterozygous IGF-1 receptor mutation associated with hypoglycemia. Endocr. Connect. 2017, 6, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Wallborn, T.; Wüller, S.; Klammt, J.; Kruis, T.; Kratzsch, J.; Schmidt, G.; Schlicke, M.; Müller, E.; van de Leur, H.S.; Kiess, W.; et al. A heterozygous mutation of the insulin-like growth factor-I receptor causes retention of the nascent protein in the endoplasmic reticulum and results in intrauterine and postnatal growth retardation. J. Clin. Endocr. Metab. 2010, 95, 2316–2324. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P. Overview of the IGF-I system. Hormone Res. 2006, 65, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Laron, Z. Natural history of the classical form of primary growth hormone resistance (Laron syndrome). J. Pediatr. Endocrinol. Metab. 1999, 12, 231–249. [Google Scholar]
- Laron, Z.; Pertzelan, A.; Mannheimer, S. Genetic pituitary dwarfism with high serum concentration of growth hormone-a new inborn error of metabolism? Isr. J. Med. Sci. 1966, 2, 152–155. [Google Scholar]
- Laron, Z. Extensive personal experience. Laron syndrome (primary growth hormone resistance or insensitivity): The personal experience 1958–2003. J. Clin. Endocrinol. Metab. 2004, 89, 1031–1044. [Google Scholar] [CrossRef]
- Godowski, P.J.; Leung, D.W.; Meacham, L.R.; Galgani, J.P.; Hellmiss, R.; Keret, R.; Rotwein, P.S.; Parks, J.S.; Laron, Z.; Wood, W.I. Characterization of the human growth hormone receptor gene and demonstration of a partial gene deletion in two patients with Laron-type dwarfism. Proc. Natl. Acad. Sci. USA 1989, 86, 8083–8087. [Google Scholar] [CrossRef]
- Ayling, R.M.; Ross, R.; Towner, P.; Von Laue, S.; Finidori, J.; Moutoussamy, S.; Buchanan, C.R.; Clayton, P.E.; Norman, M.R. A dominant-negative mutation of the growth hormone receptor causes familial short stature. Nat. Genet. 1997, 16, 13–14. [Google Scholar] [CrossRef]
- Duquesnoy, P.; Sobrier, M.L.; Duriez, B.; Dastot, F.; Buchanan, C.R.; Savage, M.O.; Preece, M.A.; Craescu, C.T.; Blouquit, Y.; Goosens, M.; et al. A single amino acid substitution in the exoplasmic domain of the human growth hormone receptor confers familial GH resistance (Laron syndrome) with positive GH-binding activity by abolishing receptor homodimerization. EMBO J. 1994, 13, 1386–1395. [Google Scholar] [CrossRef] [PubMed]
- Iida, K.; Takahashi, Y.; Kaji, H.; Nose, O.; Okimura, Y.; Hiromi, A.; Chihara, K. Growth hormone insensitivity syndrome with high serum GH-binding protein levels caused by a heterozygous splice site mutation of the GH receptor gene producing a lack of intracellular domain. J. Clin. Endocrinol. Metab. 1998, 83, 531–537. [Google Scholar] [PubMed]
- Shevah, O.; Nunez, O.; Rubinstein, M.; Laron, Z. Intronic mutation in the growth hormone receptor gene in a Peruvian girl with Laron syndrome. J. Pediatr. Endocrinol. Metab. 2002, 15, 1039–1040. [Google Scholar] [CrossRef] [PubMed]
- Rosenbloom, A.L.; Guevara-Aguirre, J.; Rosenfeld, R.G.; Francke, U. Growth hormone receptor deficiency in Ecuador. J. Clin. Endocrinol. Metab. 1999, 84, 4436–4443. [Google Scholar] [CrossRef] [PubMed]
- Fang, P.; Girgis, R.; Little, B.M.; Pratt, K.L.; Guevara-Aguirre, J.; Hwa, V.; Rosenfeld, R.G. Growth hormone insensitivity and IGF-I deficiency in Inuit subjects and an Ecuadorian cohort: Functional studies of two codon 180 GH receptor gene mutations. J. Clin. Endocrinol. Metab. 2008, 93, 1030–1037. [Google Scholar] [CrossRef] [PubMed]
- Berg, M.A.; Guevara-Aguirre, J.; Rosenbloom, A.L.; Rosenfeld, R.G.; Francke, U. Mutation creating a new splice site in the growth hormone receptor genes of 37 Ecuadorean patients with Laron syndrome. Hum. Mutat. 1992, 1, 124–134. [Google Scholar] [CrossRef]
- Gonçalves, F.T.; Fridman, C.; Pinto, E.M.; Guevara-Aguirre, J.; Shevah, O.; Rosenbloom, A.L.; Hwa, V.; Cassorla, F.; Rosenfeld, R.G.; Lins, T.S.S.; et al. The E180 splice mutation in the GHR gene causing Laron syndrome: Witness of a Sephardic Jewish exodus from the Iberian Peninsula to the New World? Am. J. Med. Genet. 2014, 164A, 1204–1208. [Google Scholar] [CrossRef]
- Shevah, O.; Laron, Z. Genetic analysis of the pedigrees and molecular defects of the GH-receptor gene in the Israeli cohort of patients with Laron syndrome. Pediatr. Endocrinol. Rev. 2006, (Suppl. 3), 489–497. [Google Scholar]
- Laron, Z. Lessons from 50 years of study of Laron syndrome. Endocr. Pract. 2015, 21, 1395–1402. [Google Scholar] [CrossRef]
- Rosenfeld, R.G. The future of growth-promoting therapy. Growth Horm. IGF Res. 2016, 28, 43–45. [Google Scholar] [CrossRef]
- Rosenbloom, A. A half-century of studies of growth hormone insensitivity/Laron syndrome: A historical perspective. Growth Horm. IGF Res. 2016, 28, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Laron, Z.; Ginsberg, S.; Lilos, P.; Arbiv, M.; Vaisman, N. Long-term IGF-I treatment of children with Laron Syndrome increases adiposity. Growth Horm. IGF Res. 2006, 16, 61–64. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.M.; Stampfer, M.J.; Giovannucci, E.; Gann, P.H.; Ma, J.; Wilkinson, P.; Hennekens, C.H.; Pollak, M. Plasma insulin-like growth factor-I and prostate cancer risk: A prospective study. Science 1998, 279, 563–566. [Google Scholar] [CrossRef] [PubMed]
- Hankinson, S.E.; Willett, W.C.; Colditz, G.A.; Hunter, D.J.; Michaud, D.S.; Deroo, B.; Rosner, B.; Speizer, F.E.; Pollak, M. Circulating concentrations of insulin-like growth factor-I and risk of breast cancer. Lancet 1998, 351, 1393–1396. [Google Scholar] [CrossRef]
- Renehan, A.G.; Zwahlen, M.; Zwahlen, M.; Minder, C.; O’Dwyer, S.T.; Shalet, S.M.; Egger, M. Insulin-like growth factor-I, IGF binding protein-3, and cancer risk: Systematic review and meta-regression analysis. Lancet 2004, 363, 1346–1353. [Google Scholar] [CrossRef]
- Pollak, M.N. Insulin-like growth factors and neoplasia. Novartis Found. Symp. 2004, 262, 84–98. [Google Scholar] [PubMed]
- Steuerman, R.; Shevah, O.; Laron, Z. Congenital IGF1 deficiency tends to confer protection against post-natal development of malignancies. Eur. J. Endocrinol. 2011, 164, 485–489. [Google Scholar] [CrossRef] [PubMed]
- Shevah, O.; Laron, Z. Patients with congenital deficiency of IGF-I seem protected from the development of malignancies: A preliminary report. Growth Horm. IGF Res. 2007, 17, 54–57. [Google Scholar] [CrossRef]
- Guevara-Aguirre, J.; Balasubramanian, P.; Guevara-Aguirre, M.; Wei, M.; Madia, F.; Cheng, C.W.; Hwang, D.; Martin-Montalvo, A.; Saavedra, J.; Ingles, S.; et al. Growth hormone receptor deficiency is associated with a major reduction in pro-aging signaling, cancer, and diabetes in humans. Sci. Transl. Med. 2011, 3, 70ra13. [Google Scholar] [CrossRef]
- Wang, Z.; Prins, G.S.; Coschigano, K.T.; Kopchick, J.J.; Green, J.E.; Ray, V.H.; Hedayat, S.; Christov, K.T.; Unterman, T.G.; Swanson, S.M. Disruption of growth hormone signaling retards early stages of prostate carcinogenesis in the C3(1)/T antigen mouse. Endocrinology 2005, 146, 5188–5196. [Google Scholar] [CrossRef]
- Moore, T.; Carbajal, S.; Beltran, L.; Perkins, S.N.; Yakar, S.; LeRoith, D.; Hursting, S.D.; Digiovanni, J. Reduced susceptibility to two-stage skin carcinogenesis in mice with low circulating insulin-like growth factor-I levels. Cancer Res. 2008, 68, 3680–3688. [Google Scholar] [CrossRef] [PubMed]
- Laron, Z.; Kauli, R.; Lapkina, L.; Werner, H. IGF-I deficiency, longevity and cancer protection of patients with Laron syndrome. Mutat. Res. Rev. Mutat. Res. 2017, 772, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Green, J.; Cairns, B.J.; Casabonne, D.; Wright, F.L.; Reeves, G.; Beral, V. Million Women Study Collaborators. Height and cancer incidence in the Million Women Study: Prospective cohort, and meta-analysis of prospective studies of height and total cancer risk. Lancet Oncol. 2011, 12, 785–794. [Google Scholar] [CrossRef]
- Lapkina-Gendler, L.; Rotem, I.; Pasmanik-Chor, M.; Gurwitz, D.; Sarfstein, R.; Laron, Z.; Werner, H. Identification of signaling pathways associated with cancer protection in Laron syndrome. Endocr. Relat. Cancer 2016, 23, 399–410. [Google Scholar] [CrossRef]
- Moscat, J.; Diaz-Meco, M.T. p62 at the crossroads of autophagy, apoptosis, and cancer. Cell 2009, 137, 1001–1004. [Google Scholar] [CrossRef]
- Rosenfeldt, M.T.; Ryan, K.M. The multiple roles of autophagy in cancer. Carcinogenesis 2011, 32, 955–963. [Google Scholar] [CrossRef]
- Baserga, R.; Peruzzi, F.; Reiss, K. The IGF-1 receptor in cancer biology. Int. J. Cancer 2003, 107, 873–877. [Google Scholar] [CrossRef]
- Werner, H. The pathophysiological significance of IGF-I receptor overexpression: New insights. Pediatr. Endocrinol. Rev. 2009, 7, 2–5. [Google Scholar]
- Werner, H.; Shalita-Chesner, M.; Abramovitch, S.; Idelman, G.; Shaharabani-Gargir, L.; Glaser, T. Regulation of the insulin-like growth factor-I receptor gene by oncogenes and antioncogenes: Implications in human cancer. Mol. Genet. Metab. 2000, 71, 315–320. [Google Scholar] [CrossRef]
- Werner, H. Tumor suppressors govern insulin-like growth factor signaling pathways: Implications in metabolism and cancer. Oncogene 2012, 31, 2703–2714. [Google Scholar] [CrossRef]
- Werner, H.; Lapkina-Gendler, L.; Nagaraj, K.; Sarfstein, R.; Laron, Z. Genome-wide profiling of congenital IGF1 deficient patients: Translational implications in cancer prevention and metabolism. Transl. Med. Rep. 2017, 1, 6657. [Google Scholar] [CrossRef]
- Vidal, A.C.; Tucker, C.; Schildkraut, J.M.; Richardson, R.M.; McPhail, M.; Freedland, S.J.; Hoyo, C.; Grant, D.J. Novel associations of UDP-glucuronosyltransferase 2B gene variants with prostate cancer risk in a multiethnic study. BMC Cancer 2013, 13, 556. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Basit, A.; Busch, D.; Yabut, K.; Bhatt, D.K.; Drozdzik, M.; Ostrowski, M.; Li, A.; Collins, C.; Oswald, S.; et al. Quantitative characterization of UDP-glucuronosyltransferase 2B17 in human liver and intestine and its role in testosterone first-pass metabolism. Biochem. Pharmacol. 2018, 156, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, M.; Hatakeyama, S.; Oyamada, K.; Oda, Y.; Nishimura, T.; Nakayama, K.I. Large-scale analysis of the human ubiquitin-related proteome. Proteomics 2005, 5, 4145–4151. [Google Scholar] [CrossRef]
- Chen, K.S.; DeLuca, H.F. Isolation and characterization of a novel cDNA from HL-60 cells treated with 1,25-dihydroxyvitamin D-3. Biochim. Biophys. Acta Gene Struct. Expr. 1994, 1219, 26–32. [Google Scholar] [CrossRef]
- Patwari, P.; Higgins, L.J.; Chutkow, W.A.; Yoshioka, J.; Lee, R.T. The interaction of thioredoxin with Txnip: Evidence for formation of a mixed disulfide by disulfide exchange. J. Biol. Chem. 2006, 281, 21884–21891. [Google Scholar] [CrossRef]
- DeBalsi, K.L.; Wong, K.E.; Koves, T.R.; Slentz, D.H.; Seiler, S.E.; Wittmann, A.H.; Ilkayeva, O.R.; Stevens, R.D.; Perry, C.G.; Lark, D.; et al. Targeted metabolomics connects thioredoxin-interacting protein (TXNIP) to mitochondrial fuel selection and regulation of specific oxidoreductase enzymes in skeletal muscle. J. Biol. Chem. 2014, 289, 8106–8120. [Google Scholar] [CrossRef]
- Goldberg, S.F.; Miele, M.E.; Hatta, N.; Takata, M.; Paquette-Straub, C.; Freedman, L.P.; Welch, D.R. Melanoma metastasis suppression by chromosome 6: Evidence for a pathway regulated by CRSP3 and TXNIP. Cancer Res. 2003, 63, 432–440. [Google Scholar]
- Blouet, C.; Schwartz, G.J. Nutrient sensing hypothalamic TXNIP links nutrient excess to energy imbalance in mice. J. Neurosci. 2011, 31, 6019–6027. [Google Scholar] [CrossRef]
- Wu, N.; Zheng, B.; Shaywitz, A.; Dagon, Y.; Tower, C.; Bellinger, G.; Shen, C.; Wen, J.; Asara, J.; McGraw, T.; et al. AMPK-dependent degradation of TXNIP upon energy stress leads to enhanced glucose uptake via GLUT1. Mol. Cell 2013, 49, 1167–1175. [Google Scholar] [CrossRef]
- Huy, H.; Song, H.; Kim, M.; Kim, W.; Kim, D.; Byun, J.; Lee, J.; Park, Y.; Kim, T.; Yoon, S.; et al. TXNIP regulates AKT-mediated cellular senescence by direct interaction under glucose-mediated metabolic stress. Aging Cell 2018, 17, e12836. [Google Scholar] [CrossRef] [PubMed]
- Nagaraj, K.; Lapkina-Gendler, L.; Sarfstein, R.; Gurwitz, D.; Pasmanik-Chor, M.; Laron, Z.; Yakar, S.; Werner, H. Identification of thioredoxin-interacting protein (TXNIP) as a downstream target for IGF1 action. Proc. Natl. Acad. Sci. USA 2018, 115, 1045–1050. [Google Scholar] [CrossRef] [PubMed]
- Baxter, R.C. IGF binding proteins in cancer: Mechanistic and clinical insights. Nature Rev. Cancer 2014, 14, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Bach, L.A. Insulin-Like Growth Factor Binding Proteins--an Update. Pediatr. Endocrinol. Rev. 2015, 13, 521–530. [Google Scholar] [PubMed]
- Oh, Y. IGF-independent regulation of breast cancer growth by IGF binding proteins. Breast Cancer Res. Treat. 1998, 47, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Roddam, A.; Allen, N.; Appleby, P.; Key, T.; Ferrucci, L.; Carter, H.; Metter, E.J.; Chen, C.; Weiss, N.S.; Fitzpatrick, A.; et al. Insulin-like growth factors, their binding proteins, and prostate cancer risk: Analysis of individual patient data from 12 prospective studies. Ann. Intern. Med. 2008, 149, 461–471. [Google Scholar] [PubMed]
- Belobrajdic, D.; Priebe, I.; Forbes, B.; Flyvbjerg, A.; Chen, J.; Cosgrove, L.; Frystyk, J.; Saunders, I. Assessing the potential usefulness of IGF-related peptides and adiponectin for predicting disease risk. Growth Horm. IGF Res. 2008, 18, 198–204. [Google Scholar] [CrossRef]
- Rollison, D.; Giuliano, A.; Risendal, B.; Sweeney, C.; Boulware, D.; Laronga, C.; Baumgartner, K.; Byers, T.; Slattery, M. Serum insulin-like growth factor (IGF)-1 and IGF binding protein-3 in relation to breast cancer among Hispanic and white, non-Hispanic women in the US Southwest. Breast Cancer Res. Treat. 2010, 121, 661–669. [Google Scholar] [CrossRef]
- Liu, B.; Lee, K.; Anzo, M.; Zhang, B.; Zi, X.; Tao, Y.; Shiry, L.; Pollak, M.; Lin, S.; Cohen, P. Insulin-like growth factor-binding protein-3 inhibition of prostate cancer growth involves suppression of angiogenesis. Oncogene 2007, 26, 1811–1819. [Google Scholar] [CrossRef]
- Somri, L.; Sarfstein, R.; Lapkina-Gendler, L.; Nagaraj, K.; Laron, Z.; Bach, L.A.; Werner, H. Differential expression of IGFBPs in Laron syndrome-derived lymphoblastoid cell lines: Potential correlation with reduced cancer incidence. Growth Horm. IGF Res. 2018, 39, 6–12. [Google Scholar] [CrossRef]
- Wex, H.; Vorwerk, P.; Mohnike, K.; Bretschneider, D.; Kluba, U.; Aumann, V.; Blum, W.F.; Mittler, U. Elevated serum levels of IGFBP-2 found in children suffering from acute leukaemia is accompanied by the occurrence of IGFBP-2 mRNA in the tumour clone. Br. J. Cancer 1998, 78, 515–520. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Xie, L.F.; Tsaprailis, G.; Chen, Q.M. Proteomic identification of insulin-like growth factor-binding protein-6 induced by sublethal H2O2 stress from human diploid fibroblasts. Mol. Cell Proteom. 2005, 4, 1273–1283. [Google Scholar] [CrossRef]
- Liso, A.; Castellani, S.; Massenzio, F.; Trotta, R.; Pucciarini, A.; Bigerna, B.; De Luca, P.; Zoppoli, P.; Castiglione, F.; Palumbo, M.C.; et al. Human monocyte-derived dendritic cells exposed to hyperthermia show a distinct gene expression profile and selective upregulation of IGFBP6. Oncotarget 2017, 8, 60826–60840. [Google Scholar] [CrossRef] [PubMed]
- Yasuoka, H.; Yamaguchi, Y.; Feghali-Bostwick, C.A. The pro-fibrotic factor IGFBP-5 induces lung fibroblast and mononuclear cell migration. Am. J. Respir. Cell. Mol. Biol. 2009, 41, 179–188. [Google Scholar] [CrossRef] [PubMed]
- King, H.; Aleksic, T.; Haluska, P.; Macaulay, V.M. Can we unlock the potential of IGF-1R inhibition in cancer therapy? Cancer Treat. Rev. 2014, 40, 1096–1105. [Google Scholar] [CrossRef]
- Bruchim, I.; Attias, Z.; Werner, H. Targeting the IGF1 axis in cancer proliferation. Exp. Opinion Ther. Targets 2009, 13, 1179–1192. [Google Scholar] [CrossRef]
- Ekyalongo, R.C.; Yee, D. Revisiting the IGF-1R as a breast cancer target. NP J. Precis. Oncol. 2017, 1, 14. [Google Scholar] [CrossRef]
- Cohen-Sinai, T.; Cohen, Z.; Werner, H.; Berger, R. Identification of BRCA1 as a potential biomarker for insulin-like growth factor-1 receptor targeted therapy in breast cancer. Front. Endocrinol. 2017, 8, 148. [Google Scholar] [CrossRef]
- Werner, H.; Lapkina-Gendler, L.; Laron, Z. Fifty years on: New lessons from Laron syndrome. Isr. Med. Assoc. J. 2017, 19, 6–7. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecular defects leading to congenital IGF1 deficiency |
| Growth hormone (GH)-releasing hormone receptor (GHRH-R) defect |
| GH gene deletion (isolated GH deficiency, IGHD) |
| GH receptor (GH-R) gene deficiency (Laron syndrome) |
| IGF1 gene deletion |
| Defects of post-GH-R signaling (e.g., STAT5 defects) |
| Acid labile subunit (ALS) mutations |
| PPA2 protein mutations |
| Laron Syndrome | First-Degree Relatives | Further Relatives | |
|---|---|---|---|
| Total number (n) | 230 | 218 | 113 |
| Number of malignancies | 0 | 18 | 25 |
| Prevalence of malignancy | 0.0% | 8.3% | 22.1% |
| Pathway/Function | Number of Genes |
|---|---|
| Cell adhesion | 12 |
| Egf-like domain | 5 |
| G-protein coupled receptor protein signaling pathway | 15 |
| Cell migration and motility | 8 |
| Toll-like receptor signaling pathway | 4 |
| Immune response | 11 |
| Regulation of cytokine production | 5 |
| Jak-STAT signaling pathway | 4 |
| Apoptosis | 3 |
| Oxidation reduction | 4 |
| Metabolic pathways | 13 |
| Pathways in cancer | 6 |
| Metabolism of xenobiotics by cytochrome P450 | 3 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Werner, H.; Lapkina-Gendler, L.; Achlaug, L.; Nagaraj, K.; Somri, L.; Yaron-Saminsky, D.; Pasmanik-Chor, M.; Sarfstein, R.; Laron, Z.; Yakar, S. Genome-Wide Profiling of Laron Syndrome Patients Identifies Novel Cancer Protection Pathways. Cells 2019, 8, 596. https://doi.org/10.3390/cells8060596
Werner H, Lapkina-Gendler L, Achlaug L, Nagaraj K, Somri L, Yaron-Saminsky D, Pasmanik-Chor M, Sarfstein R, Laron Z, Yakar S. Genome-Wide Profiling of Laron Syndrome Patients Identifies Novel Cancer Protection Pathways. Cells. 2019; 8(6):596. https://doi.org/10.3390/cells8060596
Chicago/Turabian StyleWerner, Haim, Lena Lapkina-Gendler, Laris Achlaug, Karthik Nagaraj, Lina Somri, Danielle Yaron-Saminsky, Metsada Pasmanik-Chor, Rive Sarfstein, Zvi Laron, and Shoshana Yakar. 2019. "Genome-Wide Profiling of Laron Syndrome Patients Identifies Novel Cancer Protection Pathways" Cells 8, no. 6: 596. https://doi.org/10.3390/cells8060596
APA StyleWerner, H., Lapkina-Gendler, L., Achlaug, L., Nagaraj, K., Somri, L., Yaron-Saminsky, D., Pasmanik-Chor, M., Sarfstein, R., Laron, Z., & Yakar, S. (2019). Genome-Wide Profiling of Laron Syndrome Patients Identifies Novel Cancer Protection Pathways. Cells, 8(6), 596. https://doi.org/10.3390/cells8060596