Knock Down of Plakophillin 2 Dysregulates Adhesion Pathway through Upregulation of miR200b and Alters the Mechanical Properties in Cardiac Cells

 , , ,
, , , 
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Immunostaining
2.2. Morphology and Mechanical Properties Assessment Using Atomic Force Microscopy
2.3. mRNA and microRNA Targets Analysis
2.4. Quantitative PCR
2.5. Lentiviral Vectors
2.6. Cell–ECM Interaction Measured by AFM
2.7. Statistics
3. Results
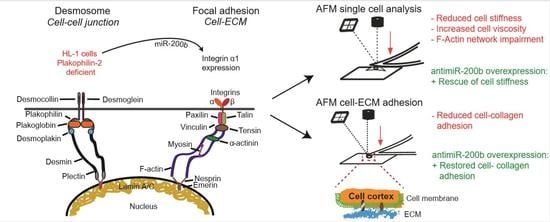
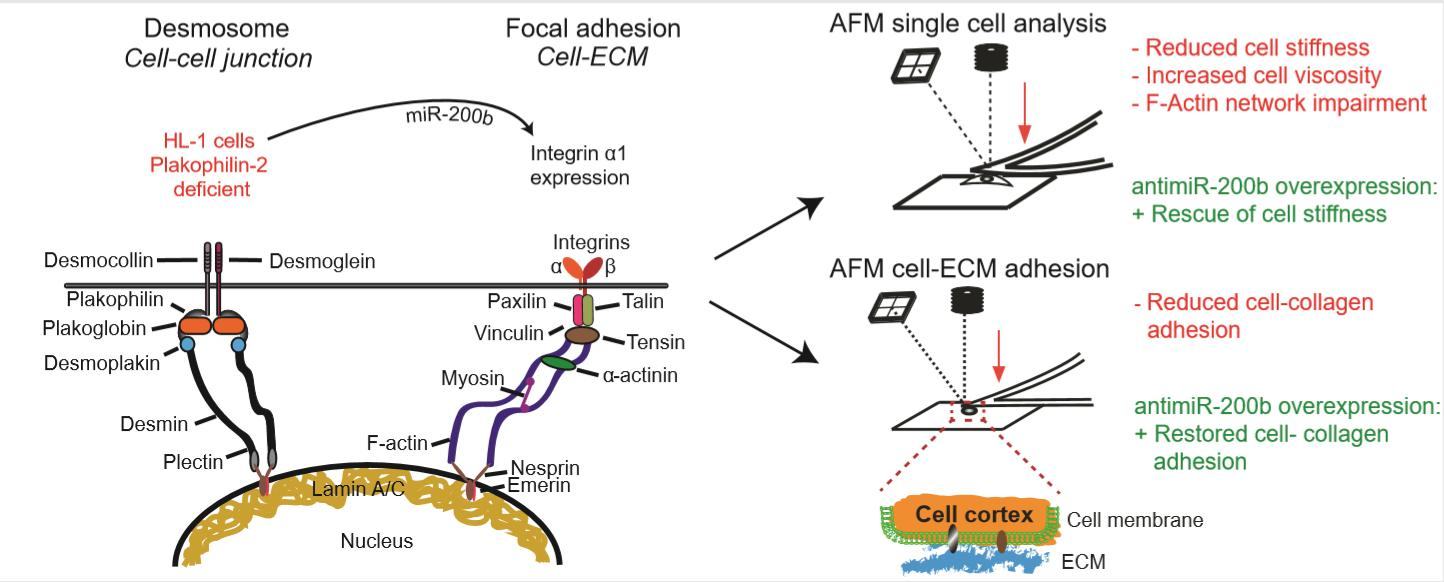
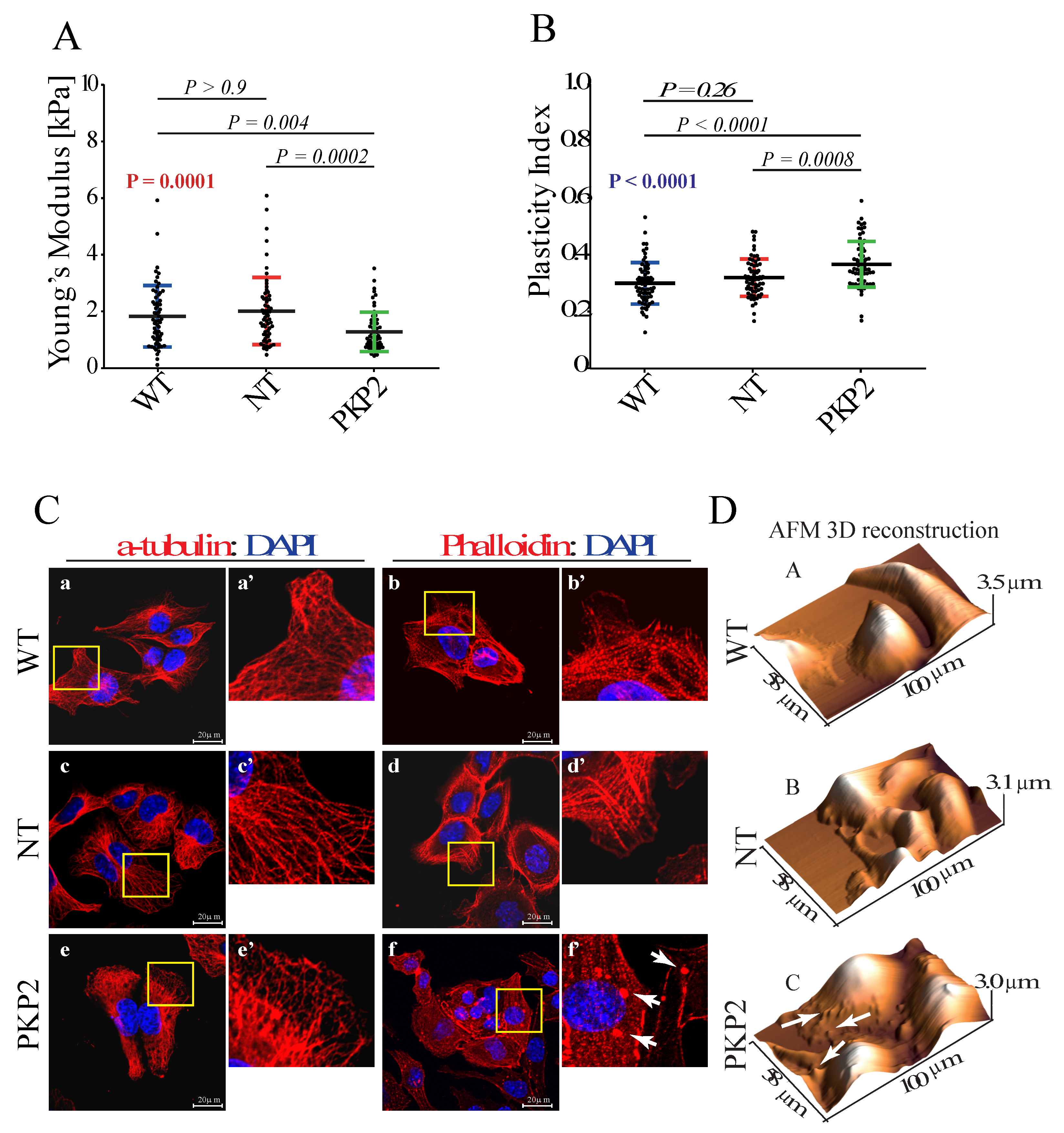
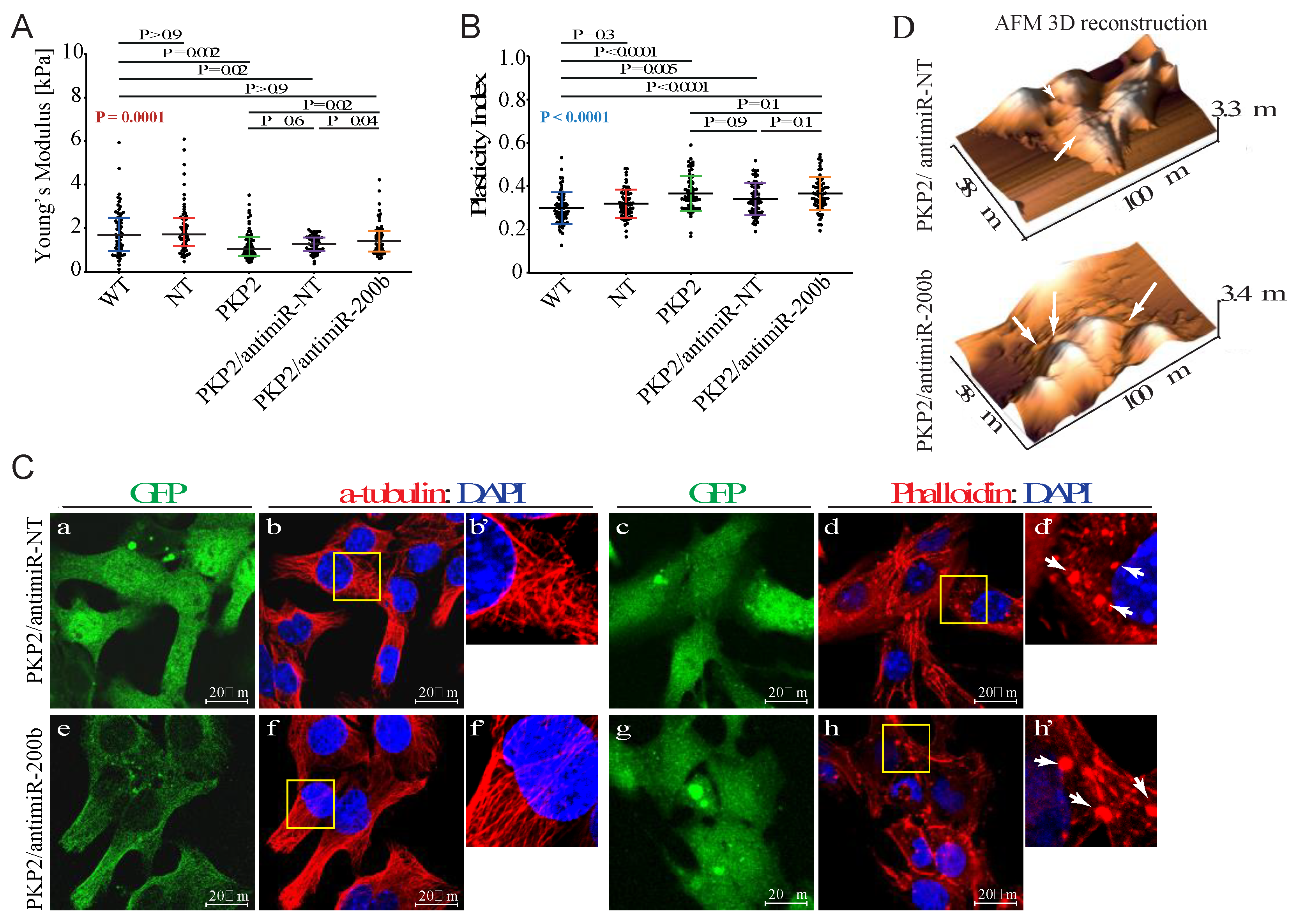
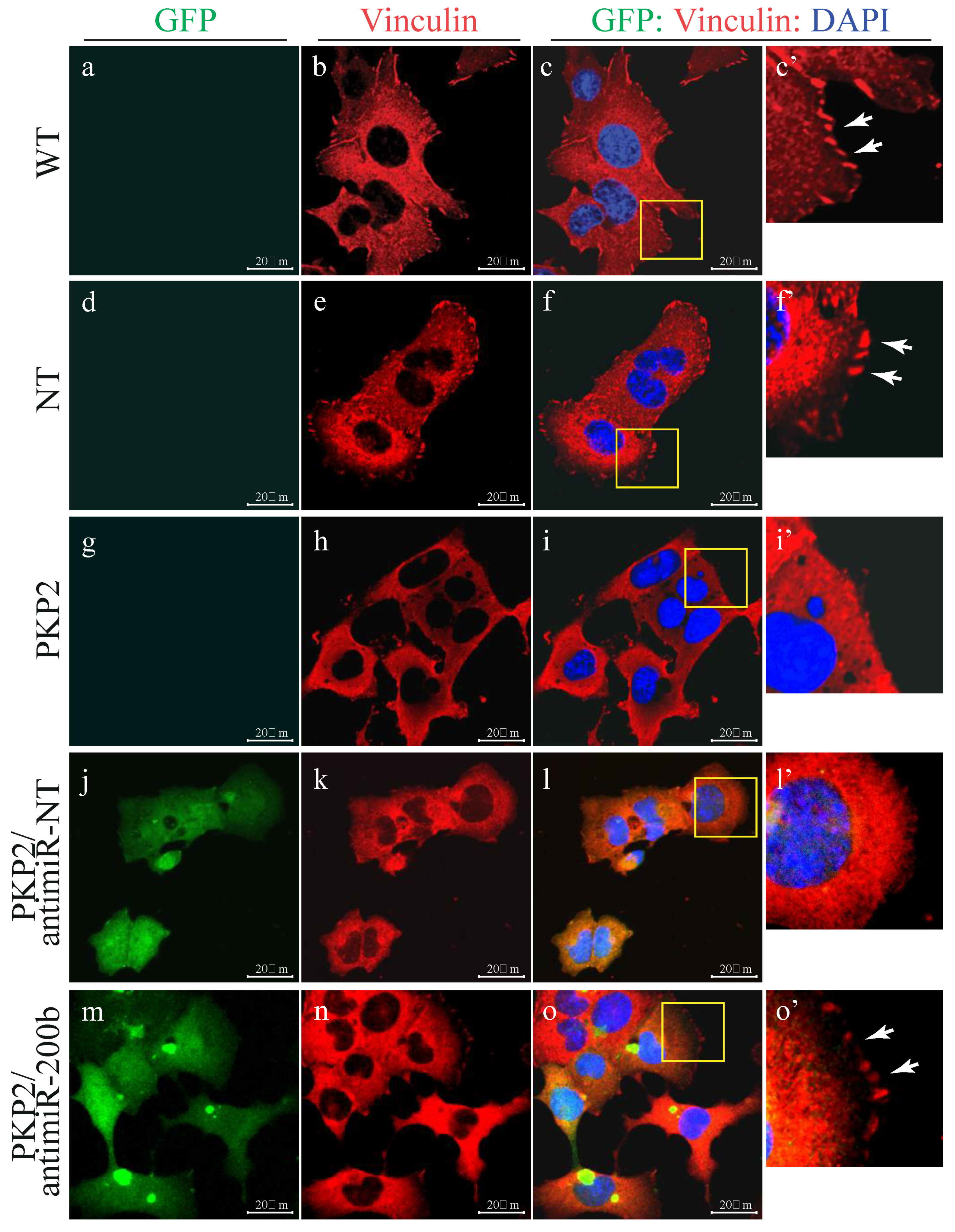
3.1. Knock Down of PKP-2 Affects Mechanical Properties and Morphology of HL-1 Cells
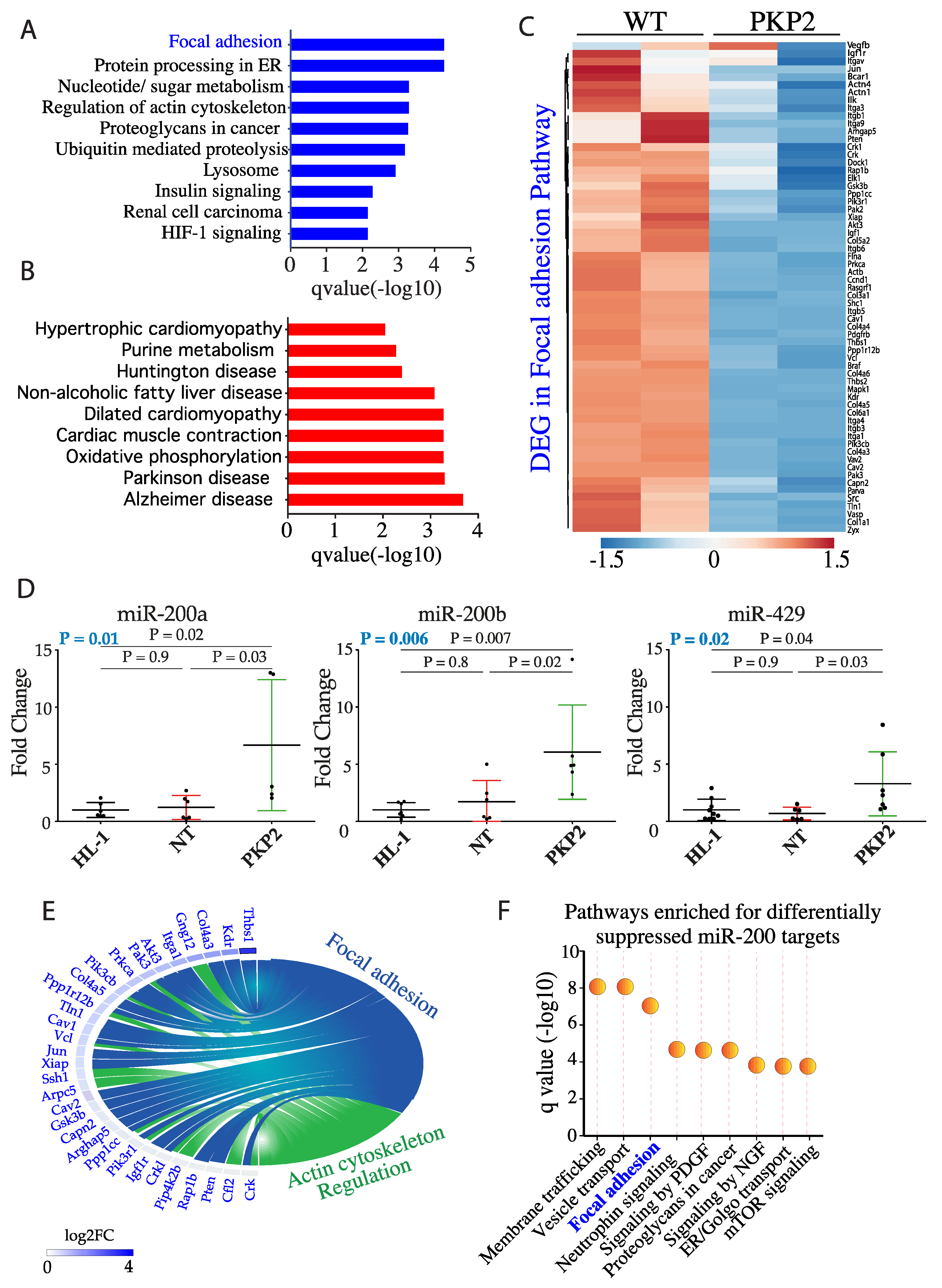
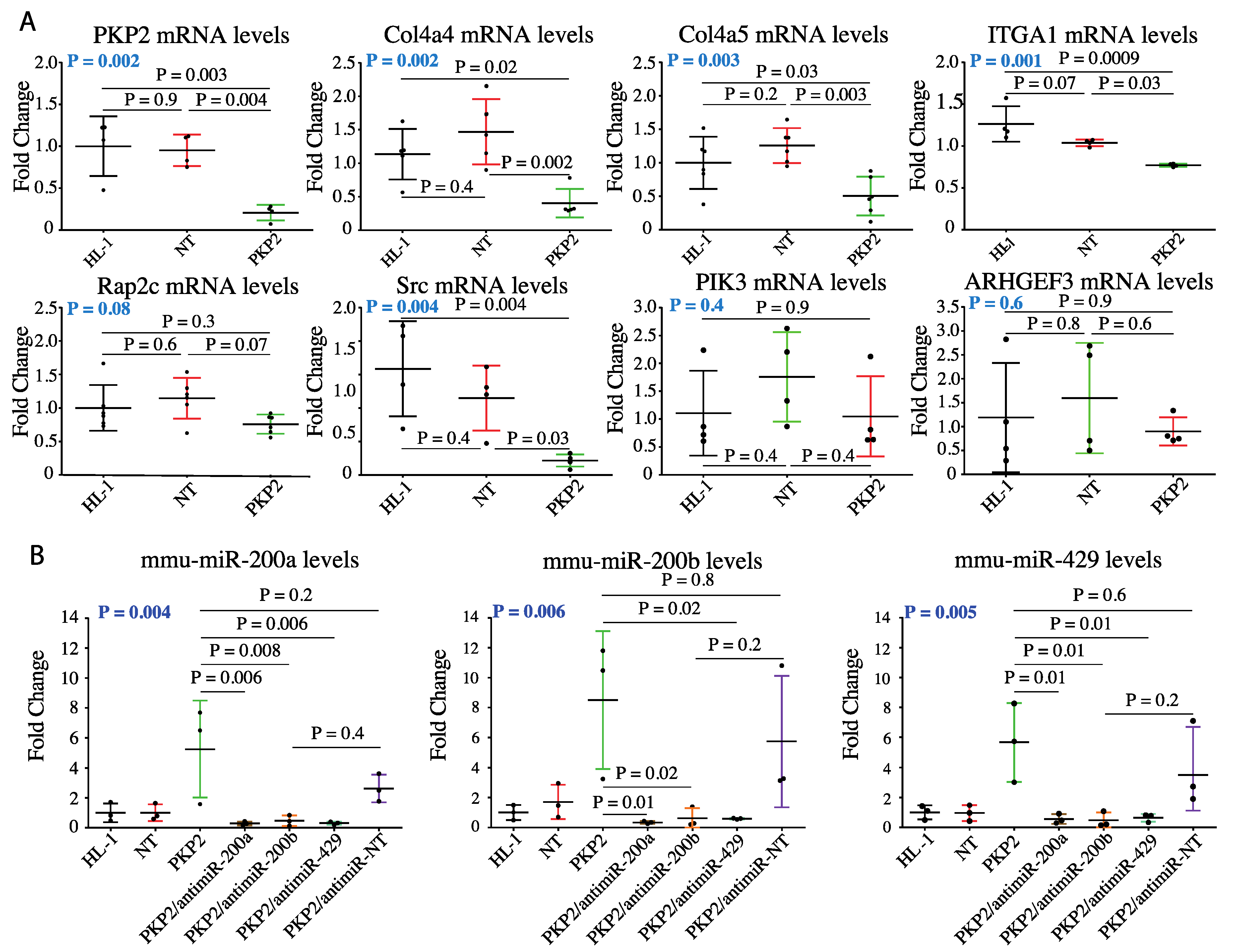
3.2. PKP-2 Knockdown Affects Focal Adhesion Pathway
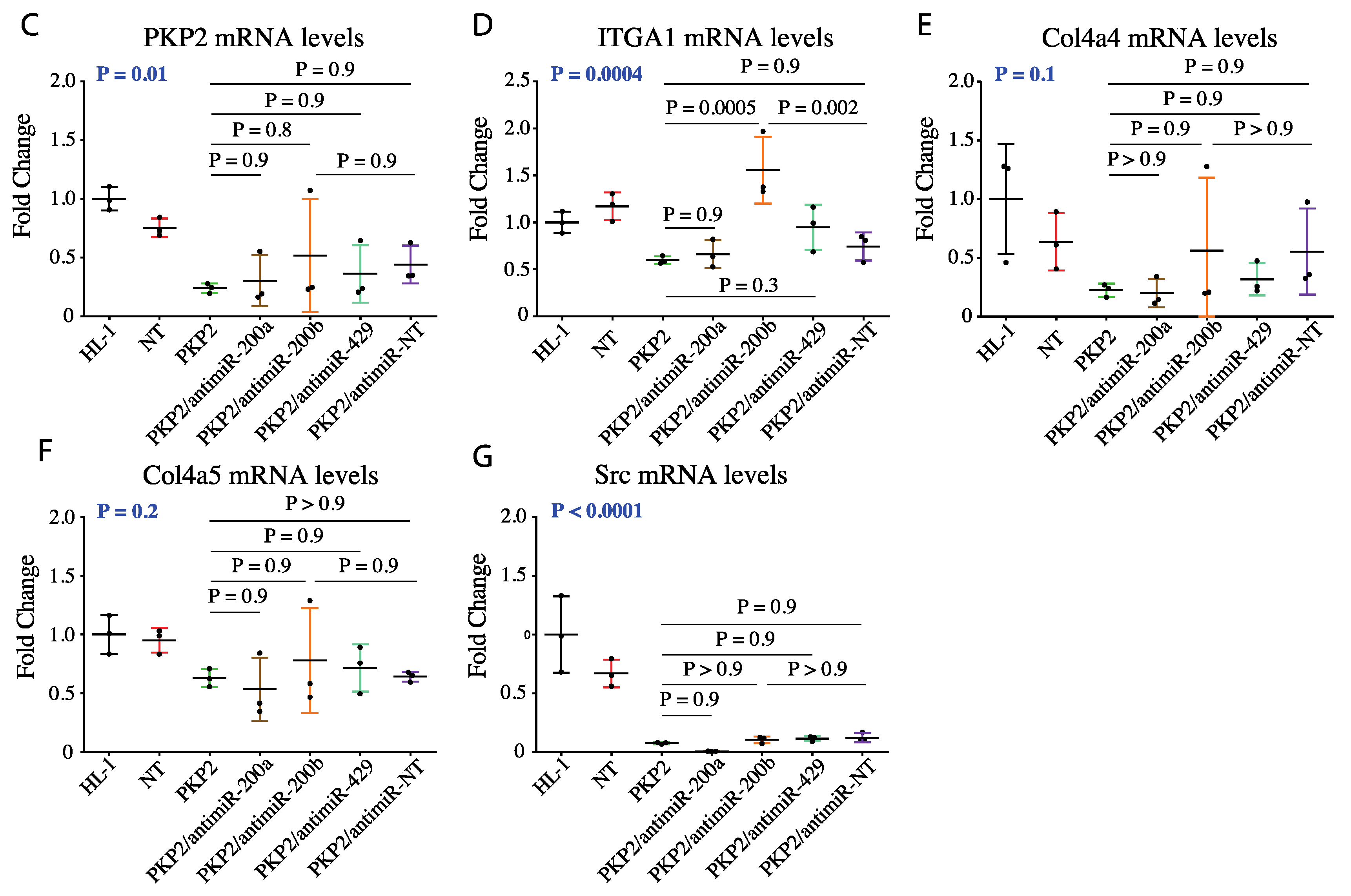
3.3. DEGs in the Focal Adhesion Pathways are Targets of miR200b
3.4. Knock Down of miR200b Rescued Itga1 Levels
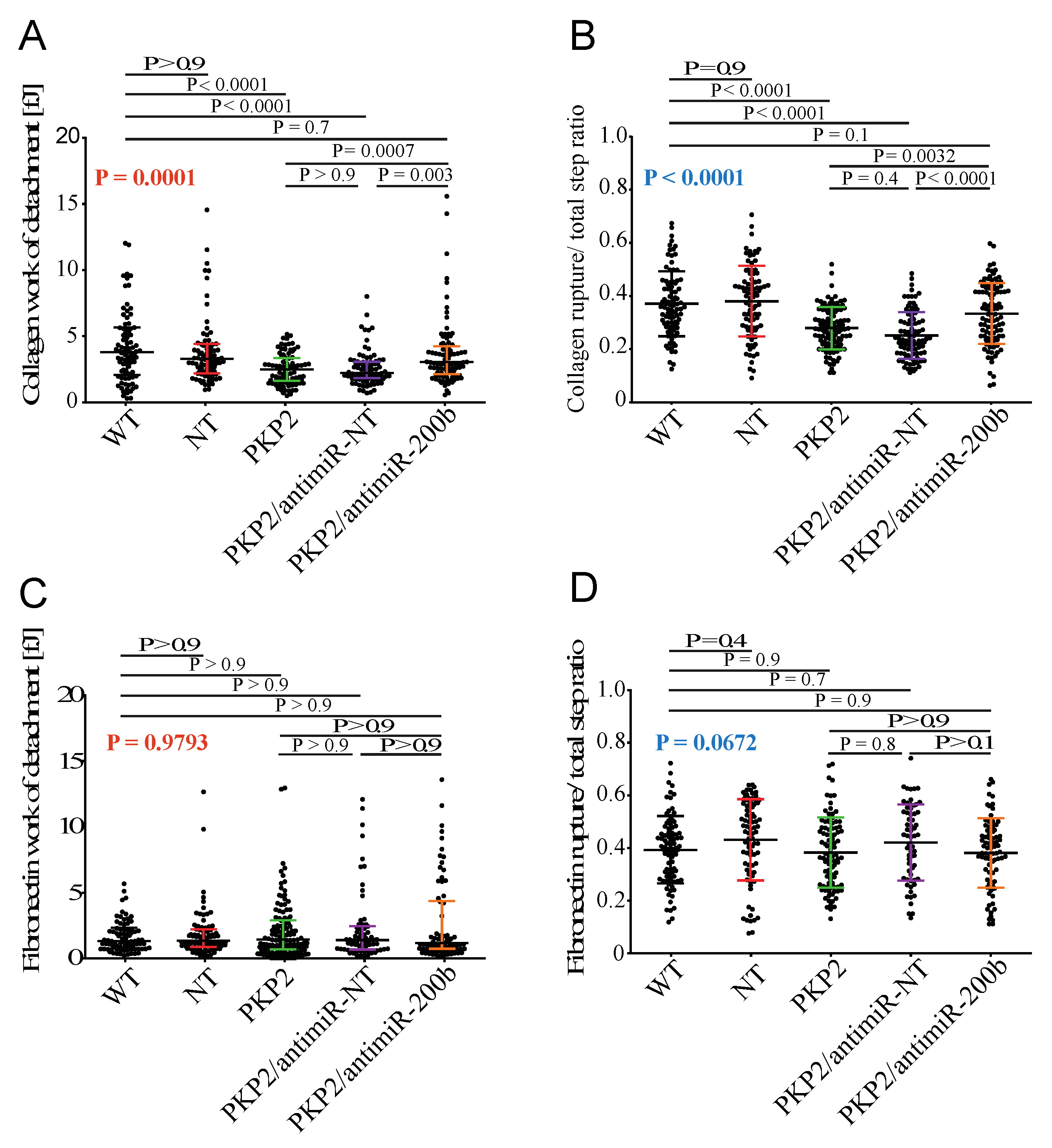
3.5. Downregulation of miR-200b Partially Rescues Impaired Mechanical Properties of PKP-2 Deficient HL-1 Cells
3.6. Downregulation of miR-200b Rescues Cell–ECM Adhesion in the HL-1PKP2-shRNA Cells
4. Discussion
Study Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fournier, A.K.; Campbell, L.E.; Castagnino, P.; Liu, W.F.; Chung, B.M.; Weaver, V.M.; Chen, C.S.; Assoian, R.K. Rac-dependent cyclin D1 gene expression regulated by cadherin- and integrin-mediated adhesion. J. Cell Sci. 2008, 121, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Koetsier, J.L.; Amargo, E.V.; Todorovic, V.; Green, K.J.; Godsel, L.M. Plakophilin 2 affects cell migration by modulating focal adhesion dynamics and integrin protein expression. J. Investig. Dermatol. 2014, 134, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Rico, C.; Pincet, F.; Thiery, J.P.; Dufour, S. Integrins stimulate E-cadherin-mediated intercellular adhesion by regulating Src-kinase activation and actomyosin contractility. J. Cell Sci. 2010, 123, 712–722. [Google Scholar] [CrossRef] [PubMed]
- Sakai, T.; Larsen, M.; Yamada, K.M. Fibronectin requirement in branching morphogenesis. Nature 2003, 423, 876–881. [Google Scholar] [CrossRef]
- Wang, H.; Radjendirane, V.; Wary, K.K.; Chakrabarty, S. Transforming growth factor beta regulates cell-cell adhesion through extracellular matrix remodeling and activation of focal adhesion kinase in human colon carcinoma Moser cells. Oncogene 2004, 23, 5558–5561. [Google Scholar] [CrossRef]
- Corrado, D.; Basso, C.; Judge, D.P. Arrhythmogenic Cardiomyopathy. Circ. Res. 2017, 121, 784–802. [Google Scholar] [CrossRef]
- Garcia-Gras, E.; Lombardi, R.; Giocondo, M.J.; Willerson, J.T.; Schneider, M.D.; Khoury, D.S.; Marian, A.J. Suppression of canonical Wnt/beta-catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy. J. Clin. Investig. 2006, 116, 2012–2021. [Google Scholar] [CrossRef]
- Chen, S.N.; Gurha, P.; Lombardi, R.; Ruggiero, A.; Willerson, J.T.; Marian, A.J. The hippo pathway is activated and is a causal mechanism for adipogenesis in arrhythmogenic cardiomyopathy. Circ. Res. 2014, 114, 454–468. [Google Scholar] [CrossRef]
- Gurha, P.; Chen, X.; Lombardi, R.; Wilerson, J.; Marian, A.J. Knock Down of Plakophilin 2 Downregulates miR-184 Through CpG Hypermethylation and Suppression of the E2F1 Pathway and Leads to Enhanced Adipogenesis In Vitro. Circ. Res. 2016, 119, 731–750. [Google Scholar] [CrossRef]
- Claycomb, W.C.; Lanson, N.A., Jr.; Stallworth, B.S.; Egeland, D.B.; Delcarpio, J.B.; Bahinski, A.; Izzo, N.J., Jr. HL-1 cells: A cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc. Natl. Acad. Sci. UAS 1998, 95, 2979–2984. [Google Scholar] [CrossRef]
- Puzzi, L.; Borin, D.; Martinelli, V.; Mestroni, L.; Kelsell, D.P.; Sbaizero, O. Cellular biomechanics impairment in keratinocytes is associated with a C-terminal truncated desmoplakin: An atomic force microscopy investigation. Micron 2018, 106, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Borin, D.; Puzzi, L.; Martinelli, V.; Cibinel, M.; Lapasin, R.; Sbaizero, O. An engineering insight into the relationship of selective cytoskeletal impairment and biomechanics of HeLa cells. Micron 2017, 102, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Matzke, R.; Jacobson, K.; Radmacher, M. Direct, high-resolution measurement of furrow stiffening during division of adherent cells. Nat. Cell Biol. 2001, 3, 607–610. [Google Scholar] [CrossRef] [PubMed]
- Hermanowicz, P.; Sarna, M.; Burda, K.; Gabrys, H. AtomicJ: An open source software for analysis of force curves. Rev. Sci. Instrum. 2014, 85, 063703. [Google Scholar] [CrossRef]
- Klymenko, O.; Wiltowska-Zuber, J.; Lekka, M.; Kwiatek, W.M. Energy Dissipation in the AFM Elasticity Measurements. Acta Phys. Pol. A 2009, 115, 4. [Google Scholar] [CrossRef]
- Agarwal, V.; Bell, G.W.; Nam, J.W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015, 4, e05005. [Google Scholar] [CrossRef]
- Li, J.H.; Liu, S.; Zhou, H.; Qu, L.H.; Yang, J.H. starBase v2.0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014, 42, D92–D97. [Google Scholar] [CrossRef]
- Dweep, H.; Sticht, C.; Pandey, P.; Gretz, N. miRWalk--database: Prediction of possible miRNA binding sites by “walking” the genes of three genomes. J. Biomed. Inform. 2011, 44, 839–847. [Google Scholar] [CrossRef]
- Bracken, C.P.; Li, X.; Wright, J.A.; Lawrence, D.M.; Pillman, K.A.; Salmanidis, M.; Anderson, M.A.; Dredge, B.K.; Gregory, P.A.; Tsykin, A.; et al. Genome-wide identification of miR-200 targets reveals a regulatory network controlling cell invasion. EMBO J. 2014, 33, 2040–2056. [Google Scholar] [CrossRef]
- Bharadwaj, M.; Strohmeyer, N.; Colo, G.P.; Helenius, J.; Beerenwinkel, N.; Schiller, H.B.; Fassler, R.; Muller, D.J. alphaV-class integrins exert dual roles on alpha5beta1 integrins to strengthen adhesion to fibronectin. Nat. Commun. 2017, 8, 14348. [Google Scholar] [CrossRef]
- Andolfi, L.; Bourkoula, E.; Migliorini, E.; Palma, A.; Pucer, A.; Skrap, M.; Scoles, G.; Beltrami, A.P.; Cesselli, D.; Lazzarino, M. Investigation of adhesion and mechanical properties of human glioma cells by single cell force spectroscopy and atomic force microscopy. PLoS ONE 2014, 9, e112582. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.; Celik, E.; Rico, F.; Moy, V.T. Elongated membrane tethers, individually anchored by high affinity alpha4beta1/VCAM-1 complexes, are the quantal units of monocyte arrests. PLoS ONE 2013, 8, e64187. [Google Scholar]
- Friedrichs, J.; Helenius, J.; Muller, D.J. Quantifying cellular adhesion to extracellular matrix components by single-cell force spectroscopy. Nat. Protoc. 2010, 5, 1353–1361. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, B.D.; Grashoff, C.; Schwartz, M.A. Dynamic molecular processes mediate cellular mechanotransduction. Nature 2011, 475, 316–323. [Google Scholar] [CrossRef]
- Grashoff, C.; Hoffman, B.D.; Brenner, M.D.; Zhou, R.; Parsons, M.; Yang, M.T.; McLean, M.A.; Sligar, S.G.; Chen, C.S.; Ha, T.; et al. Measuring mechanical tension across vinculin reveals regulation of focal adhesion dynamics. Nature 2010, 466, 263–266. [Google Scholar] [CrossRef]
- Huveneers, S.; Oldenburg, J.; Spanjaard, E.; van der Krogt, G.; Grigoriev, I.; Akhmanova, A.; Rehmann, H.; de Rooij, J. Vinculin associates with endothelial VE-cadherin junctions to control force-dependent remodeling. J. Cell Biol. 2012, 196, 641–652. [Google Scholar] [CrossRef]
- Matthews, B.D.; Overby, D.R.; Mannix, R.; Ingber, D.E. Cellular adaptation to mechanical stress: Role of integrins, Rho, cytoskeletal tension and mechanosensitive ion channels. J. Cell Sci. 2006, 119, 508–518. [Google Scholar] [CrossRef]
- Ingber, D.E. Tensegrity and mechanotransduction. J. Bodyw. Mov. 2008, 12, 198–200. [Google Scholar] [CrossRef]
- Stamenovic, D. Effects of cytoskeletal prestress on cell rheological behavior. Acta Biomater. 2005, 1, 255–262. [Google Scholar] [CrossRef]
- Vang, S.; Corydon, T.J.; Børglum, A.D.; Scott, M.D.; Frydman, J.; Mogensen, J.; Gregersen, N.; Bross, P. Actin mutations in hypertrophic and dilated cardiomyopathy cause inefficient protein folding and perturbed filament formation. FEBS J. 2005, 272, 2037–2049. [Google Scholar] [CrossRef]
- Haas, R.H. Mitochondrial Dysfunction in Aging and Diseases of Aging. Biology 2019, 8, 48. [Google Scholar] [CrossRef] [PubMed]
- Yuan, B.; Wan, P.; Chu, D.; Nie, J.; Cao, Y.; Luo, W.; Lu, S.; Chen, J.; Yang, Z. A cardiomyocyte-specific Wdr1 knockout demonstrates essential functional roles for actin disassembly during myocardial growth and maintenance in mice. Am. J. Pathol. 2014, 184, 1967–1980. [Google Scholar] [CrossRef] [PubMed]
- Stanley, B.A.; Graham, D.R.; James, J.; Mitsak, M.; Tarwater, P.M.; Robbins, J.; Van Eyk, J.E. Altered myofilament stoichiometry in response to heart failure in a cardioprotective α-myosin heavy chain transgenic rabbit model. Proteom. Clin. Appl. 2011, 5, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Grossmann, K.S.; Grund, C.; Huelsken, J.; Behrend, M.; Erdmann, B.; Franke, W.W.; Birchmeier, W. Requirement of plakophilin 2 for heart morphogenesis and cardiac junction formation. J. Cell Biol. 2004, 167, 149–160. [Google Scholar] [CrossRef]
- Wang, N.; Ingber, D.E. Control of cytoskeletal mechanics by extracellular matrix, cell shape, and mechanical tension. Biophys. J. 1994, 66, 2181–2189. [Google Scholar] [CrossRef]
- Shah, P.; Bristow, M.R.; Port, J.D. MicroRNAs in Heart Failure, Cardiac Transplantation, and Myocardial Recovery: Biomarkers with Therapeutic Potential. Curr. Heart Fail. Rep. 2017, 14, 454–464. [Google Scholar] [CrossRef]
- Ungewiss, C.; Rizvi, Z.H.; Roybal, J.D.; Peng, D.H.; Gold, K.A.; Shin, D.H.; Creighton, C.J.; Gibbons, D.L. The microRNA-200/Zeb1 axis regulates ECM-dependent beta1-integrin/FAK signaling, cancer cell invasion and metastasis through CRKL. Sci. Rep. 2016, 6, 18652. [Google Scholar] [CrossRef]
- Zhang, H.F.; Zhang, K.; Liao, L.D.; Li, L.Y.; Du, Z.P.; Wu, B.L.; Wu, J.Y.; Xu, X.E.; Zeng, F.M.; Chen, B.; et al. miR-200b suppresses invasiveness and modulates the cytoskeletal and adhesive machinery in esophageal squamous cell carcinoma cells via targeting Kindlin-2. Carcinogenesis 2014, 35, 292–301. [Google Scholar] [CrossRef]
- Carl, P.; Schillers, H. Elasticity measurement of living cells with an atomic force microscope: Data acquisition and processing. Pflug. Arch. Eur. J. Physiol. 2008, 457, 551–559. [Google Scholar] [CrossRef]
- Guz, N.; Dokukin, M.; Kalaparthi, V.; Sokolov, I. If cell mechanics can be described by elastic modulus: Study of different models and probes used in indentation experiments. Biophys. J. 2014, 107, 564–575. [Google Scholar] [CrossRef]
- Dokukin, M.; Ablaeva, Y.; Kalaparthi, V.; Seluanov, A.; Gorbunova, V.; Sokolov, I. Pericellular Brush and Mechanics of Guinea Pig Fibroblast Cells Studied with AFM. Biophys. J. 2016, 111, 236–246. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sokolov, I.; Dokukin, M.E. AFM Indentation Analysis of Cells to Study Cell Mechanics and Pericellular Coat. Methods Mol. Biol. 2018, 1814, 449–468. [Google Scholar] [PubMed]
- Kossivas, F.; Michaelides, M.; Hadjisavvas, A.; Kyprianou, A.; Kyriacou, K.; Constantinides, C. Elasticity Measurements from Left Ventricular Murine HL-1 Cardiomyocytes using Atomic Force Microscopy. FASEB J. 2015, 29, 799.8. [Google Scholar]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Puzzi, L.; Borin, D.; Gurha, P.; Lombardi, R.; Martinelli, V.; Weiss, M.; Andolfi, L.; Lazzarino, M.; Mestroni, L.; Marian, A.J.; et al. Knock Down of Plakophillin 2 Dysregulates Adhesion Pathway through Upregulation of miR200b and Alters the Mechanical Properties in Cardiac Cells. Cells 2019, 8, 1639. https://doi.org/10.3390/cells8121639
Puzzi L, Borin D, Gurha P, Lombardi R, Martinelli V, Weiss M, Andolfi L, Lazzarino M, Mestroni L, Marian AJ, et al. Knock Down of Plakophillin 2 Dysregulates Adhesion Pathway through Upregulation of miR200b and Alters the Mechanical Properties in Cardiac Cells. Cells. 2019; 8(12):1639. https://doi.org/10.3390/cells8121639
Chicago/Turabian StylePuzzi, Luca, Daniele Borin, Priyatansh Gurha, Raffaella Lombardi, Valentina Martinelli, Marek Weiss, Laura Andolfi, Marco Lazzarino, Luisa Mestroni, Ali J. Marian, and et al. 2019. "Knock Down of Plakophillin 2 Dysregulates Adhesion Pathway through Upregulation of miR200b and Alters the Mechanical Properties in Cardiac Cells" Cells 8, no. 12: 1639. https://doi.org/10.3390/cells8121639
APA StylePuzzi, L., Borin, D., Gurha, P., Lombardi, R., Martinelli, V., Weiss, M., Andolfi, L., Lazzarino, M., Mestroni, L., Marian, A. J., & Sbaizero, O. (2019). Knock Down of Plakophillin 2 Dysregulates Adhesion Pathway through Upregulation of miR200b and Alters the Mechanical Properties in Cardiac Cells. Cells, 8(12), 1639. https://doi.org/10.3390/cells8121639