Hypoxia and Hypoxia-Inducible Factors in Kidney Injury and Repair


Abstract
1. Introduction
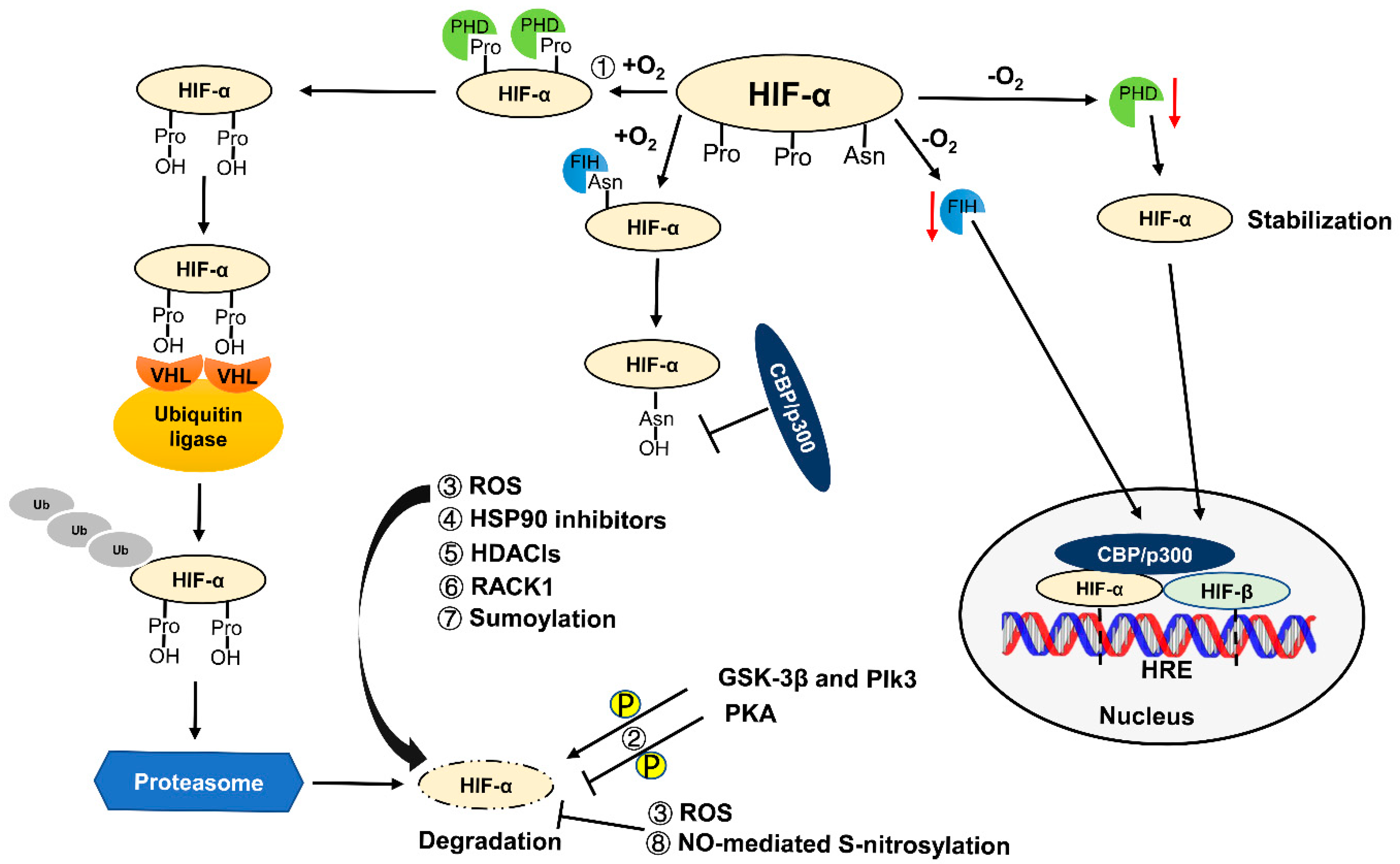
2. Regulation of HIF
3. Expression Patterns and Functions of HIFs
4. Well-Known HIF Target Genes and Their Functions in Kidney
4.1. Erythropoietin (EPO)
4.2. Vascular Endothelial Growth Factor (VEGF)
5. HIF in AKI and Mechanisms of HIF Signaling in AKI
5.1. HIF in IR-Induced AKI
5.2. HIF in Cisplatin-Induced AKI
5.3. HIF in Sepsis-Associated AKI
5.4. HIF in AKI Induced by Other Causes
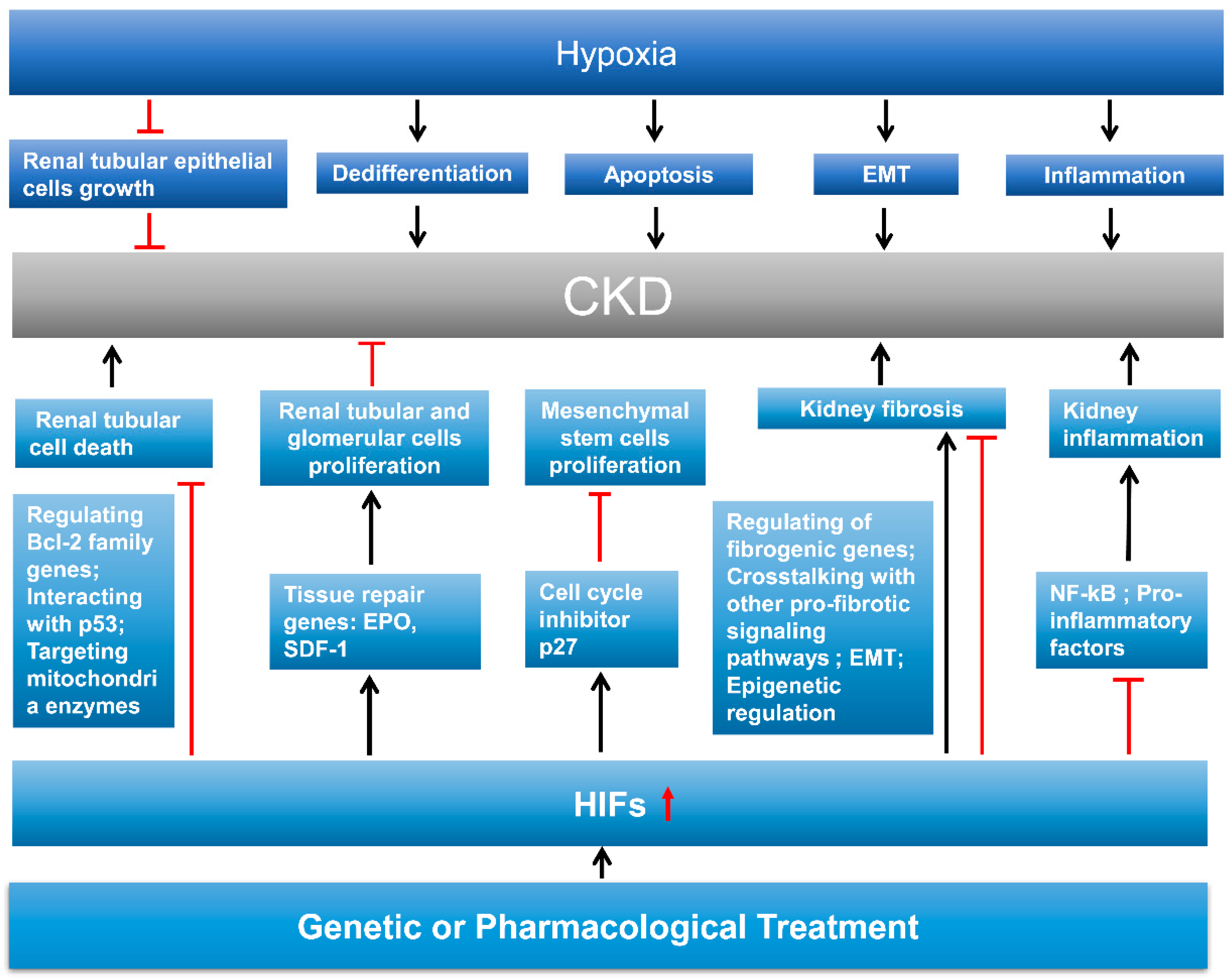
6. Role of HIF in Kidney Repair
6.1. Integral Introduction of Kidney Repair
6.2. HIF in Kidney Cell Death, Dedifferentiation, and Proliferation
6.3. HIF in Kidney Fibrosis
6.4. HIF in Kidney Inflammation
7. Therapeutic Potential of HIF in AKI and CKD
Author Contributions
Funding
Conflicts of Interest
References
- Mehta, R.L.; Burdmann, E.A.; Cerda, J.; Feehally, J.; Finkelstein, F.; Garcia-Garcia, G.; Godin, M.; Jha, V.; Lameire, N.H.; Levin, N.W.; et al. Recognition and management of acute kidney injury in the International Society of Nephrology 0by25 Global Snapshot: A multinational cross-sectional study. Lancet 2016, 387, 2017–2025. [Google Scholar] [CrossRef]
- Agarwal, A.; Dong, Z.; Harris, R.; Murray, P.; Parikh, S.M.; Rosner, M.H.; Kellum, J.A.; Ronco, C. Cellular and Molecular Mechanisms of AKI. J. Am. Soc. Nephrol. 2016, 27, 1288–1299. [Google Scholar] [CrossRef] [PubMed]
- Linkermann, A.; Chen, G.; Dong, G.; Kunzendorf, U.; Krautwald, S.; Dong, Z. Regulated cell death in AKI. J. Am. Soc. Nephrol. 2014, 25, 2689–2701. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Wei, Q.; Liu, J.; Yi, M.; Liu, Y.; Liu, H.; Sun, L.; Peng, Y.; Liu, F.; Venkatachalam, M.A.; et al. AKI on CKD: Heightened injury, suppressed repair, and the underlying mechanisms. Kidney Int. 2017, 92, 1071–1083. [Google Scholar] [CrossRef] [PubMed]
- Heung, M.; Steffick, D.E.; Zivin, K.; Gillespie, B.W.; Banerjee, T.; Hsu, C.Y.; Powe, N.R.; Pavkov, M.E.; Williams, D.E.; Saran, R.; et al. Acute Kidney Injury Recovery Pattern and Subsequent Risk of CKD: An Analysis of Veterans Health Administration Data. Am. J. Kidney Dis. 2016, 67, 742–752. [Google Scholar] [CrossRef] [PubMed]
- Ferenbach, D.A.; Bonventre, J.V. Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat. Rev. Nephrol. 2015, 11, 264–276. [Google Scholar] [CrossRef] [PubMed]
- Basile, D.P.; Bonventre, J.V.; Mehta, R.; Nangaku, M.; Unwin, R.; Rosner, M.H.; Kellum, J.A.; Ronco, C. Progression after AKI: Understanding Maladaptive Repair Processes to Predict and Identify Therapeutic Treatments. J. Am. Soc. Nephrol. 2016, 27, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. HIF-1, O2, and the 3 PHDs: How animal cells signal hypoxia to the nucleus. Cell 2001, 107, 1–3. [Google Scholar] [CrossRef]
- Ohashi, R.; Shimizu, A.; Masuda, Y.; Kitamura, H.; Ishizaki, M.; Sugisaki, Y.; Yamanaka, N. Peritubular capillary regression during the progression of experimental obstructive nephropathy. J. Am. Soc. Nephrol. 2002, 13, 1795–1805. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, M.; Tanaka, T.; Yamamoto, T.; Noiri, E.; Miyata, T.; Inagi, R.; Fujita, T.; Nangaku, M. Hypoperfusion of peritubular capillaries induces chronic hypoxia before progression of tubulointerstitial injury in a progressive model of rat glomerulonephritis. J. Am. Soc. Nephrol. 2004, 15, 1574–1581. [Google Scholar] [CrossRef] [PubMed]
- Conde, E.; Alegre, L.; Blanco-Sanchez, I.; Saenz-Morales, D.; Aguado-Fraile, E.; Ponte, B.; Ramos, E.; Saiz, A.; Jimenez, C.; Ordonez, A.; et al. Hypoxia inducible factor 1-α (HIF-1 α) is induced during reperfusion after renal ischemia and is critical for proximal tubule cell survival. PLoS ONE 2012, 7, e33258. [Google Scholar] [CrossRef] [PubMed]
- Rosenberger, C.; Pratschke, J.; Rudolph, B.; Heyman, S.N.; Schindler, R.; Babel, N.; Eckardt, K.U.; Frei, U.; Rosen, S.; Reinke, P. Immunohistochemical detection of hypoxia-inducible factor-1α in human renal allograft biopsies. J. Am. Soc. Nephrol. 2007, 18, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Basile, D.P.; Donohoe, D.L.; Roethe, K.; Mattson, D.L. Chronic renal hypoxia after acute ischemic injury: Effects of l-arginine on hypoxia and secondary damage. Am. J. Physiol. Ren. Physiol. 2003, 284, F338–F348. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Colgan, S.P.; Shelley, C.S. Hypoxia: The Force that Drives Chronic Kidney Disease. Clin. Med. Res. 2016, 14, 15–39. [Google Scholar] [CrossRef] [PubMed]
- Hirakawa, Y.; Tanaka, T.; Nangaku, M. Renal Hypoxia in CKD; Pathophysiology and Detecting Methods. Front. Physiol. 2017, 8, 99. [Google Scholar] [CrossRef] [PubMed]
- Gruber, M.; Hu, C.J.; Johnson, R.S.; Brown, E.J.; Keith, B.; Simon, M.C. Acute postnatal ablation of HIF-2α results in anemia. Proc. Natl. Acad. Sci. USA 2007, 104, 2301–2306. [Google Scholar] [CrossRef] [PubMed]
- Rius, J.; Guma, M.; Schachtrup, C.; Akassoglou, K.; Zinkernagel, A.S.; Nizet, V.; Johnson, R.S.; Haddad, G.G.; Karin, M. NF-κB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1α. Nature 2008, 453, 807–811. [Google Scholar] [CrossRef] [PubMed]
- Haase, V.H. Hypoxia-inducible factors in the kidney. Am. J. Physiol. Ren. Physiol. 2006, 291, F271–F281. [Google Scholar] [CrossRef] [PubMed]
- Wenger, R.H.; Stiehl, D.P.; Camenisch, G. Integration of oxygen signaling at the consensus HRE. Sci. STKE 2005, 2005, re12. [Google Scholar] [CrossRef] [PubMed]
- Nangaku, M.; Eckardt, K.U. Hypoxia and the HIF system in kidney disease. J. Mol. Med. 2007, 85, 1325–1330. [Google Scholar] [CrossRef] [PubMed]
- Kapitsinou, P.P.; Sano, H.; Michael, M.; Kobayashi, H.; Davidoff, O.; Bian, A.; Yao, B.; Zhang, M.Z.; Harris, R.C.; Duffy, K.J.; et al. Endothelial HIF-2 mediates protection and recovery from ischemic kidney injury. J. Clin. Investig. 2014, 124, 2396–2409. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yu, X.; Zhang, Y.; Ding, G.; Zhu, C.; Huang, S.; Jia, Z.; Zhang, A. Hypoxia-inducible factor prolyl hydroxylase inhibitor roxadustat (FG-4592) protects against cisplatin-induced acute kidney injury. Clin. Sci. 2018, 132, 825–838. [Google Scholar] [CrossRef] [PubMed]
- Fahling, M.; Mathia, S.; Paliege, A.; Koesters, R.; Mrowka, R.; Peters, H.; Persson, P.B.; Neumayer, H.H.; Bachmann, S.; Rosenberger, C. Tubular von Hippel-Lindau knockout protects against rhabdomyolysis-induced AKI. J. Am. Soc. Nephrol. 2013, 24, 1806–1819. [Google Scholar] [CrossRef] [PubMed]
- Higgins, D.F.; Kimura, K.; Bernhardt, W.M.; Shrimanker, N.; Akai, Y.; Hohenstein, B.; Saito, Y.; Johnson, R.S.; Kretzler, M.; Cohen, C.D.; et al. Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J. Clin. Investig. 2007, 117, 3810–3820. [Google Scholar] [CrossRef] [PubMed]
- Jaakkola, P.; Mole, D.R.; Tian, Y.M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-α to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Oxygen sensing, homeostasis, and disease. N. Engl. J. Med. 2011, 365, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Soilleux, E.J.; Turley, H.; Tian, Y.M.; Pugh, C.W.; Gatter, K.C.; Harris, A.L. Use of novel monoclonal antibodies to determine the expression and distribution of the hypoxia regulatory factors PHD-1, PHD-2, PHD-3 and FIH in normal and neoplastic human tissues. Histopathology 2005, 47, 602–610. [Google Scholar] [CrossRef] [PubMed]
- Schodel, J.; Klanke, B.; Weidemann, A.; Buchholz, B.; Bernhardt, W.; Bertog, M.; Amann, K.; Korbmacher, C.; Wiesener, M.; Warnecke, C.; et al. HIF-prolyl hydroxylases in the rat kidney: Physiologic expression patterns and regulation in acute kidney injury. Am. J. Pathol. 2009, 174, 1663–1674. [Google Scholar] [CrossRef] [PubMed]
- Berra, E.; Benizri, E.; Ginouves, A.; Volmat, V.; Roux, D.; Pouyssegur, J. HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1α in normoxia. EMBO J. 2003, 22, 4082–4090. [Google Scholar] [CrossRef] [PubMed]
- Appelhoff, R.J.; Tian, Y.M.; Raval, R.R.; Turley, H.; Harris, A.L.; Pugh, C.W.; Ratcliffe, P.J.; Gleadle, J.M. Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J. Biol. Chem. 2004, 279, 38458–38465. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G., Jr.; Ratcliffe, P.J. Oxygen sensing by metazoans: The central role of the HIF hydroxylase pathway. Mol. Cell 2008, 30, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Mahon, P.C.; Hirota, K.; Semenza, G.L. FIH-1: A novel protein that interacts with HIF-1α and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev. 2001, 15, 2675–2686. [Google Scholar] [CrossRef] [PubMed]
- Lando, D.; Peet, D.J.; Whelan, D.A.; Gorman, J.J.; Whitelaw, M.L. Asparagine hydroxylation of the HIF transactivation domain a hypoxic switch. Science 2002, 295, 858–861. [Google Scholar] [CrossRef] [PubMed]
- Dayan, F.; Roux, D.; Brahimi-Horn, M.C.; Pouyssegur, J.; Mazure, N.M. The oxygen sensor factor-inhibiting hypoxia-inducible factor-1 controls expression of distinct genes through the bifunctional transcriptional character of hypoxia-inducible factor-1α. Cancer Res. 2006, 66, 3688–3698. [Google Scholar] [CrossRef] [PubMed]
- Koivunen, P.; Hirsila, M.; Gunzler, V.; Kivirikko, K.I.; Myllyharju, J. Catalytic properties of the asparaginyl hydroxylase (FIH) in the oxygen sensing pathway are distinct from those of its prolyl 4-hydroxylases. J. Biol. Chem. 2004, 279, 9899–9904. [Google Scholar] [CrossRef] [PubMed]
- Schodel, J.; Bohr, D.; Klanke, B.; Schley, G.; Schlotzer-Schrehardt, U.; Warnecke, C.; Kurtz, A.; Amann, K.; Eckardt, K.U.; Willam, C. Factor inhibiting HIF limits the expression of hypoxia-inducible genes in podocytes and distal tubular cells. Kidney Int. 2010, 78, 857–867. [Google Scholar] [CrossRef] [PubMed]
- Kietzmann, T.; Mennerich, D.; Dimova, E.Y. Hypoxia-Inducible Factors (HIFs) and Phosphorylation: Impact on Stability, Localization, and Transactivity. Front. Cell Dev. Biol. 2016, 4, 11. [Google Scholar] [CrossRef] [PubMed]
- Mennerich, D.; Dimova, E.Y.; Kietzmann, T. Direct phosphorylation events involved in HIF-α regulation: The role of GSK-3beta. Hypoxia 2014, 2, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Dai, W.; Li, C. Polo-like kinase 3, hypoxic responses, and tumorigenesis. Cell Cycle 2017, 16, 2032–2036. [Google Scholar] [CrossRef] [PubMed]
- Bullen, J.W.; Tchernyshyov, I.; Holewinski, R.J.; DeVine, L.; Wu, F.; Venkatraman, V.; Kass, D.L.; Cole, R.N.; Van Eyk, J.; Semenza, G.L. Protein kinase A-dependent phosphorylation stimulates the transcriptional activity of hypoxia-inducible factor 1. Sci. Signal. 2016, 9, ra56. [Google Scholar] [CrossRef] [PubMed]
- Mylonis, I.; Chachami, G.; Samiotaki, M.; Panayotou, G.; Paraskeva, E.; Kalousi, A.; Georgatsou, E.; Bonanou, S.; Simos, G. Identification of MAPK phosphorylation sites and their role in the localization and activity of hypoxia-inducible factor-1α. J. Biol. Chem. 2006, 281, 33095–33106. [Google Scholar] [CrossRef] [PubMed]
- Mylonis, I.; Kourti, M.; Samiotaki, M.; Panayotou, G.; Simos, G. Mortalin-mediated and ERK-controlled targeting of HIF-1α to mitochondria confers resistance to apoptosis under hypoxia. J. Cell Sci. 2017, 130, 466–479. [Google Scholar] [CrossRef] [PubMed]
- Karagiota, A.; Kourti, M.; Simos, G.; Mylonis, I. HIF-1α-derived cell-penetrating peptides inhibit ERK-dependent activation of HIF-1 and trigger apoptosis of cancer cells under hypoxia. Cell. Mol. Life Sci. 2019, 76, 809–825. [Google Scholar] [CrossRef] [PubMed]
- Kalousi, A.; Mylonis, I.; Politou, A.S.; Chachami, G.; Paraskeva, E.; Simos, G. Casein kinase 1 regulates human hypoxia-inducible factor HIF-1. J. Cell Sci. 2010, 123, 2976–2986. [Google Scholar] [CrossRef] [PubMed]
- Pangou, E.; Befani, C.; Mylonis, I.; Samiotaki, M.; Panayotou, G.; Simos, G. HIF-2α phosphorylation by CK1δ promotes erythropoietin secretion in liver cancer cells under hypoxia. J. Cell Sci. 2016, 129, 4213–4226. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.T. Mitochondria and cellular oxygen sensing in the HIF pathway. Biochem. J. 2008, 409, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Kietzmann, T.; Gorlach, A. Reactive oxygen species in the control of hypoxia-inducible factor-mediated gene expression. Semin. Cell Dev. Biol. 2005, 16, 474–486. [Google Scholar] [CrossRef] [PubMed]
- Zou, A.P.; Cowley, A.W., Jr. Reactive oxygen species and molecular regulation of renal oxygenation. Acta Physiol. Scand. 2003, 179, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Gorlach, A.; Kietzmann, T. Superoxide and derived reactive oxygen species in the regulation of hypoxia-inducible factors. Methods Enzymol. 2007, 435, 421–446. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-inducible factors: Coupling glucose metabolism and redox regulation with induction of the breast cancer stem cell phenotype. EMBO J. 2017, 36, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Lacher, S.E.; Levings, D.C.; Freeman, S.; Slattery, M. Identification of a functional antioxidant response element at the HIF1A locus. Redox Biol. 2018, 19, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Gorlach, A.; Dimova, E.Y.; Petry, A.; Martinez-Ruiz, A.; Hernansanz-Agustin, P.; Rolo, A.P.; Palmeira, C.M.; Kietzmann, T. Reactive oxygen species, nutrition, hypoxia and diseases: Problems solved? Redox Biol. 2015, 6, 372–385. [Google Scholar] [CrossRef] [PubMed]
- Dehne, N.; Brune, B. Sensors, transmitters, and targets in mitochondrial oxygen shortage-a hypoxia-inducible factor relay story. Antioxid. Redox Signal 2014, 20, 339–352. [Google Scholar] [CrossRef] [PubMed]
- Shvetsova, A.N.; Mennerich, D.; Keratar, J.M.; Hiltunen, J.K.; Kietzmann, T. Non-electron transfer chain mitochondrial defects differently regulate HIF-1α degradation and transcription. Redox Biol. 2017, 12, 1052–1061. [Google Scholar] [CrossRef] [PubMed]
- Bonello, S.; Zahringer, C.; BelAiba, R.S.; Djordjevic, T.; Hess, J.; Michiels, C.; Kietzmann, T.; Gorlach, A. Reactive oxygen species activate the HIF-1α promoter via a functional NFkappaB site. Arterioscler. Thromb. Vas. Biol. 2007, 27, 755–761. [Google Scholar] [CrossRef] [PubMed]
- Al Taleb, Z.; Petry, A.; Chi, T.F.; Mennerich, D.; Gorlach, A.; Dimova, E.Y.; Kietzmann, T. Differential transcriptional regulation of hypoxia-inducible factor-1α by arsenite under normoxia and hypoxia: Involvement of Nrf2. J. Mol. Med. 2016, 94, 1153–1166. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, J.S.; Jung, Y.J.; Mimnaugh, E.G.; Martinez, A.; Cuttitta, F.; Neckers, L.M. Hsp90 regulates a von Hippel Lindau-independent hypoxia-inducible factor-1 α-degradative pathway. J. Biol. Chem. 2002, 277, 29936–29944. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Lin, Z.; Liang, D.; Fath, D.; Sang, N.; Caro, J. Histone deacetylase inhibitors induce VHL and ubiquitin-independent proteasomal degradation of hypoxia-inducible factor 1α. Mol. Cell. Biol. 2006, 26, 2019–2028. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.V.; Baek, J.H.; Zhang, H.; Diez, R.; Cole, R.N.; Semenza, G.L. RACK1 competes with HSP90 for binding to HIF-1α and is required for O2-independent and HSP90 inhibitor-induced degradation of HIF-1α. Mol. Cell 2007, 25, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Sonveaux, P.; Rabbani, Z.N.; Liu, S.; Yan, B.; Huang, Q.; Vujaskovic, Z.; Dewhirst, M.W.; Li, C.Y. Regulation of HIF-1α stability through S-nitrosylation. Mol. Cell 2007, 26, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Kang, X.; Zhang, S.; Yeh, E.T. SUMO-specific protease 1 is essential for stabilization of HIF1α during hypoxia. Cell 2007, 131, 584–595. [Google Scholar] [CrossRef] [PubMed]
- Minamishima, Y.A.; Kaelin, W.G., Jr. Reactivation of hepatic EPO synthesis in mice after PHD loss. Science 2010, 329, 407. [Google Scholar] [CrossRef] [PubMed]
- Wiesener, M.S.; Jurgensen, J.S.; Rosenberger, C.; Scholze, C.K.; Horstrup, J.H.; Warnecke, C.; Mandriota, S.; Bechmann, I.; Frei, U.A.; Pugh, C.W.; et al. Widespread hypoxia-inducible expression of HIF-2α in distinct cell populations of different organs. FASEB J. 2003, 17, 271–273. [Google Scholar] [CrossRef] [PubMed]
- Rosenberger, C.; Mandriota, S.; Jurgensen, J.S.; Wiesener, M.S.; Horstrup, J.H.; Frei, U.; Ratcliffe, P.J.; Maxwell, P.H.; Bachmann, S.; Eckardt, K.U. Expression of hypoxia-inducible factor-1α and -2α in hypoxic and ischemic rat kidneys. J. Am. Soc. Nephrol. 2002, 13, 1721–1732. [Google Scholar] [CrossRef] [PubMed]
- Koh, M.Y.; Powis, G. Passing the baton: The HIF switch. Trends Biochem. Sci. 2012, 37, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Bartoszewska, S.; Kochan, K.; Piotrowski, A.; Kamysz, W.; Ochocka, R.J.; Collawn, J.F.; Bartoszewski, R. The hypoxia-inducible miR-429 regulates hypoxia-inducible factor-1α expression in human endothelial cells through a negative feedback loop. FASEB J. 2015, 29, 1467–1479. [Google Scholar] [CrossRef] [PubMed]
- Holmquist-Mengelbier, L.; Fredlund, E.; Lofstedt, T.; Noguera, R.; Navarro, S.; Nilsson, H.; Pietras, A.; Vallon-Christersson, J.; Borg, A.; Gradin, K.; et al. Recruitment of HIF-1α and HIF-2α to common target genes is differentially regulated in neuroblastoma: HIF-2α promotes an aggressive phenotype. Cancer Cell 2006, 10, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Uchida, T.; Rossignol, F.; Matthay, M.A.; Mounier, R.; Couette, S.; Clottes, E.; Clerici, C. Prolonged hypoxia differentially regulates hypoxia-inducible factor (HIF)-1α and HIF-2α expression in lung epithelial cells: Implication of natural antisense HIF-1α. J. Biol. Chem. 2004, 279, 14871–14878. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.R.; Dai, A.G.; Hu, R.C. [Reciprocal regulation between hypoxia-inducible factor-1α and its prolyl hydroxylases in hypoxic pulmonary hypertension rats]. Chin. J. Tuberc. Respir. Dis. 2006, 29, 668–673. [Google Scholar]
- Foxler, D.E.; Bridge, K.S. A HIF-LIMD1 negative feedback mechanism mitigates the pro-tumorigenic effects of hypoxia. EMBO Mol. Med. 2018, 10, e8304. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, R.; Zhang, H.; Kim, J.W.; Shimoda, L.; Dang, C.V.; Semenza, G.L. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 2007, 129, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Thompson, C.B. Into Thin Air: How We Sense and Respond to Hypoxia. Cell 2016, 167, 9–11. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wei, Q.; Guo, C.; Dong, G.; Liu, Y.; Tang, C.; Dong, Z. Hypoxia, HIF, and Associated Signaling Networks in Chronic Kidney Disease. Int. J. Mol. Sci. 2017, 18, 950. [Google Scholar] [CrossRef] [PubMed]
- Skuli, N.; Liu, L.; Runge, A.; Wang, T.; Yuan, L.; Patel, S.; Iruela-Arispe, L.; Simon, M.C.; Keith, B. Endothelial deletion of hypoxia-inducible factor-2α (HIF-2α) alters vascular function and tumor angiogenesis. Blood 2009, 114, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Skuli, N.; Majmundar, A.J.; Krock, B.L.; Mesquita, R.C.; Mathew, L.K.; Quinn, Z.L.; Runge, A.; Liu, L.; Kim, M.N.; Liang, J.; et al. Endothelial HIF-2α regulates murine pathological angiogenesis and revascularization processes. J. Clin. Investig. 2012, 122, 1427–1443. [Google Scholar] [CrossRef] [PubMed]
- Duan, C. Hypoxia-inducible factor 3 biology: Complexities and emerging themes. Am. J. Physiol. Cell Physiol. 2016, 310, C260–C269. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Wiesener, M.; Bernhardt, W.; Eckardt, K.U.; Warnecke, C. The human HIF (hypoxia-inducible factor)-3α gene is a HIF-1 target gene and may modulate hypoxic gene induction. Biochem. J. 2009, 424, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Heyman, S.N.; Rosen, S.; Rosenberger, C. Hypoxia-inducible factors and the prevention of acute organ injury. Crit. Care 2011, 15, 209. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.J.; Wang, L.Y.; Chodosh, L.A.; Keith, B.; Simon, M.C. Differential roles of hypoxia-inducible factor 1α (HIF-1α) and HIF-2α in hypoxic gene regulation. Mol. Cell. Biol. 2003, 23, 9361–9374. [Google Scholar] [CrossRef] [PubMed]
- Morita, M.; Ohneda, O.; Yamashita, T.; Takahashi, S.; Suzuki, N.; Nakajima, O.; Kawauchi, S.; Ema, M.; Shibahara, S.; Udono, T.; et al. HLF/HIF-2α is a key factor in retinopathy of prematurity in association with erythropoietin. EMBO J. 2003, 22, 1134–1146. [Google Scholar] [CrossRef] [PubMed]
- Rankin, E.B.; Higgins, D.F.; Walisser, J.A.; Johnson, R.S.; Bradfield, C.A.; Haase, V.H. Inactivation of the arylhydrocarbon receptor nuclear translocator (Arnt) suppresses von Hippel-Lindau disease-associated vascular tumors in mice. Mol. Cell. Biol. 2005, 25, 3163–3172. [Google Scholar] [CrossRef] [PubMed]
- Warnecke, C.; Zaborowska, Z.; Kurreck, J.; Erdmann, V.A.; Frei, U.; Wiesener, M.; Eckardt, K.U. Differentiating the functional role of hypoxia-inducible factor (HIF)-1α and HIF-2α (EPAS-1) by the use of RNA interference: Erythropoietin is a HIF-2α target gene in Hep3B and Kelly cells. FASEB J. 2004, 18, 1462–1464. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.L.; Semenza, G.L. General involvement of hypoxia-inducible factor 1 in transcriptional response to hypoxia. Proc. Natl. Acad. Sci. USA 1993, 90, 4304–4308. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L.; Wang, G.L. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol. Cell. Biol. 1992, 12, 5447–5454. [Google Scholar] [CrossRef] [PubMed]
- Fandrey, J. Oxygen-dependent and tissue-specific regulation of erythropoietin gene expression. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 286, R977–R988. [Google Scholar] [CrossRef] [PubMed]
- Jelkmann, W. Molecular biology of erythropoietin. Int. Med. 2004, 43, 649–659. [Google Scholar] [CrossRef]
- Scortegagna, M.; Ding, K.; Zhang, Q.; Oktay, Y.; Bennett, M.J.; Bennett, M.; Shelton, J.M.; Richardson, J.A.; Moe, O.; Garcia, J.A. HIF-2α regulates murine hematopoietic development in an erythropoietin-dependent manner. Blood 2005, 105, 3133–3140. [Google Scholar] [CrossRef] [PubMed]
- Vesey, D.A.; Cheung, C.; Pat, B.; Endre, Z.; Gobe, G.; Johnson, D.W. Erythropoietin protects against ischaemic acute renal injury. Nephrol. Dial. Transplant. 2004, 19, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Forsythe, J.A.; Jiang, B.H.; Iyer, N.V.; Agani, F.; Leung, S.W.; Koos, R.D.; Semenza, G.L. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell. Biol. 1996, 16, 4604–4613. [Google Scholar] [CrossRef] [PubMed]
- Markovic-Mueller, S.; Stuttfeld, E.; Asthana, M.; Weinert, T.; Bliven, S.; Goldie, K.N.; Kisko, K.; Capitani, G.; Ballmer-Hofer, K. Structure of the Full-length VEGFR-1 Extracellular Domain in Complex with VEGF-A. Structure 2017, 25, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Keir, L.S.; Firth, R.; Aponik, L.; Feitelberg, D.; Sakimoto, S.; Aguilar, E.; Welsh, G.I.; Richards, A.; Usui, Y.; Satchell, S.C.; et al. VEGF regulates local inhibitory complement proteins in the eye and kidney. J. Clin. Investig. 2017, 127, 199–214. [Google Scholar] [CrossRef] [PubMed]
- Eremina, V.; Sood, M.; Haigh, J.; Nagy, A.; Lajoie, G.; Ferrara, N.; Gerber, H.P.; Kikkawa, Y.; Miner, J.H.; Quaggin, S.E. Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal diseases. J. Clin. Investig. 2003, 111, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Eremina, V.; Jefferson, J.A.; Kowalewska, J.; Hochster, H.; Haas, M.; Weisstuch, J.; Richardson, C.; Kopp, J.B.; Kabir, M.G.; Backx, P.H.; et al. VEGF inhibition and renal thrombotic microangiopathy. N. Engl. J. Med. 2008, 358, 1129–1136. [Google Scholar] [CrossRef] [PubMed]
- Bastin, A.J.; Ostermann, M.; Slack, A.J.; Diller, G.P.; Finney, S.J.; Evans, T.W. Acute kidney injury after cardiac surgery according to Risk/Injury/Failure/Loss/End-stage, Acute Kidney Injury Network, and Kidney Disease: Improving Global Outcomes classifications. J. Crit. Care 2013, 28, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Kriegel, A.J.; Liu, Y.; Usa, K.; Mladinov, D.; Liu, H.; Fang, Y.; Ding, X.; Liang, M. Delayed ischemic preconditioning contributes to renal protection by upregulation of miR-21. Kidney Int. 2012, 82, 1167–1175. [Google Scholar] [CrossRef] [PubMed]
- Bernhardt, W.M.; Campean, V.; Kany, S.; Jurgensen, J.S.; Weidemann, A.; Warnecke, C.; Arend, M.; Klaus, S.; Gunzler, V.; Amann, K.; et al. Preconditional activation of hypoxia-inducible factors ameliorates ischemic acute renal failure. J. Am. Soc. Nephrol. 2006, 17, 1970–1978. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, M.; Makino, Y.; Tanaka, T.; Tanaka, H.; Ishizaka, N.; Noiri, E.; Fujita, T.; Nangaku, M. Induction of renoprotective gene expression by cobalt ameliorates ischemic injury of the kidney in rats. J. Am. Soc. Nephrol. 2003, 14, 1825–1832. [Google Scholar] [CrossRef] [PubMed]
- Hill, P.; Shukla, D.; Tran, M.G.; Aragones, J.; Cook, H.T.; Carmeliet, P.; Maxwell, P.H. Inhibition of hypoxia inducible factor hydroxylases protects against renal ischemia-reperfusion injury. J. Am. Soc. Nephrol. 2008, 19, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Schley, G.; Turkoglu, G.; Burzlaff, N.; Amann, K.U.; Willam, C.; Eckardt, K.U.; Bernhardt, W.M. The protective effect of prolyl-hydroxylase inhibition against renal ischaemia requires application prior to ischaemia but is superior to EPO treatment. Nephrol. Dial. Transplant. 2012, 27, 929–936. [Google Scholar] [CrossRef] [PubMed]
- Kapitsinou, P.P.; Jaffe, J.; Michael, M.; Swan, C.E.; Duffy, K.J.; Erickson-Miller, C.L.; Haase, V.H. Preischemic targeting of HIF prolyl hydroxylation inhibits fibrosis associated with acute kidney injury. Am. J. Physiol. Ren. Physiol. 2012, 302, F1172–F1179. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Song, N.; Zhang, X.; Jiao, X.; Hu, J.; Liang, M.; Teng, J.; Ding, X. Renal Protection Mediated by Hypoxia Inducible Factor-1α Depends on Proangiogenesis Function of miR-21 by Targeting Thrombospondin 1. Transplantation 2017, 101, 1811–1819. [Google Scholar] [CrossRef] [PubMed]
- Jamadarkhana, P.; Chaudhary, A.; Chhipa, L.; Dubey, A.; Mohanan, A.; Gupta, R.; Deshpande, S. Treatment with a novel hypoxia-inducible factor hydroxylase inhibitor (TRC160334) ameliorates ischemic acute kidney injury. Am. J. Nephrol. 2012, 36, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Bi, L.Y.; Zhao, D.A.; Yang, D.S.; Guo, J.G.; Liang, B.; Zhang, R.X.; Zhao, J.L.; Bai, H.T.; Li, S.J. Effects of autologous SCF- and G-CSF-mobilized bone marrow stem cells on hypoxia-inducible factor-1 in rats with ischemia-reperfusion renal injury. Genet. Mol. Res. 2015, 14, 4102–4112. [Google Scholar] [CrossRef] [PubMed]
- Conde, E.; Gimenez-Moyano, S.; Martin-Gomez, L.; Rodriguez, M.; Ramos, M.E.; Aguado-Fraile, E.; Blanco-Sanchez, I.; Saiz, A.; Garcia-Bermejo, M.L. HIF-1α induction during reperfusion avoids maladaptive repair after renal ischemia/reperfusion involving miR127-3p. Sci. Rep. 2017, 7, 41099. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Huang, H.; Cheng, J.; Liu, J.; Zhao, H.; Vizcaychipi, M.P.; Ma, D. Pre-treatment with isoflurane ameliorates renal ischemic-reperfusion injury in mice. Life Sci. 2011, 88, 1102–1107. [Google Scholar] [CrossRef] [PubMed]
- Kojima, I.; Tanaka, T.; Inagi, R.; Kato, H.; Yamashita, T.; Sakiyama, A.; Ohneda, O.; Takeda, N.; Sata, M.; Miyata, T.; et al. Protective role of hypoxia-inducible factor-2α against ischemic damage and oxidative stress in the kidney. J. Am. Soc. Nephrol. 2007, 18, 1218–1226. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Liu, Y.; Liu, P.; Hao, J.; Liang, M.; Mi, Q.S.; Chen, J.K.; Dong, Z. MicroRNA-489 Induction by Hypoxia-Inducible Factor-1 Protects against Ischemic Kidney Injury. J. Am. Soc. Nephrol. 2016, 27, 2784–2796. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Sun, H.; Song, S.; Liu, Y.; Liu, P.; Livingston, M.J.; Wang, J.; Liang, M.; Mi, Q.S.; Huo, Y.; et al. MicroRNA-668 represses MTP18 to preserve mitochondrial dynamics in ischemic acute kidney injury. J. Clin. Investig. 2018, 128, 5448–5464. [Google Scholar] [CrossRef] [PubMed]
- Song, N.; Zhang, T.; Xu, X.; Lu, Z.; Yu, X.; Fang, Y.; Hu, J.; Jia, P.; Teng, J.; Ding, X. miR-21 Protects Against Ischemia/Reperfusion-Induced Acute Kidney Injury by Preventing Epithelial Cell Apoptosis and Inhibiting Dendritic Cell Maturation. Front. Physiol. 2018, 9, 790. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, K.; Wei, Q.; Pabla, N.; Dong, G.; Mi, Q.S.; Liang, M.; Mei, C.; Dong, Z. MicroRNA-687 Induced by Hypoxia-Inducible Factor-1 Targets Phosphatase and Tensin Homolog in Renal Ischemia-Reperfusion Injury. J. Am. Soc. Nephrol. 2015, 26, 1588–1596. [Google Scholar] [CrossRef] [PubMed]
- Mathia, S.; Rudigier, L.J.; Kasim, M.; Kirschner, K.M. A dual role of miR-22 in rhabdomyolysis-induced acute kidney injury. Acta Physiol. 2018, 224, e13102. [Google Scholar] [CrossRef] [PubMed]
- Pabla, N.; Dong, Z. Cisplatin nephrotoxicity: Mechanisms and renoprotective strategies. Kidney Int. 2008, 73, 994–1007. [Google Scholar] [CrossRef] [PubMed]
- Winston, J.A.; Safirstein, R. Reduced renal blood flow in early cisplatin-induced acute renal failure in the rat. Am. J. Physiol. 1985, 249, F490–F496. [Google Scholar] [CrossRef] [PubMed]
- Weidemann, A.; Bernhardt, W.M.; Klanke, B.; Daniel, C.; Buchholz, B.; Campean, V.; Amann, K.; Warnecke, C.; Wiesener, M.S.; Eckardt, K.U.; et al. HIF activation protects from acute kidney injury. J. Am. Soc. Nephrol. 2008, 19, 486–494. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Kojima, I.; Ohse, T.; Inagi, R.; Miyata, T.; Ingelfinger, J.R.; Fujita, T.; Nangaku, M. Hypoxia-inducible factor modulates tubular cell survival in cisplatin nephrotoxicity. Am. J. Physiol. Ren. Physiol. 2005, 289, F1123–F1133. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.W.; Li, Z.Z.; Wang, W.; Jiang, Y.; Cheng, J.; Lu, S.; Zhang, J.Y. Enhanced renoprotective effect of HIF-1α modified human adipose-derived stem cells on cisplatin-induced acute kidney injury in vivo. Sci. Rep. 2015, 5, 10851. [Google Scholar] [CrossRef] [PubMed]
- Bao, H.; Ge, Y.; Wang, Z.; Zhuang, S.; Dworkin, L.; Peng, A.; Gong, R. Delayed administration of a single dose of lithium promotes recovery from AKI. J. Am. Soc. Nephrol. 2014, 25, 488–500. [Google Scholar] [CrossRef] [PubMed]
- Doi, K. Role of kidney injury in sepsis. J. Intensive Care 2016, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.C.; Ma, M.C.; Chien, C.T.; Wu, M.S.; Sun, W.K.; Chen, C.F. Hypoxic preconditioning attenuates lipopolysaccharide-induced oxidative stress in rat kidneys. J. Physiol. 2007, 582, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Schaalan, M.F.; Mohamed, W.A. Determinants of hepcidin levels in sepsis-associated acute kidney injury: Impact on pAKT/PTEN pathways? J. Immunotoxicol. 2016, 13, 751–757. [Google Scholar] [CrossRef] [PubMed]
- He, K.; Chen, X.; Han, C.; Xu, L.; Zhang, J.; Zhang, M.; Xia, Q. Lipopolysaccharide-induced cross-tolerance against renal ischemia-reperfusion injury is mediated by hypoxia-inducible factor-2α-regulated nitric oxide production. Kidney Int. 2014, 85, 276–288. [Google Scholar] [CrossRef] [PubMed]
- Stoyanoff, T.R.; Rodriguez, J.P.; Todaro, J.S.; Colavita, J.P.M.; Torres, A.M.; Aguirre, M.V. Erythropoietin attenuates LPS-induced microvascular damage in a murine model of septic acute kidney injury. Biomed. Pharmacother. 2018, 107, 1046–1055. [Google Scholar] [CrossRef] [PubMed]
- Ogura, Y.; Jesmin, S.; Yamaguchi, N.; Oki, M.; Shimojo, N.; Islam, M.M.; Khatun, T.; Kamiyama, J.; Sakuramoto, H.; Hagiya, K.; et al. Potential amelioration of upregulated renal HIF-1α-endothelin-1 system by landiolol hydrochloride in a rat model of endotoxemia. Life Sci. 2014, 118, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.M.; You, S.J.; Lee, Y.M.; Oh, S.W.; Ahn, S.Y.; Kim, S.; Chin, H.J.; Chae, D.W.; Na, K.Y. Hypoxia-inducible factor activation protects the kidney from gentamicin-induced acute injury. PLoS ONE 2012, 7, e48952. [Google Scholar] [CrossRef] [PubMed]
- Rosenberger, C.; Heyman, S.N.; Rosen, S.; Shina, A.; Goldfarb, M.; Griethe, W.; Frei, U.; Reinke, P.; Bachmann, S.; Eckardt, K.U. Up-regulation of HIF in experimental acute renal failure: Evidence for a protective transcriptional response to hypoxia. Kidney Int. 2005, 67, 531–542. [Google Scholar] [CrossRef] [PubMed]
- Heyman, S.N.; Reichman, J.; Brezis, M. Pathophysiology of radiocontrast nephropathy: A role for medullary hypoxia. Investig. Radiol. 1999, 34, 685–691. [Google Scholar] [CrossRef]
- Heyman, S.N.; Rosenberger, C.; Rosen, S. Regional alterations in renal haemodynamics and oxygenation: A role in contrast medium-induced nephropathy. Nephrol. Dial. Transplant. 2005, 20 (Suppl. 1), i6–i11. [Google Scholar] [CrossRef]
- Grgic, I.; Campanholle, G.; Bijol, V.; Wang, C.; Sabbisetti, V.S.; Ichimura, T.; Humphreys, B.D.; Bonventre, J.V. Targeted proximal tubule injury triggers interstitial fibrosis and glomerulosclerosis. Kidney Int. 2012, 82, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Shu, S.; Zhu, J.; Liu, Z.; Tang, C.; Cai, J.; Dong, Z. Endoplasmic reticulum stress is activated in post-ischemic kidneys to promote chronic kidney disease. EBioMedicine 2018, 37, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Cleveland, R.P.; Koch, C.J.; Schelling, J.R. Hypoxia induces renal tubular epithelial cell apoptosis in chronic renal disease. Lab. Investig. 1999, 79, 1089–1099. [Google Scholar] [PubMed]
- Tanaka, T.; Hanafusa, N.; Ingelfinger, J.R.; Ohse, T.; Fujita, T.; Nangaku, M. Hypoxia induces apoptosis in SV40-immortalized rat proximal tubular cells through the mitochondrial pathways, devoid of HIF1-mediated upregulation of Bax. Biochem. Biophys. Res. Commun. 2003, 309, 222–231. [Google Scholar] [CrossRef]
- Manotham, K.; Tanaka, T.; Matsumoto, M.; Ohse, T.; Inagi, R.; Miyata, T.; Kurokawa, K.; Fujita, T.; Ingelfinger, J.R.; Nangaku, M. Transdifferentiation of cultured tubular cells induced by hypoxia. Kidney Int. 2004, 65, 871–880. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.L.; Lv, L.L.; Tang, T.T.; Wang, B.; Feng, Y.; Zhou, L.T.; Cao, J.Y.; Tang, R.N.; Wu, M.; Liu, H.; et al. HIF-1α inducing exosomal microRNA-23a expression mediates the cross-talk between tubular epithelial cells and macrophages in tubulointerstitial inflammation. Kidney Int. 2018, 95, 388–404. [Google Scholar] [CrossRef] [PubMed]
- Lendahl, U.; Lee, K.L.; Yang, H.; Poellinger, L. Generating specificity and diversity in the transcriptional response to hypoxia. Nat. Rev. Genet. 2009, 10, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T. Expanding roles of the hypoxia-response network in chronic kidney disease. Clin. Exp. Nephrol. 2016, 20, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Zuk, A.; Bonventre, J.V. Acute Kidney Injury. Ann. Rev. Med. 2016, 67, 293–307. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Miyata, T.; Inagi, R.; Kurokawa, K.; Adler, S.; Fujita, T.; Nangaku, M. Hypoxia-induced apoptosis in cultured glomerular endothelial cells: Involvement of mitochondrial pathways. Kidney Int. 2003, 64, 2020–2032. [Google Scholar] [CrossRef] [PubMed]
- Sendoel, A.; Hengartner, M.O. Apoptotic cell death under hypoxia. Physiology 2014, 29, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Matsumoto, M.; Inagi, R.; Miyata, T.; Kojima, I.; Ohse, T.; Fujita, T.; Nangaku, M. Induction of protective genes by cobalt ameliorates tubulointerstitial injury in the progressive Thy1 nephritis. Kidney Int. 2005, 68, 2714–2725. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Kojima, I.; Ohse, T.; Ingelfinger, J.R.; Adler, S.; Fujita, T.; Nangaku, M. Cobalt promotes angiogenesis via hypoxia-inducible factor and protects tubulointerstitium in the remnant kidney model. Lab. Investig. 2005, 85, 1292–1307. [Google Scholar] [CrossRef] [PubMed]
- Bonventre, J.V. Maladaptive proximal tubule repair: Cell cycle arrest. Nephron Clin. Pract. 2014, 127, 61–64. [Google Scholar] [CrossRef] [PubMed]
- Geng, H.; Lan, R.; Wang, G.; Siddiqi, A.R.; Naski, M.C.; Brooks, A.I.; Barnes, J.L.; Saikumar, P.; Weinberg, J.M.; Venkatachalam, M.A. Inhibition of autoregulated TGFbeta signaling simultaneously enhances proliferation and differentiation of kidney epithelium and promotes repair following renal ischemia. Am. J. Pathol. 2009, 174, 1291–1308. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Besschetnova, T.Y.; Brooks, C.R.; Shah, J.V.; Bonventre, J.V. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat. Med. 2010, 16, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Witzgall, R.; Brown, D.; Schwarz, C.; Bonventre, J.V. Localization of proliferating cell nuclear antigen, vimentin, c-Fos, and clusterin in the postischemic kidney. Evidence for a heterogenous genetic response among nephron segments, and a large pool of mitotically active and dedifferentiated cells. J. Clin. Investig. 1994, 93, 2175–2188. [Google Scholar] [CrossRef] [PubMed]
- Witzgall, R.; O’Leary, E.; Gessner, R.; Ouellette, A.J.; Bonventre, J.V. Kid-1, a putative renal transcription factor: Regulation during ontogeny and in response to ischemia and toxic injury. Mol. Cell. Biol. 1993, 13, 1933–1942. [Google Scholar] [CrossRef] [PubMed]
- Ishibe, S.; Cantley, L.G. Epithelial-mesenchymal-epithelial cycling in kidney repair. Curr. Opin. Nephrol. Hypertens. 2008, 17, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Qing, G.; Skuli, N.; Mayes, P.A.; Pawel, B.; Martinez, D.; Maris, J.M.; Simon, M.C. Combinatorial regulation of neuroblastoma tumor progression by N-Myc and hypoxia inducible factor HIF-1α. Cancer Res. 2010, 70, 10351–10361. [Google Scholar] [CrossRef] [PubMed]
- Moore, E.; Bellomo, R. Erythropoietin (EPO) in acute kidney injury. Ann. Intensive Care 2011, 1, 3. [Google Scholar] [CrossRef] [PubMed]
- Ceradini, D.J.; Kulkarni, A.R.; Callaghan, M.J.; Tepper, O.M.; Bastidas, N.; Kleinman, M.E.; Capla, J.M.; Galiano, R.D.; Levine, J.P.; Gurtner, G.C. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat. Med. 2004, 10, 858–864. [Google Scholar] [CrossRef] [PubMed]
- Koshiji, M.; Huang, L.E. Dynamic balancing of the dual nature of HIF-1α for cell survival. Cell Cycle 2004, 3, 853–854. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Vaidya, M. Hypoxia inhibits mesenchymal stem cell proliferation through HIF1α-dependent regulation of P27. Mol. Cell. Biochem. 2016, 415, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhu, Q.; Li, P.L.; Dhaduk, R.; Zhang, F.; Gehr, T.W.; Li, N. Silencing of hypoxia-inducible factor-1α gene attenuates chronic ischemic renal injury in two-kidney, one-clip rats. Am. J. Physiol. Ren. Physiol. 2014, 306, F1236–F1242. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.R.; You, S.J.; Lee, Y.M.; Chin, H.J.; Chae, D.W.; Oh, Y.K.; Joo, K.W.; Han, J.S.; Na, K.Y. Activation of hypoxia-inducible factor attenuates renal injury in rat remnant kidney. Nephrol. Dial. Transplant. 2010, 25, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Fang, Y.; Liu, H.; Zhu, J.; Zou, J.; Xu, X.; Jiang, S.; Ding, X. The balance of beneficial and deleterious effects of hypoxia-inducible factor activation by prolyl hydroxylase inhibitor in rat remnant kidney depends on the timing of administration. Nephrol. Dial. Transplant. 2012, 27, 3110–3119. [Google Scholar] [CrossRef] [PubMed]
- Ohtomo, S.; Nangaku, M.; Izuhara, Y.; Takizawa, S.; Strihou, C.; Miyata, T. Cobalt ameliorates renal injury in an obese, hypertensive type 2 diabetes rat model. Nephrol. Dial. Transplant. 2008, 23, 1166–1172. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Gilbert, V.; Liu, Q.; Kapitsinou, P.P.; Unger, T.L.; Rha, J.; Rivella, S.; Schlondorff, D.; Haase, V.H. Myeloid cell-derived hypoxia-inducible factor attenuates inflammation in unilateral ureteral obstruction-induced kidney injury. J. Immunol. 2012, 188, 5106–5115. [Google Scholar] [CrossRef] [PubMed]
- Kimura, K.; Iwano, M.; Higgins, D.F.; Yamaguchi, Y.; Nakatani, K.; Harada, K.; Kubo, A.; Akai, Y.; Rankin, E.B.; Neilson, E.G.; et al. Stable expression of HIF-1α in tubular epithelial cells promotes interstitial fibrosis. Am. J. Physiol. Ren. Physiol. 2008, 295, F1023–F1029. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Wang, Z.; Xia, M.; Li, P.L.; Van Tassell, B.W.; Abbate, A.; Dhaduk, R.; Li, N. Silencing of hypoxia-inducible factor-1α gene attenuated angiotensin II-induced renal injury in Sprague-Dawley rats. Hypertension 2011, 58, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Luo, G.; Fang, Q.; Sun, Z. Stable expression of hypoxia-inducible factor-1α in human renal proximal tubular epithelial cells promotes epithelial to mesenchymal transition. Transplant. Proc. 2014, 46, 130–134. [Google Scholar] [CrossRef] [PubMed]
- Kong, T.; Eltzschig, H.K.; Karhausen, J.; Colgan, S.P.; Shelley, C.S. Leukocyte adhesion during hypoxia is mediated by HIF-1-dependent induction of beta2 integrin gene expression. Proc. Natl. Acad. Sci. USA 2004, 101, 10440–10445. [Google Scholar] [CrossRef] [PubMed]
- Cramer, T.; Yamanishi, Y.; Clausen, B.E.; Forster, I.; Pawlinski, R.; Mackman, N.; Haase, V.H.; Jaenisch, R.; Corr, M.; Nizet, V.; et al. HIF-1α is essential for myeloid cell-mediated inflammation. Cell 2003, 112, 645–657. [Google Scholar] [CrossRef]
- Yamaguchi, J.; Tanaka, T.; Eto, N.; Nangaku, M. Inflammation and hypoxia linked to renal injury by CCAAT/enhancer-binding protein δ. Kidney Int. 2015, 88, 262–275. [Google Scholar] [CrossRef] [PubMed]
- Anders, H.J.; Ryu, M. Renal microenvironments and macrophage phenotypes determine progression or resolution of renal inflammation and fibrosis. Kidney Int. 2011, 80, 915–925. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| AKI Model | Approach for HIF Activation/Inhibition | Which HIF Was Activated/Inhibited | Effects on Kidney Injury | Mechanisms | References |
|---|---|---|---|---|---|
| IRI in mice | 15 min renal ischemic pre-conditioning | HIF-1 was activated | Attenuate AKI | Increasing the expression of miR-21 | [95] |
| uIRI in rat | Carbon monoxide | HIF-1 and HIF-2 were activated | Attenuate AKI | Alleviating apoptosis and macrophage infiltration | [96] |
| uIRI in mice | PHD inhibitor | HIF-1 and HIF-2 were activated | Attenuate AKI | Alleviating apoptosis and macrophage infiltration | [98] |
| IRI in rat | PHD inhibitor | HIF-1 and HIF-2 were activated | Attenuate AKI | Upregulating HIF target genes, including EPO | [99] |
| uIRI in mice | PHD inhibitor | HIF-1 and HIF-2 were activated | Attenuate AKI | Reducing VCAM1 | [21] |
| IRI in mice | PHD inhibitor | HIF-1 and HIF-2 were activated | Attenuate AKI and renal fibrosis | Reducing inflammation | [100] |
| uIRI in rat | Cobalt chloride | HIF-1 was activated | Attenuate AKI | Inducing renoprotective gene expression | [97] |
| IRI in mice | Cobalt chloride | HIF-1 was activated | Attenuate AKI | Upregulating VEGF and miR-21 | [101] |
| IRI in rat | HIF-1α siRNA | HIF-1 was inhibited | Aggravate AKI | [11] | |
| IRI in mice | HIF-1α(+/−) or HIF-2α(+/−) mice | HIF-1 or HIF-2 was inhibited | Aggravate AKI | [98] | |
| IRI in mice | HIF-2α knockdown mice | HIF-2 was inhibited | Aggravate AKI | Enhancing oxidative stress | [106] |
| uIRI in mice | EC-specific PHD2−/− mice | Endothelial HIF was activated | Attenuate kidney injury | Reducing VCAM1 | [21] |
| uIRI in mice | EC-specific HIF2α−/− mice with PHD inhibitor | HIF-1 was activated | Ineffective in attenuating AKI | [21] | |
| Cisplatin-AKI in rat | Cobalt | HIF-1 was activated | Attenuate AKI | Inhibiting mitochondrial signaling pathways | [115] |
| Cisplatin-AKI in mice | PHD inhibitor | HIF-1 was activated | Attenuate AKI | Upregulating HIF target genes | [22] |
| LPS-AKI in rat | Landiolol hydrochloride | Ameliorate the upregulation of HIF-1 | Attenuate AKI | Normalizing inflammatory cytokines | [123] |
| Rhabdomyolysis-AKI in mice | Pax8-rtTA–based inducible VHL-KO | Renal tubules HIF was activated | Attenuate AKI | Metabolic sHIFt toward anaerobic energy metabolism | [23] |
| Gentamicin-AKI in rat | Cobalt | HIF-1 was activated | Attenuate AKI | Reducing apoptosis and macrophage infiltration | [124] |
| Multi-insult-AKI in rat(contrast medium, NOS inhibitor, and COX inhibitor) | Furosemide | HIF-1 was activated | Attenuate AKI | Upregulating HO-1 | [125] |
| AKI Model | Approach for HIF Activation/Inhibition | Which HIF was Activated/Inhibited | Effects on Kidney Repair | Mechanisms | References |
|---|---|---|---|---|---|
| IRI in rat | PHD inhibitor | HIF-1 was activated | Attenuate AKI | Inducing HSP70 | [102] |
| IRI in rat | SCF and G-CSF | HIF-1 was activated | Attenuate AKI | Upregulating VEGF and EPO | [103] |
| IRI in rat | HIF-1α siRNA | HIF-1 was inhibited | Aggravate AKI and renal fibrosis | Downregulating miR-127-3p and inducing its target gene Bcl6 | [104] |
| IRI in rat | PHD inhibitor | HIF-1 and HIF-2 were activated | Ineffective in attenuating AKI | [99] | |
| IRI in mice | PHD inhibitor | HIF-1 and HIF-2 were activated | Ineffective in attenuating AKI and renal fibrosis | [100] | |
| IRI in rat | HIF-1α siRNA | HIF-1 was inhibited | Aggravate AKI | [11] | |
| uIRI in mice | EC-specific HIF-1a HIF-2α−/− mice | Endothelial HIF-1 and HIF-2 were inhibited | Impair kidney recovery and worsen renal fibrosis | Activating VCAM1 | [21] |
| uIRI in mice | EC-specific HIF-1α−/− or HIF2α−/− mice | Endothelial HIF-1 or HIF-2 was inhibited | Inactivation of endothelial HIF-2 but not HIF-1 impairs kidney recovery | Activating VCAM1 | [21] |
| Cisplatin-AKI in mice | Lentivirus-mediated HIF-1α-transfected hASCs | HIF-1 was activated | Attenuate AKI | Upregulating HO-1 | [116] |
| LPS-AKI in mice | EPO | HIF-1 was inhibited | Attenuate AKI | Promoting angiogenesis | [122] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shu, S.; Wang, Y.; Zheng, M.; Liu, Z.; Cai, J.; Tang, C.; Dong, Z. Hypoxia and Hypoxia-Inducible Factors in Kidney Injury and Repair. Cells 2019, 8, 207. https://doi.org/10.3390/cells8030207
Shu S, Wang Y, Zheng M, Liu Z, Cai J, Tang C, Dong Z. Hypoxia and Hypoxia-Inducible Factors in Kidney Injury and Repair. Cells. 2019; 8(3):207. https://doi.org/10.3390/cells8030207
Chicago/Turabian StyleShu, Shaoqun, Ying Wang, Meiling Zheng, Zhiwen Liu, Juan Cai, Chengyuan Tang, and Zheng Dong. 2019. "Hypoxia and Hypoxia-Inducible Factors in Kidney Injury and Repair" Cells 8, no. 3: 207. https://doi.org/10.3390/cells8030207
APA StyleShu, S., Wang, Y., Zheng, M., Liu, Z., Cai, J., Tang, C., & Dong, Z. (2019). Hypoxia and Hypoxia-Inducible Factors in Kidney Injury and Repair. Cells, 8(3), 207. https://doi.org/10.3390/cells8030207